
Abstract
Peptidases and esterases are hydrolytic enzymes; 2–3% of all gene products are assigned to this group alone. They are therefore, an important group of target proteins for the design of new medicines and have a special importance for structure-based drug design. This is reflected in the fact that about 14% of all known human peptidases are presently being investigated as possible target structures for drug therapy.
You have full access to this open access chapter, Download reference work entry PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Peptidases and esterases are hydrolytic enzymes; 2–3% of all gene products are assigned to this group alone. They are therefore, an important group of target proteins for the design of new medicines and have a special importance for structure-based drug design. This is reflected in the fact that about 14% of all known human peptidases are presently being investigated as possible target structures for drug therapy.
The function of these enzymes is the cleavage of peptide or ester bonds for which a nucleophile is needed for the attack on the carbonyl group of the amide or ester bond to be cleaved. A large number of proteins use the OH or SH groups of a serine, threonine, or cysteine for this purpose. In the following chapters, we will see other cleaving enzymes that use a different mechanism. During the cleavage reaction of the hydrolases discussed in this chapter, a temporary covalent bond between substrate and enzyme is formed. This intermediate, the so-called acyl–enzyme form, occurs with serine, threonine, and cysteine proteases, but lipases, esterases, transpeptidases, and β-lactamases also use this reaction mechanism. The design of inhibitors for these enzymes that act via an acyl–enzyme intermediate shall be discussed. In the following two chapters, peptidases that use a water molecule for the primary attack on the peptide bond to be hydrolyzed shall be discussed: the aspartic and metallopeptidases. Depending on whether they cleave the amino acid chain at the N or C terminus or in the center, the peptidases are classified as amino-, carboxy-, or endopeptidases. Some of these proteases are relatively unspecific, whereas others are highly specific and only cleave very particular substrates. These latter enzymes have the best chances that a selective therapeutic inhibitor can be found causing only few side effects. Bacteria and viruses have also produced their own peptidases, the inhibition of which can be exploited for chemotherapeutic treatment. Because these proteins are not endogenous in humans, and therefore also have no function in us, their inhibition should lead to therapeutic success without risking severe side effects.
1 Serine-Dependent Hydrolases
Serine proteases are the most extensive and best-studied class of peptidases. They are closely related to the esterases and lipases (hydrolases) that hydrolyze ester bonds. This enzyme class serves the human body in diverse ways. Some serine proteases, such as, the digestive enzymes trypsin and chymotrypsin, cleave a broad spectrum of peptides and proteins. Others such as the coagulation enzymes thrombin and factor Xa are highly selective and only cleave very particular substrates. Frequently, proteases are expressed in a non-active precursor form, the so-called zymogens. To transform these into their active form, in many cases sequence segments of the zymogen polypeptide chain are cleaved that otherwise serve as endogenous inhibitors of the activated enzyme. The release of the active form can either occur by autocatalysis (e.g., trypsin) or by other activating proteases (e.g., the coagulation cascade). An active site serine side chain plays a decisive role in the catalytic mechanism of serine proteases, esterases, and lipases. It is characterized by an extraordinarily high chemical reactivity. In chymotrypsin, only this serine reacts with diisopropylfluorophosphate (DFP), whereas 27 other serine residues in the enzyme remain unmodified. Upon chemical transformation with DFP, the enzyme completely loses its catalytic activity.
2 Structure and Function of Serine Proteases
The digestive enzyme chymotrypsin was the first serine protease for which the 3D structure was determined, by David Blow in Cambridge, England. The numbering of the amino acids in serine proteases of the chymotrypsin type is based on the sequence of chymotrypsin. The spatial structures of a large variety of serine proteases are now available, of which a few are listed in Table 23.1. The structures show an extraordinarily pronounced similarity in the active site even for proteases that have entirely different folding patterns (Sect. 14.7, compare trypsin with subtilisin). This so-called catalytic triad of Ser–His–Asp is characteristic of serine proteases. In some of these enzymes, the aspartate can be replaced by a glutamate whereas some transpeptidases and β-lactamases display a lysine in place of the histidine in the active site.
As these three amino acids are very far apart from one another in the sequence, the protein must fold appropriately to bring the three side chains into spatial proximity to one another. The catalytic serine, found at position 195 in the trypsin-like proteases, carries out the actual attack on the amide bond that is being cleaved (Fig.23.1). The oxygen atom of an unactivated hydroxyl group would not be reactive enough for this step. Its nucleophilicity which describes its tendency to attack an electron-poor carbonyl carbon atom, is enhanced by the neighboring histidine side chain. The imidazole side chain of this histidine can accept a proton from the serine hydroxyl group, enabling a nucleophilic attack of the now negatively charged oxygen atom on the partially positively charged carbon atom of the amide carbonyl group. The neighboring aspartate can accept a proton from the histidine imidazole ring, and release it again. In this way, it compensates the positive charge that is formed on the histidine residue. To stabilize the transition state formed upon attack on the carbonyl group, serine proteases have another characteristic structural motif, the so-called oxyanion hole. This is a small pocket next to the side chain of Ser195 composed of two main-chain NH groups (Fig.23.1). In a few cases, the terminal amide groups of asparagine or glutamine can accomplish this task. The function of the oxyanion hole is to stabilize the negative charge formed on the tetrahedral transition state and to distort the geometry of the attacked carbonyl carbon atom from a trigonal-planar to a tetrahedral configuration. The formed transition state collapses with release of the C-terminal cleavage product which carries a free amino group at its end. The N-terminal cleavage product remains covalently bound to the protease to give an acyl–enzyme intermediate. In a subsequent step, a nucleophilic attack by a water molecule again leads to a tetrahedral transition state. This finally collapses with release of the N-terminal cleavage product. The catalytic enzyme is then ready for the next transformation.


Catalytic mechanism of serine proteases. (a) The peptide substrate binds to the enzyme in specific pockets on either side of the cleavage site. (b) The oxygen atom of the serine side chain carries out a nucleophilic attack. This is fostered by the neighboring histidine side chain, which, supported by an aspartate residue, accepts a proton from the hydroxyl group. (c) The transition state collapses with formation of an acyl–enzyme intermediate. (d) This is hydrolyzed by the attack of a water molecule to release the N-terminal cleavage product.
What happens if the amino acids serine, histidine, and aspartic acid of the catalytic triad of a serine protease are individually or collectively exchanged for amino acids without similar functional groups? In 1988, Paul Carter and James Wells prepared various mutants of the bacterial serine protease subtilisin (Sect. 14.7) at Genentech. Exchange of the catalytic serine or histidine for alanine leads to a reduction in the catalytic activity by more than six orders of magnitude. Surprisingly, exchange of the aspartic acid, the only function of which is to exchange a proton with histidine, reduced the catalytic activity by more than four orders of magnitude. The combined exchange of multiple amino acids of the catalytic triad led to no further reduction in the catalytic activity. The threefold-alanine mutant, in which the catalytic triad is completely removed, still cleaves the peptide substrate more than 1,000 times faster than the pure buffer solution! The substrate remaining binding sites and the oxyanion hole, the structure and properties of which stabilize the tetrahedral transition state, are responsible for this acceleration.
Now it is certainly not difficult to destroy the binding site of an enzyme or its catalytic activity. It is, however, more difficult to purposefully alter its specificity or function. The subtilisin mutants in which the histidine was exchanged for an alanine, cleave substrates with the sequence -Phe–Ala–X–Phe- (X = Ala or Gln, for example) six orders of magnitude more slowly than the unaltered subtilisin with one exception: A substrate with the sequence -Phe–Ala–His–Phe- is cleaved only four orders of magnitude more slowly. The histidine of the substrate takes over the role of the histidine in the catalytic site to a certain extent! This process is called substrate-supported catalysis. The transformation is indeed still rather slow, but the specificity of this mutant is distinctly enhanced: The -Phe–Ala–His–Phe- sequence is cleaved 200 times faster than any of the other -Phe–Ala–X–Phe- sequences.
3 The S1 Pocket of Serine Proteases Determines Specificity
Proteases recognize polypeptide chains as substrates. For this task they use a series of more-or-less-pronounced binding pockets on their surface, as described in Chap. 14, “Three-Dimensional Structure of Biomolecules”. These are structurally and electronically complementary to the side chains of the substrate. As a consequence, the polypeptide chain of the substrate will be immobilized on the surface in the vicinity of the catalytic site. The crevices on the surface look very different depending on the protease. Surface portions of four different serine proteases from the trypsin family are shown in Fig.23.2. A comparison of the different serine proteases with different substrate specificities (Fig.23.3) shows that, above all, the structures of the S1 pockets of these enzymes are different. The S1 pocket is largely formed of the sequence segments from 189–195 and 214–220. Significant differences are unique to the side chains of the amino acid at the positions 189, 216, and 226. In chymotrypsin, these are Ser189, Gly216, and Gly226. They tailor the depth and form of this pocket to accommodate the aromatic side chains of the amino acids phenylalanine, tyrosine, and tryptophan. Correspondingly, chymotrypsin preferentially cleaves peptide chains after one of these three amino acids. Trypsin also has a deep, spacious S1 pocket that is flanked by Gly216 and Gly226. The negatively charged carboxylate group of Asp189 on the floor of the pocket is decisive for the recognition of long, positively charged side chains in the amino acids lysine and arginine in the substrate. In elastase, the S1 pocket is shaped by the amino acids Val216 and Thr226. Because of this, the pocket is significantly smaller. It can only accommodate amino acids with short hydrophobic side chains such as alanine and valine. Amino acids with large groups are no longer accommodated. The amino acid 189, a serine, is buried. The substrate specificity of the described serine proteases is primarily achieved by the recognition of the amino acid in the P 1 position. The neighboring pockets, however, are also important for substrate binding and selectivity. It is remarkable that the substrate-binding pockets of serine proteases recognizing the N-terminal part of the substrate (unprimed side, S1–S4 pockets; Sect. 14.5) are more prominently established. The pockets on the unprimed side that anchors the C-terminal part of the substrate are much less well developed. Because the N-terminal cleavage product remains temporarily covalently bound to the protease as an acyl–enzyme complex, this part of the substrate is bound particularly selectively.


The surface of the trypsin-like serine proteases trypsin, thrombin, factor VIIa, and factor Xa display deep pockets in the area of the catalytic site. To emphasize this surface structure better, the color of the surface changes from blue to green to red with increasing depth. The exposed physicochemical properties in the crevices determine the substrate selectivity of the protease. The preferred cleavage sequences are indicated in the structures, in which XXX represents an arbitrary amino acid at this position.


Comparison of the S1 pockets of chymotrypsin, trypsin, and elastase. The binding pocket of chymotrypsin is tailored for large, lipophilic side chains. The S1 pocket of trypsin binds amino acids with positively charged side chains through its negatively charged Asp189 residue. Because of the spatial filling of the side chains of Thr216 and Val226, elastase has a relatively small S1 pocket and therefore binds small hydrophobic amino acids such as alanine and valine.
These structural characteristics establish how a conceivable competitive inhibitor of a serine protease should look: It is decisive that the S 1 pocket is filled as well as possible. The chemical constitution of the parts of the inhibitor that bind in this region must be complementary to the S1 pocket. In some cases, the occupancy of the S1 pocket alone is sufficient to generate a selective serine protease inhibitor with a respectable binding affinity. Accordingly, in 1967 Marcos Mares-Guia and Elliott Shaw described small-molecule trypsin inhibitors with micromolar binding affinity that only occupy the S1 pocket. It is not difficult to see that all the molecules 23.1–23.4 in Fig.23.4 imitate the basic amino acids arginine or lysine in the P1 position of the substrate.


The molecules 23.1–23.4 that bind in the S1 pocket of trypsin are micromolar inhibitors. All of these molecules contain a strongly basic group that is protonated under physiological conditions; therefore a positive charge is available to form a salt bridge to the negatively charged side chain of Asp189. The thrombin inhibitors 23.5 and 23.6 contain an additional functional group that can form a covalent bond to the catalytically active serine.
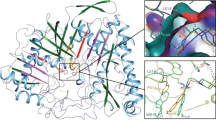
A first approach to the design of serine protease inhibitors could be based on the search for an appropriate group for the occupation of the S1 pocket, to then be coupled with a chemically reactive group that binds to the catalytic serine. The different groups that have been described in the literature for this purpose are summarized in Table 23.2. Even natural products follow this principle. The macrocyclic pentapeptide thrombin inhibitor cyclotheonamide A from the marine sponge Theonella sp. contains an α-keto function next to an amide bond. As the X-ray structure shows, this ketone group forms a tetrahedral hemiacetal structure with the OH group of the catalytic serine (Fig.23.5).


Crystal structure of the inhibitor cyclotheonamide with thrombin. The inhibitor forms a covalent bond to the catalytic serine with its α-keto group to form a hemiketal structure. The now negatively charged oxygen is stabilized by two hydrogen bonds in an oxyanion hole.
If the sequence of the peptide substrate of the serine protease is known, the N-terminal amino acid prior to the cleavage site can be coupled with one of the groups from Table 23.2 to produce a compound that will most likely be an inhibitor. An example of this is the elastase inhibitor N-(methylsuccinoyl)-Ala–Ala–Pro–Val-CF3 (23.21 in Fig.23.14), which is derived from the substrate sequence Pro–Val. In favorable cases, the P1 equivalent alone is sufficient, for example, in the trypsin and thrombin inhibitors 23.5 and 23.6, respectively (Fig.23.4). However, the usually high chemical reactivity of the functional groups in covalently binding serine protease inhibitors, which is necessary to interact with the catalytically active serine, can be problematic. Because of their reactivity, such groups can also undergo undesirable reactions with serine residues from other enzymes and therefore cause side effects. The design of highly potent and selective inhibitors requires at least the occupancy of the S2, S3, and S4 pockets. An additional structural characteristic that all serine proteases share in common should also be mentioned: Their substrates are bound to the peptide backbone via two antiparallel-oriented hydrogen bonds. This orientation of the two hydrogen-bonding partners leads to a pleated-sheet-like geometry. In most inhibitors, an attempt is made to imitate this hydrogen-bonding pattern (Fig.23.6).


General binding mode of a peptide chain that is to be cleaved (gray carbon atoms) in the catalytic site of a serine protease. The amide bond to be cleaved is shown in yellow. The substrate’s P1 (light-blue) and P2 groups (green) are shown with a surface; they bind in the S1 and S2 pockets of the protein. Two antiparallel-oriented hydrogen bonds (green) are formed to the main chain. The H-bonds to the oxyanion hole are in purple, and the direction of the nucleophilic attack of the Ser195 oxygen on the carbonyl carbon is indicated in blue.
4 Seeking Small-Molecule Thrombin Inhibitors
The serine protease thrombin plays a central role in the control of blood coagulation. Thrombin is at the end of a complex, highly regulated cascade of serine proteases. An injury to the arterial vascular system leads to the situation that membrane-bound tissue factor that is found outside the vessel comes into contact with the precursor of the serine protease, factor VII, in blood. The precursor is activated to factor VIIa, and induces the coagulation cascade. Different factors are released along the cascade, which are activated by proteases from the previous step from their zymogen form. Finally, the cascade leads to the release of “von Willebrand factor,” which binds to thrombocytes, and in doing so initiates the formation of a blood clot. In addition to extrinsic activation, there is also an intrinsic coagulation pathway. It is initiated by reduced blood flow or pathologically altered vasculature. In this case, the coagulation cascade is started to form a platelet aggregate, which is then stabilized by a fibrin network. Factor X is found in one of the last steps in which the two pathways merge. All of the different steps involve proteases that represent conceivable target structures for a drug therapy. Until now, in particular developments for the enzymes thrombin, factor Xa, and factor VIIa have been tackled. This has already led to development candidates and marketed products for the first two.
Thrombin transforms the inactive fibrinogen into reactive fibrin. It forms a polymer together with aggregated platelets in which different blood cells are trapped. A thrombus is formed, which is further cross-linked and stabilized by transglutaminase factor XIII (Sect. 23.8). This is an essential protective mechanism of the body to ensure wound closure. In particular diseases or situations, for example, after surgery, after a heart attack, or to prevent stroke in patients with atrial fibrillation, it is necessary to reduce the coagulation capacity of the blood. For this reason, there is great interest in the development of selective, and above all, orally available coagulation cascade inhibitors. Thrombin cleaves fibrinogen between the amino acids arginine and glycine. This sequence served as a starting point for the development of the first synthetic thrombin inhibitors that therefore possessed either an Arg or an Arg-analogous building block.
In this section, three different approaches for the development of thrombin inhibitors shall be presented: substrate analogues, benzamidine, and structurally significantly modified analogues.
One approach for the design of thrombin inhibitors is provided by the P3…P3′ substrate sequence Gly–Val–Arg–Gly–Pro–Arg of fibrinogen. In the early 1970s, the Japanese group of Hamao Umezawa established that peptide aldehydes with C-terminal arginine residues that are isolated from bacteria are potent inhibitors of some trypsin-like serine proteases. The tripeptide aldehydes that were investigated by Sándor Bajusz were derived from the amino acids P3–P1 or P3′–P1′, that are the three amino acids “before” and “after” the cleavage site. The relative binding affinity of a few peptide aldehydes are summarized in Table 23.3. Interestingly, the direct comparison of Gly–Val–Arg-H and Gly–Pro–Arg-H shows that a proline in the P2 position inhibits thrombin about ninefold more strongly. The introduction of phenylalanine instead of glycine in the P3 position leads to an additional significant increase in the binding. Then, d-amino acids were investigated in position P3. Surprisingly, these led to a dramatic improvement in binding affinity. This result was not expected if one considers that the substrate sequence from P5 to P3 Gly–Gly–Gly–Val–Arg contains only achiral glycine residues without lipophilic side chains that can hardly form any interactions that would correspond to the d-Phe side chain.
When the above-described work was carried out, the spatial structure of thrombin had not yet been determined. Wolfram Bode and Milton Stubbs managed to elucidate the structure of a thrombin complex with a chemically activated fibrinopeptide, Gly–Asp–Phe–Leu–Ala–Glu–Gly–Gly–Val–Arg-CH2Cl. This peptide corresponds to the N-terminal portion from P11 to P1 that thrombin cleaves from fibrinogen. The comparison of this structure with that of d-Phe–Pro–Arg-chloromethylketone (Fig.23.7) provided an explanation for the structure–reactivity relationship found by Sándor Bajusz. The S3 pocket is filled by both ligands; in the case of the fibrinopeptide, it is achieved by the side chains of leucine and phenylalanine in the positions P8 and P9. The peptide forms a β-turn that enables the amino acids in this sequence to be positioned in the S3 pocket. The same pocket is accessed by the tripeptide through the side chain of the d-amino acid at position P3.


Comparison of the binding mode of the irreversibly binding thrombin inhibitors d-Phe–Pro–Arg-CH2Cl (dark-red carbon atoms) with that of the fibrinopeptide derivative (gray carbon atoms). Both inhibitors bind with an arginine side chain in the S1 pocket. The S2 pocket is occupied by a valine side chain of the fibrinopeptide. Its additional peptide chain is folded back so that the Leu and Phe side chains in positions P8 and P9 are oriented into the lipophilic S3 binding pocket. In the case of d-Phe–Pro–Arg-CH2Cl the phenyl ring of the d-Phe is found in this pocket too.
The compound d-Phe–Pro–Arg-H, synthesized by Bajusz is a high-affinity thrombin inhibitor (K i = 75 nm). However, the compound proved to be chemically unstable. This problem could be addressed by N-methylation of the free NH2 group. N-Methyl-d-Phe–Pro–Arg-H 23.7 (Gyki 14766/Efegatran, Fig.23.8) is chemically stable.


The inhibitor 23.7 (Gyki 14766, efegatran) contains an aldehyde group that binds reversibly to Ser195. Compounds 23.8 and 23.9 are simple derivatives of the benzamidine that non-covalently inhibit the enzyme.
Jörg Stürzebecher and Fritz Marquardt took a different route. They pursued the goal of managing inhibition without a covalent attachment. Their approach was based on the finding that aside from trypsin (K i = 18 μm), benzamidine 23.1 (Fig.23.4, Sect. 23.3) also inhibits thrombin (K i = 220 μm). The combination of the benzamidine group with a reactive group from Table 23.2 gave potent thrombin inhibitors. The first low-molecular-weight thrombin inhibitor that was clinically tested in the 1970s was p-amidinophenylpyruvic acid 23.5 (Fig.23.4, Sect. 23.3). The compound proved to be efficacious, but its selectivity was unsatisfactory. The simple benzamidine derivatives 23.8 and 23.9 (Fig.23.8) are further typical representatives with micromolar affinity for thrombin, but without selectivity compared to trypsin.
The coupling of the benzamidine groups with a peptide structure brought significant improvement. N α-(β-naphthylsulfonylglycyl)-d,l-p-amidinophenylalanylpiperidide, 23.10 (NAPAP, Fig.23.9) was the result of a more than 10-year-long systematic search for potent and selective thrombin inhibitors. NAPAP was the most potent representative of the class of low-molecular-weight thrombin inhibitors (K i = 6 nm) for a long time, but it has only modest selectivity over trypsin.


The thrombin inhibitors NAPAP 23.10, CRC 220 23.11, the latter was developed at the former Behringwerke, and 23.12 which was derived from 23.10. The two latter compounds have distinctly better affinity to thrombin and improved selectivity relative to trypsin. The IC50 values for 23.10 and 23.12 are given for the racemates. Inhibitor 23.11 was measured as an enantiopure compound.
In 1989, Wolfram Bode elucidated the crystal structure of thrombin with a bound inhibitor at the Max Planck Institute for Biochemistry in Martinsried, Germany. Initially the structure determination was accomplished with the irreversible inhibitor d-Phe–Pro–Arg-CH2Cl and with NAPAP shortly thereafter. The 3D-structure of the thrombin–NAPAP complex is shown in Fig.23.10. The racemic form was used for the co-crystallization. The result that the p-amidinophenylalanine binds to thrombin as the d-amino acid was rather surprising. The substrate is composed of l-amino acids only, therefore it was expected that p-amidinophenylalanine would also bind in the l configuration.


Structure of the thrombin–NAPAP complex. The most important interactions are outlined on the left side. The positively charged benzamidine group occupies the S1 pocket and forms a salt bridge to the negatively charged side chain of Asp189. Two hydrogen bonds are formed to the amino acid Gly216. The piperidyl and naphthyl groups together occupy the two large lipophilic pockets S2 and S3.
The groups of the ligand that form polar interactions with the protein can be directly deduced from the crystal structure. For NAPAP these are the glycine unit in the center of the molecule (double hydrogen bond to the peptide backbone) and the amidinium group in the S1 pocket for NAPAP. Omitting the positively charged amidine group will result in a loss of binding affinity because the salt bridge to Asp189 can no longer be formed. More recent work has shown however that chloro-substituted aromatic rings can also bind in the S1 pocket and form a hydrophobic interaction to Tyr228. Today, an arsenal of building blocks is available that can be used as arginine side chain mimics to fill the S1 pocket of thrombin (Fig.23.11).


Numerous building blocks have been developed that bind as a mimetic for the arginine in the thrombin’s S1 pocket.
With its naphthyl and piperidyl side chains, NAPAP largely fills the lipophilic S3 pocket and the spatially rather limited S2 pocket (Fig.23.10). However, it seems as if even larger substituents could fit in the S3 pocket. A weakness of NAPAP was its inadequate selectivity compared to the digestive enzyme trypsin. Luckily, the structures of NAPAP in complex with thrombin and also with trypsin are known (Fig.23.12). A comparison of the 3D structures shows that there is a significant difference in the binding mode between the two enzymes in the S3 pocket that leads to a 180°-flipped orientation of the naphthyl group about the bond to sulfur. In thrombin, the S3 pocket is more pronounced and is surrounded by multiple lipophilic amino acid side chains. In trypsin the top end of this pocket is open, and is spatially hardly restricted at all. Obviously its structuring is not necessary in the largely unspecific digestive enzyme. Therefore, the selectivity can be increased by occupying the S3 pocket of thrombin as optimally as possible. If the thrombin–NAPAP complex is examined in more detail, it is apparent that an additional methoxy substituent on the naphthyl ring should be suitable to enhance selectivity. In fact, inhibitor 23.12 binds 600-fold more strongly to thrombin than to trypsin.


Comparison of the 3D structures of trypsin (left) and thrombin (right), each in complex with NAPAP. The active site in thrombin is further narrowed by an additional loop from above. The depth of the pocket is, once again, color-coded (see Fig.23.2).
Compound CRC220 (23.11, Fig.23.9), which fills the hydrophobic S3 pocket much better than NAPAP was developed at the former Behringwerke in Marburg, Germany. Because of this improved filling, CRC220 inhibits thrombin almost 200-fold more effectively than trypsin.
Another approach to searching for thrombin inhibitors was taken by the researchers at Hoffmann-La Roche. Initially they concentrated on optimally filling the S1 pocket. Benzamidine was known to be a weak thrombin inhibitor that occupies the S1 pocket. It has, however, the disadvantage that it binds more strongly to trypsin (Fig.23.4). Accordingly, the researchers in Basel initially sought a small molecule that binds more strongly to thrombin than trypsin. More than 200 small molecules were tested in this narrowly focused search. Structures were chosen only if their functional groups were able to interact with the negatively charged side chain of Asp189. Guanidines, amidines, and amines were investigated. N-Amidinopiperidine (23.13, Fig.23.13) was identified as an interesting lead structure. In contrast to benzamidine, amidinopiperidine binds more strongly to thrombin (K i = 150 μm) than to trypsin (K i = 300 μm). A systematic derivatization led to 23.14, a moderately active thrombin inhibitor (K i = 0.48 μm). Based on the structural model with the protease, it appeared obvious that the replacement of the glycine unit with a d-amino acid, for example, d-Phe, should fill a lipophilic pocket and lead to a distinct increase in the affinity. The compound was quickly prepared and tested. In fact, 23.15 bound tenfold more strongly to thrombin. Other d-amino acids were then investigated and additional affinity could be achieved. High selectivity against trypsin was also encouraging; 23.16 binds 840-fold more strongly to thrombin than to trypsin. The surprise was great when the 3D structure of 23.14 in complex with thrombin was determined: The compound binds differently than predicted in the binding pocket! In contrast to the original assumption, the naphthylsulfonyl group exchanged positions with the benzyl side chain.


One approach to the structure-based design of thrombin inhibitors began with 23.13 in the S1 pocket. Compound 23.14 was derived from this lead structure. Its docking into the active site of thrombin generated the idea for the synthesis of 23.15. The systematic variation of the side chain R gave compounds with better binding affinity such as 23.16 and 23.17. The compound was tested in depth in the clinic under the name napsagatran. The compound melagatran from AstraZeneca was introduced as the double prodrug ximelagatran 23.18 as the first orally available thrombin inhibitor on the market. It is derived from the tripeptide sequence d-Phe–Pro–Arg. Another orally available inhibitor, dabigatran 23.19, was launched to market by Boehringer Ingelheim. The tricyclic inhibitor 23.30, which was developed at the ETH in Zurich, foregoes peptide character entirely.
The incorporation of a non-proteinogenic amino acid proved to be unfavorable from a synthetic point of view. Therefore, other central building blocks were sought that were synthetically more easily accessible. This work finally led to napsagatran 23.17, a highly potent and exceedingly selective substance. Because it is only intravenously applicable, however, it never found its way to a marketed product, particularly because argatroban, a marketed product for intravenous use, was discovered much earlier and was already available.
The search for low-molecular-weight, orally available thrombin inhibitors intensively occupied numerous large pharmaceutical companies for many years. It took a long time until AstraZeneca introduced Ximelagatran (23.18, Fig.23.13) to the market as the first orally available thrombin inhibitor. The compound is a double prodrug of the actual active substance melagatran. Its relation to the initial parent structures (e.g., the tripeptide sequence d-Phe–Pro–Arg) is still quite apparent. The head group of the arginine residue was replaced with a benzamidine, the five-membered ring of the proline was narrowed to a four-membered ring, and the terminal benzyl group was shortened to a cyclohexyl ring. The N terminus was substituted with a methylenecarboxylic acid group. It proved to be extremely difficult to make the thrombin inhibitors adequately bioavailable and to maintain the necessary plasma level over an acceptable length of time. With regard to the bioavailability, AstraZeneca, in collaboration with the group of Bernd Clement at the University of Kiel, pursued a double prodrug strategy: The terminal acid function was masked as an ester, and the benzamidine group was transformed into an N-hydroxyamidine. The release of the active substance, melagatran, in the body is made possible by ubiquitously present esterases and a set of three reductases. AstraZenecca withdrew Ximelagatran (Exanta®) after 2 years because some issues with liver toxicity were observed in a small number of cases after weeks of use.
Many years of thrombin research finally also led to success at Boehringer Ingelheim. The compound dabigatran (23.19, Fig.23.13) was introduced to the market in the spring of 2008 for the prevention of stroke in patients with atrial fibrillations. It also has a benzamidine anchor, and it has a pyridine group for the hydrophobic S3 pocket. A benzimidazole building block with an attached amide bond was chosen as a linker between these groups. As with ximelagatran, it uses a carboxylic acid on the N terminus. It shows distinctly less peptide character than the lead structures. A double prodrug strategy was also used for this substance to make it adequately bioavailable. In addition to the esterification of the acid group, the amidine group was masked as a carbamoyl moiety. The prodrug carries the name dabigatran (Pradaxa® in the USA and Europe and Pradax® in Canada).
The group of François Diederich at the ETH in Zurich managed to develop a thrombin inhibitor (23.20, Fig.23.13) that foregoes the peptidomimetic character entirely. Precise design in the binding pocket led to an inhibitor with a central tricyclic moiety, which was readily prepared by a 1,3-dipolar-addition reaction. With a benzamidine anchor for the S1 pocket and piperonyl moiety for the S3 pocket, this very rigid derivative advanced into the realm of nanomolar inhibitors.
5 Design of Orally Available Low-Molecular-Weight Elastase Inhibitors
Human leukocyte elastase is a serine protease that is released in the lung to destroy dead tissue and invading bacteria. The destructive potential of this enzyme is normally controlled by a series of endogenous inhibitors, such as α1 protease inhibitor or leukocyte protease inhibitor. If the equilibrium between the protease and inhibitor is shifted, for instance, because of a genetically caused underexpression of an inhibitor or by toxic substances that are taken in with the air, elastase also attacks healthy lung tissue. Cigarette smoke contains compounds that oxidize an essential methionine side chain on the endogenous α1 protease inhibitor and cause its deactivation. The chronic destruction of cells in the alveoli leads to a life-threatening disease: emphysema.
A possible approach to the pharmaceutical treatment of this disease is therefore the use of elastase inhibitors. In contrast to thrombin, elastase does not have a deep, pronounced S1 pocket with an acidic amino acid, through which a polar contact to a ligand can be made. It accepts only substrates with small hydrophobic amino acids such as valine (Fig.23.3). If a large binding contribution cannot be expected by occupying the S1 pocket, as is in the case with thrombin, the catalytic serine itself can be involved in the protein–ligand interaction by forming a reversible covalent bond with the inhibitor. Such a concept was pursued at the former ICI (today part of AstraZeneca) by starting with a trifluoromethylketone R–COCF3 as a reversible, covalent-binding serine protease inhibitor. By starting from the substrate sequence, potent elastase inhibitors were found such as 23.21 and 23.22 (Fig.23.14).


Elastase inhibitors 23.21 and 23.22 (ICI 200880) are substrate analogues. Compound 23.22 is a highly active compound, but it is not orally available.
ICI 200880 (23.22) proved to be an efficacious elastase inhibitor in clinical trials, but it lacked oral bioavailability and had a short biological half-life. The spatial structure of the related inhibitor Ac-Ala–Pro–Val-CF3 had been determined in complex with elastase. The most important interactions between elastase and the inhibitor are shown in Fig.23.15. The inhibitor binds to elastase in a β-pleated-sheet conformation in which two H-bonds to Val216 and one to Ser214 are formed. The valine side chain fills the S1 pocket, and the carbonyl group binds covalently as a hemiketal to the side chain of Ser195. The research concentrated on non-peptidic structures with functional groups that are able to form the same interactions as the peptidic inhibitors.


Comparison of the binding mode of the elastase inhibitor Ac-Ala–Pro–Val-CF3 with the postulated binding mode of the pyridone moiety (e.g., 23.23, Fig.23.16). Both compounds should be able to form a double H-bond to Val216.
By starting with the 3D structure of the protein–ligand complex, pyridones were chosen as the most promising peptidomimetic replacement. The postulated binding mode of the pyridone compared to the binding mode of the peptidic inhibitors is shown in Fig.23.15. Compounds of this chemotype were synthesized at Zeneca (now AstraZeneca) and in fact proved to be very effective elastase inhibitors. Compound 23.23 (Fig.23.16) binds to the protein with K i = 5.6 nm. This compound, however, has multiple unfavorable properties. It is not orally available and inhibits chymotrypsin (K i = 60 nm) in addition to elastase. Poor oral bioavailability was attributed to excessive lipophilicity (log P > 4), which led to low water solubility.


Design of orally available elastase inhibitors at Zeneca. The original idea to replace the Ala–Pro unit with a pyridone afforded 23.23. Later, pyrimidinones were overwhelmingly investigated. An additional nitrogen atom has been added to the heterocycle. Very potent compounds (e.g., 23.25) are found in this class. Compound 23.26 has the best in vivo properties. The p-fluorophenyl group (in 23.26) or the p-aminophenyl group (in 23.27) increases the lipophilic contact with the enzyme. The compound ONO-6818 23.28 was developed in Japan all the way to clinical trials, where it was discontinued due to abnormally elevated liver values in the treated patients. Another compound, 23.29, was clinically tested under the name sivelestat (ONO-4056). These compounds specifically transfer an acyl group to the catalytic serine and reversibly block the enzyme.
The pyrimidone class in which a carbon atom of the heterocycle was exchanged for a nitrogen atom seemed to be synthetically simpler and therefore more broadly variable. Compound 23.24 is less lipophilic (log P = 2.1) than 23.23, ten times more water soluble, and orally available. Its binding to elastase proved to be practically unchanged (K i = 6.6 nm), whereas the chymotrypsin inhibition was much less pronounced (K i = 1,000 nm). Numerous representatives of the new substance class were synthesized, and their inhibitory effects and bioavailability were tested. In doing so it was shown that the strength of the inhibitory effect and the in vivo activity did not run parallel to one another. For example, 23.25 is a highly potent elastase inhibitor in the enzyme test but is not orally available. With an oral bioavailability of 60–90%, compound 23.26 (K i = 100 nm) proved to be optimal in the animal model. The crystal structure with an analogous derivative 23.27, which carries only an additional sulfonamide group, confirmed the expected binding mode (Fig.23.17).


The crystal structure of 23.27 (Fig.23.16) in complex with elastase. The inhibitor forms two H-bonds to Val216, and one H-bond to Ser214. Furthermore, the oxyanion hole is occupied by an oxygen atom.
The Japanese company ONO Pharmaceuticals Co. developed compound 23.28, which was derived from 23.26 and carries a 1,3,4-oxadiazole ring in place of the trifluoromethyl group on the ketone and an unsubstituted phenyl ring on the pyrimidone. However, the development of ONO-6818 was discontinued in clinical phase II because of abnormally elevated liver enzyme levels. Nevertheless, ONO had success with ONO-5046, 23.29, which was developed under the name sivelestat (Elaspol®; Fig.23.16). This inhibitor reacts with elastase specifically and reversibly acylates the catalytic serine.
6 Serine Protease Inhibitors, Thrombin Was Just the Starting Point
Factor Xa and VIIa occur along the coagulation cascade prior to thrombin and are investigated as targets for antithrombotics. They both have an aspartic acid on the bottom of their deep S1 pockets, similarly to thrombin. Moreover, a narrow and deep S3 pocket that is flanked by aromatic amino acids (Tyr99, Trp215, and Phe174) is specific to factor Xa. Therefore, this pocket is ideally suited for aromatic groups on inhibitors. As already mentioned, in the mid-1990s a dogma prevailed that the S1 pocket of trypsin-like serine proteases could only accept groups with basic character. The binding of chloro-substituted aromatic portions, however, could be demonstrated for thrombin at Merck & Co. in the USA. These groups made a breakthrough for factor Xa inhibitors. Highly potent inhibitors could be developed with chlorophenyl, chloronaphthyl, or chlorothiophene groups protruding into the S1 pocket. So much affinity was gained by the accommodation of additional groups in the deep, aromatic S3 pocket that these compounds bind to the protease with single-digit-nanomolar values without the benzamidine anchor. Such a derivative was introduced by AstraZeneca (23.30, Fig.23.19). In addition to the development of compounds with chloro-substituted aromatic rings for the S1 pocket, other inhibitors with benzamidine groups were also synthesized as factor Xa inhibitors. However, it is much more difficult to achieve adequate selectivity compared to other trypsin-like serine proteases with these derivatives. Furthermore, they showed similar problems with regard to bioavailability as the thrombin inhibitors. Bayer HealthCare introduced a new factor Xa inhibitor to the market in September 2008, rivaroxaban (Xarelto®; 23.31, Fig.23.18 and 23.19), that places a chlorothiophene group in the S1 pocket.


Crystal structure of rivaroxaban 23.31 (Fig.23.19) in factor Xa. The inhibitor’s chlorothiophene group binds in the deep S1 pocket, at the end of which Tyr228 and Asp189 are found. The chlorine atom forms interactions with the aromatic rings. The phenyl ring and the terminal lactam ring of the inhibitor are found in the S3 pocket, which is enclosed by the three aromatic groups of Tyr99, Phe174, and Trp215.


Three potent factor Xa inhibitors. The chloroaromatic rings of the two first examples bind in the S1 pocket of the enzyme. Compound 23.30 was developed by AstraZeneca. Rivaroxaban 23.31 was introduced to the market in 2008 by Bayer as the first orally available factor Xa antithrombotic. Apixaban 23.32 from BMS binds at the subnanomolar level with its methoxysubstituted aromatic rings in the S1 pocket of factor Xa.
Other companies are working on inhibitors with comparable chloroaromatic groups to fill the S1 pocket. The subnanomolar-binding inhibitor apixaban 23.34 from Bristol-Myers Squibb has recently been approved for market. It foregoes the halogen group for the interaction in the S1 pocket completely. As a crystal structure shows, the p-methoxy group displaces the water molecules that are almost always in the S1 pocket. The previously mentioned compounds that lack a benzamidine group show better bioavailability and good selectivity for factor Xa. Attempts were also undertaken to develop dual inhibitors for thrombin and factor Xa. By inhibiting both proteins together, not additive, but rather synergistic antithrombotic effects are achieved that hopefully expand the therapeutic scope.
Factor VIIa is at the beginning of the extrinsic path of the coagulation cascade. This enzyme also belongs to the family of trypsin-like serine proteases, and specific inhibitors have been sought for this enzyme for years. In this case, the activation of the protease is interesting. In cases of injury, blood comes into contact with tissue. When this happens, factor VIIa and the membrane-bound tissue factor can form a complex that causes a change in the conformation of the protease’s catalytic domain. A peptide segment next to the catalytic center goes from being in an unfolded conformation into a helical structure. This leads to a change in the geometry of the catalytic site. Only in the complexed state does the protease have a structure that allows the coagulation cascade to be initiated. Although numerous nanomolar inhibitors are available by now, none of them has been able to forego the basic group on the P1 aromatic ring.
In addition to the serine proteases of the coagulation cascade, other proteases of this family have been chosen for drug development. The drug design for these target enzymes has benefitted strongly from the experience gained with the thrombin inhibitors. The concepts learned there are well transferred to the special conditions of these proteins. Tryptase, urokinase, and matriptase belong to this family. Tryptase inhibitors are being investigated for the treatment of asthma, and the two others are target structures for possible cancer therapeutics. Tryptase is a tetramer with four trypsin-like catalytic sites. These sites are separated from one another by several angstroms. To develop selective inhibitors, compounds were designed that carry two benzamidine-like anchor groups and a long enough bridge to link them together. In this way, two of the four sites in the tetrameric tryptase molecule are simultaneously blocked. The disadvantage of this design concept is that the developed inhibitors are very large. They are well over the molecular weight limit of 600 Da, which should not be exceeded for good bioavailability. The protease furin also belongs to the family of serine proteases; however, it adopts the folding of the subtilisin family (Sect. 14.7). It is involved in the maturation of proproteins. In this way, the envelope proteins of viruses are cleaved to transform them into their active form. Its involvement in the “arming” of viruses was even reported upon in the tabloid press: In the BILD-Zeitung in Germany on August 28, 2003, furin was referred to as the “the most brutal protein in the world” that “makes epidemics a deadly danger for humans and acts like a detonator on a bomb.” Furin and other closely related subtilases cleave particular basic tetrapeptide sequences C-terminally: Arg–X–(Arg/Lys)–Arg-. Many glycoproteins of lipid-enveloped viruses are cleaved at this recognition sequence and consequently activated. An example of this is the highly pathogenic avian influenza viruses that contain such cleavage sequences in hemagglutinin, one of the surface glycoproteins. Whether the viruses can be activated depends on the availability of the ubiquitously occurring furin, and this is a prerequisite for the high pathogenic potential of the avian influenza viruses. Other genetic combinations or prerequisites must be fulfilled to convert these viruses to dangerous pathogens for animals and humans. Inhibitors of furin could then contribute to the suppression of “arming” of these viruses. However, the translation of such highly charged substrates into inhibitors that meet the common rules for good bioavailability is a tremendous challenge.
In the early 1990s an interesting observation was made that the incretin hormones GIP and GLP-1, which stimulate the pancreas to release insulin after eating, are substrates for dipeptidylaminopeptidase IV (DPP IV). They are quickly degraded by this serine aminopeptidase. Because incretins were already interesting candidates for a diabetes therapy, the idea immediately occurred that the inhibition of DPP IV could be used as a principle for treating type-II diabetes (non-insulin dependent). The membrane-bound protease cleaves dipeptides from its substrate when a prolyl or alanyl group is in the second position from the N terminus. Sitagliptin (Januvia®) 23.33 (Fig.23.20) was approved in 2006 for the treatment of type-II diabetes. It blocks the protease without invoking a covalent coupling. Recently two additional compounds, vildagliptin (Galvus®) 23.34 and saxagliptin (Onglyza®) 23.35, became available for clinical use. Both use a proline-derived cyanopyrrolidine that binds reversibly and covalently to the catalytic serine.


Sitagliptin 23.33, vildagliptin 23.34, and saxagliptin 23.35 are inhibitors of the serine amino peptidase DPP IV for the treatment of type-II diabetes.
We can only wait and see whether other active substances come to the market in the next few years for the many currently worked-on serine proteases. The field has increasingly benefitted from the experience that has been collected on the individual members of this protein family, so that lead structures can be quickly discovered for new target structures.
7 Serine, a Favored Nucleophile in Degrading Enzymes
Serine peptidases use the OH group of an endogenous serine as an attacking nucleophile. The neighboring histidine residue mediates the temporary proton transfer, and the aspartate compensates for the intermediately occurring charge on the imidazole ring of the histidine. A special feature is, however, the temporary covalent bond between the N-terminal part of the substrate and the enzyme. Many other hydrolytically cleaving enzymes use an analogous principle. Esterases and lipases also have a catalytic triad. Occasionally an aspartate is exchanged for a glutamate in these enzymes. The neurotransmitter acetylcholine acts on many synapses in the vegetative nervous system and is involved in the transmission of nerve impulses. It binds to the nicotinic acetylcholine receptor, among others and activates this ion channel (Sect. 30.4). Acetylcholine must be removed to limit the duration of the transmission process and to reset the receptor to the starting point. An imbalance in this nerve impulse transmission system leads to acute and chronic movement disorders. Acetylcholinesterase is responsible for the degradation of acetylcholine. Inhibitors of this enzyme are used to treat shaking palsy (Parkinson’s disease). More recent research has also explored their potential for the treatment of Alzheimer’s disease.
Acetylcholinesterase has a catalytic triad made of serine, histidine, and glutamate. Acetylcholine (3.46, Fig. 2.11) is cleaved by the enzyme in that its acetyl group is transferred to the catalytic serine; hydrolysis releases acetic acid slowly from the esterase. The drug (S)-rivastigmine is also attacked by the catalytic serine, and its carbamoyl group is transferred. Because of the enhanced stability of the carbamoyl–enzyme complex, the esterase is then only very slowly deacylated and regenerated for the next transformation. This causes an inhibition of the target enzyme for several hours. Cholinesterase inhibitors are also used as insecticides. Active substances such as paraoxon 23.37 (Fig.23.21), parathione (E605) 23.38, propoxur 23.39, or malathione 23.40 contain phosphoric acid esters or thioesters that are virtually irreversibly transferred to the catalytic serine. Because of this inhibition, acetylcholine increases to lethal concentration in insects.


(S)-Rivastigmine 23.36 transfers a carbamoyl group to the catalytic serine in the binding pocket of acetylcholine esterase and blocks its function because the carbamoyl–esterase complex decomposes very slowly. The acetylcholine esterase inhibitors paraoxon 23.37, parathione 23.38, propoxur 23.39, or malathione 23.40 are phosphoric acids, thiophosphoric acids, or carbamine esters and are used as insecticides. They also react with the catalytic serine and form a stable covalent bond.
Analogously to the esterases, lipases also hydrolyze ester bonds. Serine, histidine, and aspartate or glutamate serves as the catalytic triad. Pancreas lipase cleaves triglycerides during the digestion of fats. Inhibitors of this enzyme, which is present in the intestines, are used to treat obesity. A significantly reduced absorption of fats and their cleavage products is the result. Orlistat (Xenecal®, 23.41; Fig.23.22), a synthetic hydrogenation product of the natural product lipstatin, carries a reactive β-lactone ring at its core in addition to a very long aliphatic side chain. Serine in the catalytic site of the lipase attacks the carbonyl group of the lactone ring and opens the strained ring by transforming to a stabilized acyl–enzyme complex. Once blocked, the enzyme is no longer able to cleave triglycerides, which translates to a reduced ability to extract calories from food.


Orlistat 23.41 is a synthetic hydrogenation product of the natural product lipstatin, which has two additional double bonds. It has a reactive β-lactone ring that reacts with the catalytic serine in the catalytic site of pancreatic lipase to form an acyl–enzyme complex with ring opening. The enzyme’s function is then blocked.
Lipases are often used for the kinetic resolution of racemates. This is usually achieved by simply transforming a racemic mixture of esters in which one of the two forms reacts faster than the other. An example was described in Sect. 5.4 in which the lipase was used not only to hydrolyze but also to form a new amide bond. For this, the intermediate acyl–enzyme complex cannot be exposed to a water molecule as a nucleophile, but rather a compound with a free amino group must be available. This transformation produces a new amide bond. Bacteria use such a transpeptidase reaction for the construction of their cell wall. This has a completely different composition than that of humans. Therefore the enzymes used for cell wall synthesis are bacteria-specific and particularly well suited as a target for a side-effect-poor drug therapy.
Cross-linking of the peptidoglycan strands is accomplished in the last step of the cell wall biosynthesis. For this, the terminal amino group of a pentaglycine chain attacks between two d-alanine residues of another peptide unit. The bond between d-Ala–d-Ala is cleaved, and a new peptide bond between d-Ala and glycine is formed. The cross-linking is mediated by a glycopeptide transpeptidase. It has a catalytic machinery that is very similar to the serine proteases. In addition to a catalytic serine, a lysine and a glutamate are also found in the reaction center, and an oxyanion hole is also present. Penicillins 23.42–23.44 and cephalosporins 23.45 (Fig.23.23) inhibit these transpeptidases. They have a spatial structure that is analogous to the d-Ala–d-Ala dipeptide, and are therefore recognized as a “false” substrate (Fig.23.23). The β-lactam ring is opened by the attacking catalytic serine, and an irreversible covalent coupling to the enzyme results. The cross-linking of the glycan strands is prevented, and the newly synthesized cell wall does not achieve adequate stability. It cannot withstand the osmotic pressure of the cell contents, and the bacterial cell is killed as a consequence.


In the last step of the bacterial cell wall synthesis, a glycopeptide transpeptidase cleaves the bond between two d-Ala–d-Ala groups and forms a new bond between d-Ala and a glycine in a peptidoglycan strand. Lactam antibiotics of the penicillin (23.42–23.44) or cephalosporin type (23.45) can block this step. The penicillin scaffold (green) is reminiscent of the d-Ala–d-Ala group (orange) and is bound analogously by the enzyme. An irreversible inhibition of the transpeptidase is achieved by a nucleophilic opening of the lactam ring with the help of the catalytic serine.
Of the first penicillins that were discovered by Alexander Fleming (Sect. 2.4), only benzyl 23.43 and phenoxymethylpenicillin 23.44 still have clinical importance (Fig.23.23). The residues on the 6-amino group of penicillic acid were exchanged to improve the pharmacokinetics, activity spectrum, and acid stability. Electronegative atoms on the α-carbon atom of the acyl function increase the stability with respect to acid-catalyzed decomposition and contribute to an improvement in the oral bioavailability.
Bacteria quickly develop resistance to penicillins. They use lactamases, which are enzymes that are structurally related to the transpeptidases. Four classes of lactamases are known, of which three have a catalytic serine in their active sites. A further class belongs to the zinc-dependent metalloenzymes (Chap. 25, “Inhibitors of Hydrolyzing Metalloenzymes”). The catalytic serine of even the β-lactamases is acylated by penicillins and related cephalosporins (Fig.23.23). Up until this step, the mechanism in the transpeptidases and the β-lactamases is identical. However, transpeptidases form very stable acyl–enzymes, whereas the covalent intermediate of the β-lactamases is quickly hydrolyzed. The antibiotic to deactivate the transpeptidase is therefore rendered inactive. β-Lactamases are probably descendants of the transpeptidases. They are widespread in nature and have evolved out of the competition between bacteria and molds. The resistance gene for β-lactamases is easily transferred between bacteria because the information is stored on an extrachromosomal plasmid. Such plasmids are transferred very quickly.
How are the β-lactamases different from the transpeptidases so that they are able to quickly dispose of the covalently bound ring-opened penicillin? The release requires a hydrolytic cleavage from the protein. For this a well-placed water molecule in the active site is needed that can initiate the nucleophilic attack on the acyl–enzyme species. Although the structural architecture of transpeptidases and β-lactamases is very similar, the sequence identity is small. Nonetheless, a transpeptidase has been equipped with the hydrolytic properties of a lactamase by selective mutagenesis! Only a few amino acid exchanges were needed for this. Above all, the hydrophobic amino acids such as phenylalanine and tryptophan are the ones that protect the acyl–enzyme complex from hydrolysis in the transpeptidase. In contrast, polar amino acids such as glutamic acid (Fig.23.24, Glu166) are found in the same positions in the lactamases. In contrast to the transpeptidase’s hydrophobic amino acids, these anchor and activate a water molecule in the correct orientation for nucleophilic attack on the acyl–enzyme complex in the lactamases. As a result, the covalent complex with the penicillin cleavage product that was formed by ring opening in the lactamases is hydrolyzed, but it remains stable in the transpeptidases.


Unsubstituted penicillic acid 23.46 is quickly cleaved by TEM-1β-lactamase (a). By adding a hydroxymethyl group to the 6-position of 23.47 a compound is obtained that forms a hydrolytically stable acyl–enzyme complex with the enzyme (b). A new crystal structure was determined with this compound (b, lower picture). The hydroxyl group is found at the position where the water molecule (orange sphere) starts its nucleophilic attack on the acyl–enzyme intermediate (a, lower part, modulated structure with the coordinates from the crystal structure of the complex with 23.47). The hydrophobic amino acids such as phenylalanine and tryptophan are found at positions 166 and 170 of the transpeptidases, which are structurally related to the β-lactamases.
How can this lactamase-caused resistance be broken, and the degradation of penicillins stopped? Unsubstituted penicillic acid 23.46 is quickly cleaved by TEM-1β-lactamase (Fig.23.24). Based on structural considerations it was proposed that a hydroxymethyl group should be added to the 6-position. This group should be located in exactly the position from where the water molecule would start its nucleophilic attack on the acyl–enzyme form. In fact, derivative 23.47 inactivates TEM-1β-lactamase. A water molecule was detected in the vicinity of the CH2OH group in the subsequently determined crystal structure, but it is too far away to successfully hydrolyze the acyl–enzyme. The hydroxyl group therefore blocks the attack of a water molecule on the ester carbonyl group of the acyl–enzyme.
The incorporation of such a hydroxymethyl group has been undertaken in important β-lactamase-resistant β-lactams such as imipenem 23.48 or meropenem 23.49 (Fig.23.25). β-Lactamases can also be irreversibly inhibited. If such an inhibitor is administered with a penicillin, the degradation of the penicillin by the lactamase is blocked, and it is available to inhibit the transpeptidase. The natural product clavulanic acid 23.50 forms an acyl–enzyme complex upon the opening of its lactam ring. By rearrangement a vinylogic urethane is formed that is resistant to hydrolysis.


β-Lactamase-resistant antibiotics of the penem and carbapenem type. Imipenem 23.48 and meropenem 23.49 are derived from the carbapenem type. The natural product clavulanic acid 23.50 opens its lactam ring and forms an acyl–enzyme complex with the serine residue. A hydrolysis-resistant vinyl urethane analogue is formed by a rearrangement.
With these examples, the spectrum of enzymes that use a serine as a nucleophile is nowhere near exhausted. Viruses need cleavage enzymes. They must cleave the polypeptide chains that were synthesized by the infected cell according to their own specifications into functional viral proteins. Viruses either use proteases of the infected host cell (cf. furin) or they employ their own viral proteases. Because error-free functioning of the latter enzymes is essential for the maturation of the new viruses and is also virus-specific, these proteases are privileged targets for drug development. Peptidases with a catalytic serine as well as cysteine (Sect. 23.8) are recognized. As we shall see in Sect. 24.3, an aspartic protease serves other viruses.
The assemblins, another group of serine peptidases, have been found in herpes viruses. The enzyme from the cytomegalovirus belongs to this group as similarly those from the varicella zoster virus and the herpes simplex virus. These proteases also use a serine and a histidine. An additional histidine forms the third amino acid in the triad. Despite a different folding pattern, this triad spatially fits very well to the trypsin triad. Even the oxyanion hole is present in this viral protease.
The hepatitis C virus belongs to the group of enveloped RNA viruses. In addition to other viral proteins, its genome contains the sequence for a serine protease (HCV-NS3/4A). It is needed for the cleavage of the initially produced polypeptide chain into functional viral proteins. Therefore the inhibition of this protease represents a therapeutic approach for fighting hepatitis infections. The infections can easily become chronic and can lead to severe liver damage as well as liver cirrhosis and hepatocellular carcinoma. Orally bioavailable inhibitors of these serine proteases such as telaprevir from the company Vertex have recently been launched to market.
The carboxy serine peptidases represent another group that is folded analogously to subtilisin (Sect. 14.7). They have a triad of serine, glutamate, and aspartate. A member of this family was recently discovered on the human cnl2 gene. Mutations in this gene lead to severe neurodegenerative diseases. An oxyanion hole is also found in this enzyme, to which, interestingly, an aspartate contributes. It is only in the protonated state, however, that it can fulfill its task as a hydrogen-bond donor and negative-charge stabilizer in the transition state. Because the enzymes of this family are active in a pH range of 3–5, the requirements for being protonated are fulfilled.
There could be many more cleavage enzymes that use a catalytic serine to discover. We can only wait and see which of the discovered peptidases will be singled out for pharmaceutical development. Their catalytic machinery adopts the same spatial architecture in all examples. Therefore, the general principles can be transferred between the individual members of the family.
8 Triads in All Variations: Threonine as a Nucleophile
Aside from serine, another amino acid also carries an aliphatic-OH group: threonine. This amino acid can also be catalytically active in a protease. The proteasome represents the central protein-shredding machine for the cell and cleaves proteins that have been marked with ubiquitin into small oligopeptides containing between 3 and 20 amino acids. The ubiquitin label is itself a highly conserved protein with 76 amino acids. As a cellular shredding machine, the proteasome plays a central role in the metabolism of proteins, cell growth, and cell demise. Therefore it is an important target structure for the treatment of cancer. It is a multiprotease complex composed of more than 30 proteins and is found in the cytoplasm as well as the nucleus (Fig.23.26). The proteasome is constructed like a large barrel with two lid regions that have regulatory function; these regions control the entry of substrates into the shredder. A threonine is found in the catalytic sites of the proteases, which have chymotrypsin-like, trypsin-like, and peptidyl-glutamyl-peptide-like substrate specificity. The OH group of this threonine adopts the role of the nucleophile. A neighboring, positively charged lysine and a balancing aspartate reinforce its nucleophilic strength. Because the threonine is the first amino acid at the N terminus, it carries a free amino group. This serves as a proton acceptor in the mechanism. Two serines and one aspartate group contribute to the stabilization of the transition state and complement the nucleophilic center.


The proteasome, a cellular shredding machine, proteolytically cleaves ubiquitinylated proteins selectively into small oligopeptides that have between 3 and 20 amino acids. The crystal structure of the 20S proteasome from yeast (subunits are shown in different colors) is shown at the left. Six of these units are inhibited by bortezomib (yellow). The boronic acid derivative bortezomib 23.51 (right, gray) reacts with the N-terminal Thr1 and forms a covalent boronic acid ester complex.
The Millenium Pharmaceuticals company, which was founded as an academic research institute, introduced bortezomib 23.51 (Velcade®) to the market in 2006; this was the first active substance that blocks the threonine protease function of the proteasome. Chemically, bortezomib is a boronic acid derivative (Fig.23.26). The inhibitor reacts with the threonine of the catalytic triad to create a covalent connection. In addition to this reactive group, a distinct peptide-like character can be seen in the molecule. The molecule is able to interact with the substrate-binding site in the proteasome with this moiety.
Another peptide analogue, carfilzomib, is in clinical testing. It carries a terminal α′β′-epoxy ketone. Upon inhibition, the threonine OH group nucleophilically attacks the keto function of the inhibitor. Next, the neighboring N-terminal amino group opens the epoxide ring. This leads to an irreversible covalent bond. Even though the epoxy ketone is very reactive, carfilzomib is a highly selective proteasome inhibitor. The vicinity of the nucleophilic Thr―OH of the first residue in the sequence and the N-terminal amino group is an unusual and exceptional combination. It is, however, necessary for the activation of the inhibitor.
At present, the proteasome is an important target structure being tested for tumor therapy. More than 20 different inhibitors are currently in development. Bortezomib is used for the treatment of multiple myeloma, a malignant disease of the bone marrow. This tumor disease is based on the malignant transformation of plasma cells, the physiological function of which is to produce antibodies for immune defense. Even though bortezomib cannot heal multiple myeloma, its use can extend life of patients for whom other therapies have failed. In multiple myeloma, the plasma cells produce massive amounts of misfolded proteins that must be digested by the proteasome. Therefore these cells need a proteasome that functions optimally; otherwise apoptosis would be induced. Blocking the proteasome function is therefore desirable for such cells. Moreover these cells are significantly more sensitive to bortezomib therapy than normal cells. Some tumor cells also activate a transcription factor, NF-κB, which controls the proliferation and survival of the tumor cells. The proteasome is critical for the activation of NF-κB because it degrades an inhibitor of this transcription factor that acts as a kind of emergency brake on NF-κB. Therefore the inhibition of the proteasome serves to keep NF-κB in its benign form, because its inhibiting binding partner is no longer being degraded. It is possible that bortezomib induces apoptosis of tumor cells in that it stabilizes cyclin-dependent kinase inhibitors (Sect. 26.2) as well as the tumor-suppressor protein p53.
Interestingly, a protease was discovered in bacteria that exists as a 14mer and the spatial structure of which is reminiscent of the proteasome. The ClpP-protein is a serine protease that is involved in the degradation of cellular proteins in bacteria. Treatment with a macrolide antibiotic can cause its function to run out of control and degrade proteins in an unregulated way. This leads to cell death in the bacteria. This principle was recognized by the company Bayer and exploited for an antibiotic therapy. The goal was not to block the protease function of the ClpP-protein, but rather to promote its uncontrolled effects through synthetic antibiotics.
9 Cysteine Proteases: Sulfur, the Big Brother of Oxygen as a Nucleophile in the Triad
In addition to the OH group of serine and threonine, the thiol group of cysteine is able to carry out a nucleophilic hydrolytic attack on amide bonds. These enzymes possess a catalytic triad, analogously to the serine proteases, and are termed cysteine proteases. The first protease of this family to have been structurally investigated in detail is papain, which is isolated from the latex of the papaya, the fruit of the papaya tree (Carica papaya). Its triad is composed of a nucleophilic cysteine as well as a histidine and an asparagine. The asparagine adopts the role of the aspartate in the serine protease. The catalytic mechanism is comparable to that of serine proteases. Even the oxyanion hole (Cys25 and Gln119) is found in proteases from the papain family. There are indications that the transition state is structurally similar to the acyl–enzyme intermediate. An attempt has been made to exchange serine for cysteine in trypsin. The binding properties for the substrate (K m value) remained virtually the same, but the catalytic rate of the reaction decreased by five orders of magnitude. Even though the structures are geometrically nearly unchanged, the experiment shows that the difference between serine and cysteine proteases is more complicated than a simple exchange of sulfur for oxygen. The fine-tuning of the structural and electronic properties is pivotal. In contrast to the trypsin-like serine proteases, the nucleophilic cysteine exists as a preformed ion pair with its neighbor, histidine.
Three families of cysteine proteases of importance as targets for drug therapy have been characterized (Table 23.4). The first group is derived from papain, and the cathepsins belong to this group. They are proteases that are involved in the degradation of the extracellular matrix proteins and the basal membrane. Inhibiting their function opens very different therapeutic possibilities, for example, for inflammation, tumor metastasis, bone resorption, and muscle atrophy, or myocardial infarct. Another group is the calcium-dependent calpains, their hydrolytic domain has a very similar folding to papain. They occur in many cells and carry out different functions. Calpains occur in higher concentrations at sites of cell damage such as after traumatic brain injury, stroke, or during the formation of cataracts in the eye. Calpains seem to be regulatory enzymes. For example, they reduce the blood flow through vessels in cases of injury to limit blood loss. During a stroke, this natural protective function unfortunately leads to the contrary situation: Activation of calpains reduces the blood flow, and parts of the brain become ischemic. Destruction of the affected brain cells is the result. Specific inhibitors could counteract the over-functioning of the calpains. Cysteine proteases of the papain family have also been discovered in parasites. The inhibition of cruzipain could be a concept to treat sleeping sickness. Falcipain, which is used to digest hemoglobin by the parasite that causes malaria, represents a promising target enzyme for malaria therapy.
The second large family of cysteine proteases includes the caspases. These are involved in the control of apoptosis, or programmed cell death. If a cell is irreparably damaged so that it can no longer be remedied by natural repair mechanisms, caspases are activated that initiate apoptosis. Misregulation of apoptosis leads to different pathological conditions that are associated with tumor disease, disruption of the immune system, or neurodegenerative damage. Inhibitors of different caspases have potential as neuroprotective agents, as active substances for tumor therapy, or for the treatment of rheumatoid arthritis.
The third family includes the viral 3C proteases, which occur in picornaviruses (human rhinovirus, poliomyelitis, or hepatitis viruses) or corona viruses (SARS). These viral proteases process the primary polypeptide chain and generate the specific viral proteins during maturation. Inhibitors of these proteases represent a concept for antiviral chemotherapy.
A special feature associated with the papain-type proteases is the stereochemistry of the nucleophilic attack. In contrast to other serine and cysteine proteases, the attack occurs from the opposite side, the so-called Si face. The S1 pocket in papain is not prominent, and the P1 group of the substrate is oriented away from the protein. In contrast, all of the neighboring pockets are much more prominent. Interestingly, some of the pockets on the C-terminal side (the primed side, S1′–S4′) of cysteine proteases are strongly structured. This can be exploited for the design of potential inhibitors. Papain prefers substrates with hydrophobic P2 and P3 groups. An aspartate is recognized as a P1 group by caspases of the second folding family. For these reasons, many inhibitors that have been developed for caspases carry a functional group with a carboxylic acid group or a corresponding mimetic at this position. The interaction with the thiol group of the catalytic cysteine is decisive for the binding of cysteine protease inhibitors to their target enzyme. It is interesting that many of the developed inhibitors try to involve the sulfur atom in a covalent bond. Reversible and irreversible head groups have been developed for this purpose. The complex of the inhibitor leupeptin 23.52, a natural product with an aldehyde function, with calpain II is shown in Fig.23.27. This group reacts with the thiol group of cysteine and forms a hemithioacetal. Leupeptin binds with high affinity to many members of the papain family. In addition to the aldehyde head group, many other functionalities (so-called warheads) that can be used to inhibit cysteine proteases are known (Fig.23.28). Such irreversible inhibitors have been developed in cases of viral proteases and have a Michael-acceptor group at their disposal (i.e., 23.53). This reactive group forms an irreversible bond with cysteine and switches the enzyme off permanently. An attempt has been made to develop inhibitors for cathepsins, calpains, and caspases that can form a reversible connection to the thiol group. Most of these structures are derived from aldehydes or ketones (23.53–23.57). From a chemical point of view, the caspase inhibitor 23.56 from Vertex is interesting. In a cyclic structure, it combines an aspartate-like side chain for the S1 pocket of the enzyme and a capped aldehyde function in the form of a cyclic acetal. The aldehyde is released as active compound from this prodrug.


The binding mode of leupeptin 23.52 in the crystal structure with calpain II. The natural product binds covalently through a terminal aldehyde function to the thiol group of Cys115 to form a thiohemiacetal. The oxyanion hole is formed by the NH group of Cys115 and the carboxamide group of Gln109.


In addition to the aldehyde head group, many other functionalities have been developed that reversibly or irreversibly couple (reactive site is marked in red) to the catalytic cysteine and in doing so block cysteine proteases. Irreversible inhibitors such as 23.52 that have a Michael-acceptor group are available for viral proteases. The two aldehydes 23.54 and 23.55 are development substances for the inhibition of calpains; 23.56 and 23.57 are caspase inhibitors. Compound 23.56 is a prodrug that releases an aspartate-like P1 side chain with ring opening and forms a thiohemiacetal with the protein with its newly generated aldehyde function.
Another group of enzymes that actually belongs to the family of transferases, but follows a cysteine-protease-like mechanism, are the transglutaminases. Nine isoenzymes have been discovered in our genome. They are constructed from four domains and contain a catalytic domain composed of a Cys–His–Asp triad. Their task is the posttranslational modification of proteins (Sect. 26.2), that is, they modify proteins after they have been synthesized in the ribosome. As one aspect, they can carry out the deamination of glutamine residues to glutamate. Furthermore, they catalyze the cross-linking of chain strands on proteins by the transaminase reaction. For this the terminal amino group of a lysine is coupled with a glutamate group with the formation of an isopeptide bond. A proteolytically stable cross-linking results so that transglutaminases can be compared to “biological glue.” Their reaction is analogous to the cysteine proteases. A nucleophilic cysteine initially forms an acyl–enzyme with the substrate’s glutamine with loss of ammonia, which is cleaved in the next step by the reactive lysine. A protein cross-link is the result. Transaminases take on many tasks in our bodies and above all they stabilize tissue proteins. In the blood coagulation cascade, the transglutaminase factor XIII stabilizes the initially formed clot by cross-linking (Sect. 23.4). Therefore inhibitors for factor XIII could be potent anticoagulants. Other transglutaminases are also being investigated as possible targets for drug development. Transglutaminase-2 (TGT2) plays an important role in celiac disease, which is a type of gluten intolerance. Patients that have this disease are sensitive to gluten, which occurs in many grain products as an adhesive protein. They develop inflammation in the mucous membranes of the small intestines, which leads to the destruction of intestinal epithelial cells and severely limits their ability to extract nutrients from food. Inhibitors of TG2 could represent a therapeutic approach. Inhibitors for transglutaminases can be developed by following analogous principles as in the case of the cysteine protease inhibitors.
To date, no cysteine protease inhibitor has managed to achieve market approval, although inhibitors for very many target structures are being developed and are in clinical trials. We can only wait and see whether the first successes will be achieved in the near future.
10 Synopsis
-
Serine proteases belong to the class of hydrolyzing enzymes that cleave amide or ester bonds. Depending on where they cleave a peptide chain, they are classified as amino-, carboxy-, or endopeptidases.
-
Three amino acids, a serine, a histidine, and an aspartic acid that reside at quite distant positions in the sequence, are folded in characteristic proximity to one another. The hydroxyl oxygen atom of the serine performs a nucleophilic attack onto the carbonyl carbon atom of the scissile peptide bond. Its nucleophilicity is enhanced by an H-bond to an adjacent imidazole moiety of a histidine.
-
The histidine accepts a proton from the nucleophilic serine OH group and is thereby transposed into a positively charged state. The neighboring aspartate residue compensates for the positive charge. The simultaneously created negative charge on the former carbonyl oxygen is stabilized by NH functions in the H-bond-donating oxyanion hole. Simultaneously, the carbon atom of the cleaving amide bond rearranges to a tetrahedral geometry.
-
Upon release of the N-terminal part of the peptide substrate the C-terminal part remains covalently bound as acyl–enzyme complex. This is finally degraded via a similar mechanism by using a water molecule as a nucleophile.
-
The residues involved can be different; in particular, the nucleophilic serine can be replaced by a threonine or cysteine. The corresponding enzymes are named threonine and cysteine proteases.
-
The peptide chain to be cleaved is primarily recognized in small binding pockets on the protease surface that accommodate the amino acid side chains on the C-terminal end adjacent to the cleavage site. Their composition determines the chemical building blocks required for inhibitor design to develop highly potent ligands for the protease.
-
A number of warhead groups are known to either reversibly or irreversibly block the catalytic serine, threonine, or cysteine residue.
-
The major contribution to binding affinity and ligand specificity is achieved through binding to the S1 pocket next to the cleavage site.
-
Blood coagulation is a highly regulated cascade of serine proteases. Potent inhibitors for antithrombotic therapy have been developed for thrombin and factor Xa, which are located in the last steps of the cascade.
-
Whereas thrombin and factor Xa exhibit deep and well-structured S1 pockets, elastase exhibits a flat S1 pocket. Binding to this pocket contributes much less to the overall affinity of an inhibitor for this protease and the developed compounds all involve the catalytic serine in a reversible covalent attachment.
-
Irreversible inhibition through covalent bond formation with the catalytic serine is followed to block lipases or transpeptidases. The covalent bond formation is achieved by the ring opening of a reactive highly strained lactone or lactam ring. The latter principle is used by the penicillins and cephalosporins.
-
The transglutaminases follow a very similar enzyme mechanism as the cysteine proteases. However, instead of cleaving a peptide bond in the main chain, they form an isopeptide bond between the terminal amino group of a lysine and the carboxylate group of a glutamate. Because these bonds cause a cross-linking between different segments of the polypeptide chain, they can be compared to biological glue making proteins more stable.
Bibliography
General Literature
Abbenante G, Fairlie DP (2005) Protease inhibitors in the clinic. Med Chem 1:71–104
Babine RE, Bender SL (1997) Molecular recognition of protein-ligand complexes: applications to drug design. Chem Rev 97:1359–1472
Berliner LJ (ed) (1992) Thrombin: structure and function. Plenum, New York
Branden C, Tooze J (1991) Introduction to protein structure. Garland, New York
Kimball SD (1995) Challenges in the development of orally bioavailable thrombin active site inhibitors. Blood Coagul Fibrinolysis 6:511–519
Shafer JA, Gould RJ (eds) (1994) Design of antithrombotic agents, vol 1, Perspectives in drug discovery and design. ESCOM Science Publishers, Leiden, pp 419–550
Steinmetzer T, Stürzebecher J (2004) Progress in the development of synthetic thrombin inhibitors as new orally active anticoagulants. Curr Med Chem 11:2297–2321
Türk B (2006) Targeting proteases: sucesses, failures and future prospects. Nat Rev Drug Discov 5:785–799
Special Literature
Gustafsson D, Bylund R et al (2004) A new oral anticoagulant: the 50-year challenge. Nat Rev Drug Discov 3:649–659
Hilpert K, Ackermann J, Banner DW, Gast A, Gubernator K, Hadvary P, Labler L, Müller K, Schmid G, Tschopp TB, van de Waterbeemd H (1994) Design and synthesis of potent and highly selective thrombin inhibitors. J Med Chem 37:3889–3901
Veale CA, Bernstein PR, Bryant C et al (1995) Nonpeptidic inhibitors of human leukocyte elastase 5 design, synthesis, and x-ray crystallography of a series of orally active 5- aminopyrimidin-6-one-containing trifluorormethyl ketones. J Med Chem 38:98–108
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this entry
Cite this entry
Klebe, G. (2013). Inhibitors of Hydrolases with an Acyl–Enzyme Intermediate. In: Klebe, G. (eds) Drug Design. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-17907-5_23
Download citation
DOI: https://doi.org/10.1007/978-3-642-17907-5_23
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-17906-8
Online ISBN: 978-3-642-17907-5
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences
