
Abstract
Background
Volcanic and tectonic activities in conjunction with Quaternary climate are the main events that shaped the geographical distribution of genetic variation of many lineages. Poeciliopsis infans is the only poeciliid species that was able to colonize the temperate highlands of central Mexico. We inferred the phylogenetic relationships, biogeographic history, and historical demography in the widespread Neotropical species P. infans and correlated this with geological events and the Quaternary glacial-interglacial climate in the highlands of central Mexico, using the mitochondrial genes Cytochrome b and Cytochrome oxidase I and two nuclear loci, Rhodopsin and ribosomal protein S7.
Results
Populations of P. infans were recovered in two well-differentiated clades. The maximum genetic distances between the two clades were 3.3% for cytb, and 1.9% for coxI. The divergence of the two clades occurred ca. 2.83 Myr. Ancestral area reconstruction revealed a complex biogeographical history for P. infans. The Bayesian Skyline Plot showed a demographic decline, although more visible for clade A, and more recently showed a population expansion in the last 0.025 Myr. Finally, the habitat suitability modelling showed that during the LIG, clade B had more areas with high probabilities of presence in comparison to clade A, whereas for the LGM, clade A showed more areas with high probabilities of presence in comparisons to clade B.
Conclusions
Poeciliopsis infans has had a complex evolutionary and biogeographic history, which, as in other co-distributed freshwater fishes, seems to be linked to the volcanic and tectonic activities during the Pliocene or early Pleistocene. Populations of P. infans distributed in lowlands showed a higher level of genetic diversity than populations distributed in highlands, which could be linked to more stable and higher temperatures in lowland areas. The fluctuations in population size through time are in agreement with the continuous fluctuations of the climate of central Mexico.
Similar content being viewed by others


Background
Volcanic and tectonic activities since the Miocene have had a substantial influence on the diversification of many New World taxa [1, 2]. Paleoclimatic events since the Pliocene, mainly from the Quaternary to the present; also have influenced the distribution of many organisms by changing the climates in boreal, temperate and tropical zones. Geologic and quaternary climatic events together are the main factors that have shaped the geographical distribution of genetic variation at the species, population, and community levels in several taxa, including fishes [3,4,5,6,7,8,9,10,11].
The beta diversity of freshwater fishes worldwide, including 80% of all freshwater species described, demonstrates that geographical isolation of drainage basins, combined with Quaternary climate changes, provides a parsimonious explanation for present-day patterns of spatial turnover in the global freshwater fish fauna [12]. Under this context, the geographical location, complex topography, geological dynamism including extensive volcanism since the Miocene and the climatic history of central Mexico, changed during the Quaternary [8, 13], have shaped a biogeographically complex area, characterized by ecological components that have allowed for the coexistence of taxa of Neotropical and Nearctic origins, as well as endemic groups [14, 15].
The biogeographic limits of this area have been largely discussed, and even differ depending on the taxa analyzed [14, 16,17,18]. In general terms, central Mexico is a high plateau bounded by the Sierra Madre Oriental to the east and by the Sierra Madre Occidental to the west and crossed by the Trans Mexican Volcanic Belt (TMVB) from west to east, with elevations up to 1400–1800 MASL [18], a region also called the Mesa Central by some authors [19]. The region is characterized by a temperate climate, thus allowing for the establishment of fish species, mainly of Neartic origin. The climatic changes during the quaternary have influenced the geographical distribution of genetic lineages of terrestrial organisms in space and time, as is the case of snakes [7], lizards [8, 20, 21], birds [9], small mammals [22,23,24], and plants [2].
The dynamic geological processes that have occurred since the Miocene have promoted the genesis and destruction of aquatic ecosystems [25] and have been considered as the primary forces that have influenced the biogeography and the complex evolution of several taxa of freshwater organisms [26,27,28,29,30,31,32]. However, most of the research has focused on understanding the complex evolutionary history of freshwater fishes of Nearctic origin such as Goodeids [28, 29, 33, 34], Cyprinids [30], Catostomids [35], and a combination of taxa [36]. Only one group of species in the TMVB that are of Neotropical origin, genus Poeciliopsis, has been previously investigated in this manner [26].
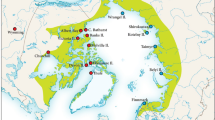
The small live-bearing topminnow Poeciliopsis infans (Woolman, 1894) is a member of the family Poeciliidae, which has more than 220 species of tropical preferences [37]. Poeciliopsis infans is the only Neotropical fish species that has colonized the temperate highlands of the TMVB, including the Lerma-Santiago Basin, headwaters of the Ameca, Armeria, Coahuayana, Balsas and Panuco Basins, as well as endorheic lakes in the region (Fig. 1) [38]. Poeciliopsis infans is co-distributed with fishes of Nearctic origin of the families Goodeidae, Catostomidae, Ictaluridae and Cyprinidae [38].

Sampling locations and the biogeographical regions where Poeciliopsis infans is distributed. The colors of the biogeographical regions corresponded with the colors used in the phylogenetic analyses. The codes of the biogeographical regions are as follows: (Mag) Magdalena Lake; (Etz) Etzatlan-San Marcos region; (Ver) Verde River; (Ame) Ameca River; (Ato) Atotonilco Lake as the number 1; (Sma) San Marcos Lake as the number 2; (Say) Sayula Lake as the number 3; (Zap) Zapotlan Lake as the number 4, these four lakes belong to Sayula region; (San) Grande de Santiago River; (Tam) Tamazula River; (Pat) Patzcuaro Lake; (Cha) Chapala Lake; (Bal) Balsas River; (Cot) Cotija Lake; (Lle) Lower Lerma Basin; (Mle) Middle Lerma Basin; (Zac) Zacapu region; and, (Cui) Cuitzeo Lake. The meters above see level (MASL) are indicated with level curves
Accordingly, since the members of the Poeciliidae are a group of Neotropical origin, most of them are adapted to tropical habitats. Furthermore, since P. infans is the only poeciliid species living in the temperate highlands of central Mexico, an area dominated by fish species of Neartic origin, we expect that the evolutionary history of P. infans could be explained by recent volcanic and tectonic activities. However, since P. infans is a species evolving in the marginal and temperate areas adjacent to the distribution of all of the other species Poeciliopsis, it may not follow the same patterns as other co-distributed species in the region.
We extensively sampled throughout the distribution of P. infans (Teleostomi: Poeciliidae), and gathered mtDNA and nDNA sequences to infer phylogeographic variation, historical biogeography, and historical demography of the widespread P. infans. These data will allow us to examine the influence of geological history and Quaternary glacial-interglacial climatic events, in space and time, in the evolutionary history of Neotropical species evolving in a predominant Nearctic area.
Methods
Sample collection
Two hundred fifty-six specimens of P. infans from throughout the range were collected (Fig. 1 and Table 1) using electrofishing equipment and trawl nets. Tissue samples (fin clips) were preserved in 95% ethanol for DNA extraction, and a maximum of five specimens per site were preserved in 5% formalin. Despite intensive collection efforts, samples were not obtained from some biogeographic regions. Fish and tissue samples were deposited in the fish collection of the Universidad Michoacana de San Nicolas de Hidalgo, Mexico (SEMARNAT registration number MICH-PEC-227-07-09; for voucher numbers see Additional file 1).
DNA extraction, PCR amplification and sequencing
Total genomic DNA was isolated with the Qiagen BioSprint Dneasy Tissue and Blood Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. Two hundred fifty-six individuals were amplified for Cytochrome b (cytb: 1083 bp) and 249 individuals for Cytochrome Oxidase Subunit I (coxI: 631 bp), for 1771 bp of mitochondrial sequence data. The first intron, a fragment of second intron, and the second exon of the gene coding for the S7 ribosomal protein (S7: 859 bp), and the gene coding for the Rhodopsin protein (RHO: 845 bp) were amplified for 1704 bp of nuclear sequence data from 201 individuals. For this subset, individuals were chosen in order to represent all biogeographic regions and all the variation shown in mitochondrial dataset. In addition, five sequences of cytb were obtained from GenBank with the following accession numbers: AF412134 of Lerma River, AF412135 of Ameca River, AF412136 of Chapala Lake, AF412137 of Santiago River and AF412138 of Panuco River.
Each fragment was individually amplified using the Polymerase Chain Reaction (PCR) in volumes of 12.5 μl, containing 4.25 μl ultrapure water, 0.5 μl of each 0.2 μM primer, 6.25 μl Dream Taq Green PCR Master Mix 2× (Thermo Scientific), and 1 μl (ca. 10–100 ng) of DNA template. The specific PCR protocols of each gene are provided in Additional file 2. The recovered PCR products were purified using ExoSAP-IT (USB Corp.) and submitted to Macrogen Inc. (The Netherlands) for sequencing. Nucleotide sequences were edited and aligned in Mega v.6.06 [39], and the chromatographs were examined by eye. For the nuclear gene (RHO), the heterozygous genotypes were phased using DNAsp v.5.10 [40] with the algorithm provided by PHASE v.2.0 [41]. Whenever sequences of S7 showed heterozygous positions defined by indels, a manual reconstruction of the two-allele phases was performed following the procedure described by [42]. The obtained sequences were deposited in GenBank under the follow access number: for cytb (MG028009 to MG028278), for coxI (MG028279 to MG028543), for RHO (MG100617 to MG100824) and for S7 (MG366191 to MG366482), (see Additional file 1).
Phylogenetic analyses and haplotype networks
Recombination of the nuclear genes (RHO, P = 1.0; S7, P = 0.23) was tested using the PHI test in Splitstree v.4.13 [43]. DNA sequences of each of the four genes (cytb, coxI, S7 and RHO) were collapsed to haplotypes using the web-based program ALTER [44].
The phylogenetic analyses were conducted with each gene separately, with concatenated datasets for mitochondrial genes, for concatenated nuclear genes, and for the four genes combined. The phylogenetic analyses with concatenated genes were performed with the available sequences of the nuclear genes (201 specimens), the sequences of mitochondrial genes were adjusted to these number, which were representative of all biogeographic regions and account the variability found in mitochondrial genes.
The performance of the phylogenies with the four concatenated genes vs. mitochondrial genes concatenated and vs. nuclear genes concatenated were assessed using Bayes Factors (BF). Bayes Factors were calculated from the estimated harmonic means of likelihood using the sump command in MrBayes. Decisions were made based on the 2ln BF criterion, with BF > or = 10 considered as strong evidence for rejecting the null hypothesis [45].
Model selection based on Akaike information criterion (AIC) and optimal partition settings were performed using PartitionFinder v.1.1.0 [46], and recovered the best partition by assigning substitution models for each gene. The parameters of each model are provided in Additional file 3.
Gene trees were constructed with Maximum Likelihood implemented in RAxMLGUI v.1.3.1 [47, 48], using the substitution model implemented for each gene. The GTR + G + I [48] substitution model was used for the concatenated genes matrix and 10,000 bootstrap replicates with the algorithm ML + rapid bootstrap. The relative stability of clades was evaluated by 1000 non-parametric bootstrap replicates [49].
Bayesian Inference was implemented in MrBayes v.3.2.1 [50], with the substitution models for each gene obtained in PartitionFinder. Analyses were run for 10 million generations, with two independent runs implementing four Markov Chain Monte Carlo (MCMC) processes and sampling every 500 generations. We evaluated the chains for convergence with the log-likelihood (-InL) values of the two independent runs using Tracer v.1.5 [51], and discarded 10% as burn-in to construct the consensus tree. Poeciliopsis prolifica was used as outgroup, based on the results of a previous study [26].
In order to determine the geographic distribution of haplotypes for all populations of P. infans for nuclear genes, we reconstructed two independent TCS haplotype networks (a phylogenetic network estimation using statistical parsimony) for RHO and S7 sequences using PopArt v.1.7 (http://popart.otago.ac.nz).
Divergence time estimation and genetic distances
The program BEAST v.1.8.1 [52] was used to estimate the most recent common ancestor for clades within P. infans. This analysis was carried out with a subset of 145 sequences that include all different haplotypes for all genes. Because the lack of fossil data for Poeciliopsis, the molecular clock was calibrated using the mutation rate of cytb in teleosts of 0.76–2.2%/million years [53,54,55]. Since the mutation rate is not available for the other genes, they were included in the analysis without calibration information.
The model parameters were unlinked across cytb, coxI, S7 and RHO genes and substitution models were set according to the selected model for each gene by PartitionFinder v. 1.1.0 [46]. We applied a lognormal relaxed clock (Uncorrelated) model on branch length [56]. We selected the tree prior Coalescent: Extended Bayesian Skyline Plot [57], and estimated a starting tree using the random method. A MCMC analysis with 50 million of generations was conducted, and sampled every 1000 generations. We assessed whether parameter values had reached effective sample size and convergence in Tracer v.1.5. [51]. Finally, the maximum clade credibility tree was built, discarding the first 10% of the trees as burn-in, using Tree Annotator v.1.8.1. [52].
Uncorrected genetic distances were calculated among the recovered groups in the phylogenetic trees for each mitochondrial gene (cytb, coxI), and between all individuals for S7 and RHO in Mega v.6.06 [39]. A bootstrapping process was performed with 1000 repetitions.
Genetic diversity and population structure
For each gene (cytb, coxI, S7 and RHO), the number of haplotypes (H), polymorphic sites (S), nucleotide (π) and haplotype (h) diversities were obtained to estimate genetic diversity levels in all populations of P. infans. To examine genetic differentiation at different hierarchical levels, as well as geographical patterns of population subdivision, an analysis of molecular variance (AMOVA) was conducted. The AMOVAs were implemented for the four separate genes and groupings according to: 1) inferred clades in phylogenetic analyses, 2) according to the biogeographic regions sensu [28] and, a third analysis was performed without a priori groupings. The analyses were conducted using 10,000 permutations to assess significance values. All genetic diversity analyses and AMOVAs were performed in Arlequin v.3.5.1 [58].
Ancestral area reconstruction
The ancestral area reconstruction for P. infans was estimated using the statistical Dispersal-Vicariance (S-DIVA) method [59], and the Dispersal-Extinction-Cladogenesis (DEC) model [60, 61] as implemented in RASP v.3.2 software [62]. These methods use statistical approaches to reconstruct biogeographic history, and allowed us to compare both results. The ultrametric and dichotomous tree obtained for cytb in BEAST was used as the tree topology on which ancestral areas were mapped. For both analyses, the maximum number of areas was limited to two. For these analyses, the distributional area of P. infans was divided into 15 biogeographic regions: (Mag) Magdalena Lake; (Etz) Etzatlán-San Marcos region; (Ver) Verde River; (Ame) Ameca River; (San) Grande de Santiago River; (Tam) Tamazula River; (Pat) Patzcuaro Lake; (Cha) Chapala Lake; (Bal) Balsas River; (Cot) Cotija Lake; (Lle) Lower Lerma River; (Mle) Middle Lerma River; (Zac) Zacapu Lake and (Cui) Cuitzeo Lake, and, within Sayula region are considered the follow Lakes: (Ato) Atotonilco, (Sma) San Marcos, (Say) Sayula and (Zap) Zapotlan [28, 29].
Historical demography
The population size fluctuations through time were tested with a Coalescent Bayesian Skyline Plot (BSP) analysis [63] as implemented in BEAST v.1.8.1 [52]. This analysis only was implemented with sequences of cytb due the higher number of available sequences. The substitution rate was the same as the divergence time analysis and the substitution model was set according to the select model by PartitionFinder v. 1.1.0 [46].
An uncorrelated relaxed clock model was set a priori, and 70 million generations were run, sampled every 1000 generations. Convergence was assessed with Tracer v.1.5 [51]. The first 10% of the states were discarded as burn-in.
Poeciliopsis infans Distribution modelling
To evaluate the concordance between the historical demography obtained in BSP analyses and the potential distribution of P. infans in the past, we carried out a species distribution modelling analyses at different temporal scales [64]. The estimations of the current and past population distribution were inferred with MaxEnt v.3.3.1 [65]. Geographical coordinates of 162 sites registered in the database of the Colección de Peces de la Universidad Michoacana de San Nicolás de Hidalgo were used as presence data (see Additional file 4). For the environmental data, we used 19 bioclimatic variables downloaded from WORLDCLIM database [66], http://www.worldclim.org, at a resolution of 30 arc-seconds for Last Inter Glacial (LIG) and 2.5 min for Last Glacial Maximum (LGM). The WORLDCLIM variables represent biologically meaningful summaries of precipitation and temperature in the present (1950–2000), and for the past, the Community Climate System Model (CCSM) for the LGM: 0.025 Myr, and the LIG: 0.15–0.10 Myr periods. To construct the models, we employed a logistic output [65], and the default settings. The value of the regularization multiplier was tested for 0.5, 1.0, 1.5 and 2.0, but the highest AUC value was for a regularization multiplier of one, and this value was used in all analyses. The model was run with 100 subsample replicates estimating mean habitat suitability values (S).
To evaluate if the performance of all distribution models was better than one, a random model was assessed using 75% of the presence data to run the model and the remaining 25% for statistical testing. In addition, the area under the receiver operating characteristic curve (AUC) was estimated to assess the accuracy of the models. A jackknife test of variables of importance was conducted to evaluate the relative importance of each climate variable [67]. Variables contributing the least to the model or those highly correlated [68] were removed for each model. The correlation of variables was evaluated through the response curves, which reflect the dependencies induced by correlations between the selected variable and other variables.
Results
Phylogenetic relationships and haplotype networks
The ambiguously aligned positions that showed the sequences of the S7 gene are see in Additional file 5. The BF comparison indicated that the analyses using the four concatenated genes provided a better explanation of the data than the mitochondrial genes concatenated or nuclear genes concatenated (BF of four genes concatenated vs mitochondrial genes concatenated = 1.87 and BF of four concatenated genes vs nuclear genes concatenated = 2.2).
The phylogenetic results for both analyses (Maximum likelihood and Bayesian Inference) recovered two well differentiated and well supported clades for each mitochondrial gene (cytb: 1083 bp and coxI: 631 bp) (Additional files 6 and 7), for the concatenated mitochondrial genes (cytb, coxI: 1771 bp) (Additional file 8), and for all concatenated genes (cytb, coxI, S7 and RHO: 3475 bp; Fig. 2).

The Bayesian inference tree of Poeciliopsis infans from concatenated sequences of two mitochondrial (cytb, coxI) and two nuclear genes (S7 and RHO). Bayesian posterior probability (> 0.9; above the branches) and maximum likelihood bootstrap values (> 80%; below the branches) are indicated. The divergence time estimations are shown with 95% HPD
In general terms, clade A clustered populations of the biogeographic regions of lowlands of TMVB, while; clade B clustered populations of the biogeographic regions of the highlands of the TMVB. However, one sample from Ameca, Magdalena and Sayula regions respectively were grouped in clade B. (Figs. 1 and 2).
Clade A clustered individuals of seven biogeographic regions is better in three sub-clades: sub-clade A1 included individuals from the Etzatlan-San Marcos region, Magdalena Lake and few individuals of Verde River; sub-clade A2 clustered individuals of the Ameca, Verde, and Grande de Santiago Rivers and the Atotonilco Lake of the Sayula region; while the sub-clade A3 clustered individuals of the Sayula, San Marcos and Zapotlan Lakes within the Sayula region, as well as samples from the Ameca River basin and Tuxpan River of the Tamazula biogeographic region. These three sub-clades were well supported in the Bayesian inference analysis, but the bootstrap support for sub-clade A1 was low (87%) (Fig. 2).
The second clade, clade B, clustered individuals of the Middle and Lower Lerma, Grande de Santiago and Balsas Rivers, as well as Cuitzeo, Patzcuaro, Chapala, Cotija and Zacapu Lakes. Also, three samples corresponding to regions that were clustered in clade A were recovered in clade B and corresponded to Magdalena and Atotonilco Lakes, and the Ameca River (Fig. 2). The phylogeny based on each nuclear gene and with both concatenated nuclear genes failed to recover resolved relationships (Additional files 9, 10 and 11).
For the haplotype networks based on the nuclear genes, two haplogroups (A and B) were recovered and these are highly congruent with the concatenated gene phylogeny. There are, however, shared haplotypes between them, with RHO network sharing the most haplotypes between groups. The first haplogroup (A), grouped individuals of Ameca, Grande de Santiago, Sayula, Magdalena, Verde and Etzatlan-San Marcos regions, for S7, one individual of the Chapala region was found in this haplogroup. The second haplogroup (B) for both nuclear genes, grouped individuals of the Lower and Middle Lerma, Grande de Santiago, Balsas, Zacapu, Cuitzeo, Patzcuaro, Cotija and Chapala regions. Some individuals of the Sayula region were grouped in the haplogroup B for both genes (see Additional file 12).
Divergence time estimation and genetic distances
The first isolation event in P. infans, separating clades A and B, was estimated to have occurred near the middle Pliocene and middle Pleistocene ca. 2.83 Myr (95% HPD: 1.25–4.41 Myr). The first separation event of the three sub-clades within clade A was estimated to have occurred during the Pleistocene period ca. 0.95 Myr (95% HPD: 0.39–1.5 Myr), whereas the rest of the isolation events were calculated in less of a million of years (Figs. 2 and 3).

a Divergence times, biogeographical events in Poeciliopsis infans, b geographic distribution of inferred clades and sub-clades across of TMVB of P. infans, and tectonic, volcanic and hydrographical features of the TMVB
The uncorrected mean genetic distances (p-distance) calculated between clade B and sub-clades A1, A2 and A3 for the mitochondrial genes ranged from 0.8–3.3% for cytb; and 0.7–1.9% for coxI (Table 2). The minimum genetic distances found for cytb were between sub-clade A1 and sub-clade A2 (0.8%), and the maximum distances were between sub-clade A3 and clade B (3.3%). Based on coxI the minimum genetic distances were observed between sub-clade A2 and sub-clade A3 (0.7%), and the maximum were between sub-clade A1 and clade B (1.9%). Between the three sub-clades of clade A the genetic distances were 0.8–1.1% for cytb and 0.7–1.3% for coxI (Table 2). For nuclear genes the genetic distances between all individuals included both alleles for each sequence, ranged between 0.0–0.5% for RHO and between 0.0–0.6% for S7.
Genetic diversity and population structure
The highest haplotype diversity was found in Cuitzeo (h = 0.81) for cytb and in the Verde River (h = 0.62) for coxI, followed by the Ameca River Basin for both genes (h = 0.75 for cytb and 0.42 for coxI), while, null haplotype diversity were found in the Patzcuaro, Cotija and Balsas regions. Atotonilco Lake, within the Sayula region and the Verde River exhibited the highest nucleotide diversity for cytb and coxI (π = 0.006, π = 0.004) respectively (Table 3).
For the nuclear genes, the highest haplotype diversity was found in the Atotonilco (h = 0.97), and Ameca (h = 0.9) for S7, and the Middle Lerma (h = 0.73) for RHO. Absence of haplotype diversity was found for the Middle Lerma for S7 and Cuitzeo for RHO. The populations of Atotonilco (π = 0.01) and the Middle Lerma (π = 0.001) exhibited the highest nucleotide diversity for S7 and RHO respectively (Table 4). When the AMOVA was performed without groups a priori, the highest variation for all genes were among populations (cytb: 91.17%, coxI: 96.07%, S7: 69.5% and RHO: 60.64%) and not within populations. The AMOVA for all genes showed a high percentage of variation when populations were grouped according to the recovered clades and sub-clades from the phylogenetic analyses (cytb: 92.24%, coxI: 91.03%, S7: 45.48% and RHO: 53.21%), but not when populations were grouped according to the biogeographic regions [18] (cytb: 31.31%, coxI: 27.09%, S7: -99.28% and RHO: 53.21) (Tables 5 and 6).
Ancestral area reconstruction
Ancestral area reconstruction using DEC and S-DIVA revealed a complex biogeographical history for P. infans, with several events of dispersal and vicariance (Figs. 3 and 4). Vicariance was most common in ancient events in comparison to dispersal events. For both analyses, the ancestral areas estimated for P. infans were Zacapu Lake and Ameca River, but with low probabilities (6.5% for DEC; 16.7% for S-DIVA). The same explanation for both analyses was found for the biogeographic history of the northwest populations of P. infans clustered in phylogenetic clade A. The biogeographical route for this clade included a dispersal event from the Ameca River toward the Sayula and Etzatlan-San Marcos regions, followed by a vicariant event that separated the populations of Ameca River with respect to Etzatlan-San Marcos and Sayula regions. A more recent dispersal event from the Ameca to the Verde River was recovered (S-DIVA > 50%, DEC > 27% of probabilities).

Ancestral area reconstruction with a DEC and b S-DIVA base in BEAST tree using the biogeographical regions proposed by Domínguez-Domínguez et al., (2006)
The biogeographical history of the southeastern clade B differs slightly in both biogeographical methods. For both methods, one dispersal event occurred from Zacapu toward Cuitzeo, followed by a vicariant event that separated these two regions. For DEC, dispersal events occurred from Zacapu toward the Lower Lerma and from here to Chapala, while there was also a recent dispersal event from Cuitzeo to the Middle Lerma. The S-DIVA differed with the DEC in that dispersal events from Zacapu were toward Chapala and once both regions were separated, a second dispersal event occurred from Chapala to the Lower Lerma (S-DIVA 100% and DEC > 60%) (Figs. 3 and 4).
Historical demography
The BSP analyses for cytb for populations of clade A showed a demographic decline for Magdalena Lake, San Marcos, Sayula and Atotonilco Lakes (belonging to Sayula region), Ameca, Grande de Santiago and Etzatlan-San Marcos regions between 0.15 and 0.1 Myrs. More recently, after a demographic decline, a population expansion was detected in the last 0.025 Myr. For the Verde River, a population reduction was detected following a more recent population expansion. For this clade, the regions Zapotlan and Tamazula were not included due to the low number of individuals (Fig. 5).

Demographic history of populations of each biogeographical region grouped in clade A using Bayesian Skyline Plots (BSP) from cytb sequences. Dotted lines represent the location of the upper bound (Max), the mean (X) and lower bound (Min) of the HPD = 95%
For clade B, all analyzed groups revealed a demographic decline in the last 0.15–0.1 Myr, followed by population expansion around ≤0.18 Myr, as found for clade A. For this clade the Balsas, Cotija and Middle Lerma basins were not included due the low number of samples (Fig. 6).

Demographic history of populations of each biogeographical region grouped in clade B using Bayesian Skyline Plots (BSP) from cytb sequences. Dotted lines represent the location of the upper bound (Max), the mean (X) and lower bound (Min) of the HPD = 95%
Poeciliopsis infans Distribution modelling
The habitat suitability modelling for populations of P. infans estimated for current (1965–1978) and past (LIG: 0.15–0.10 Myr, and LGM: 0.025–0.020 Myr) time periods, showed high precision and acceptable predictive power with all models (AUC > 0.96) [69]. For the current conditions, the two variables with the highest gain were BIO3 Isothermality (BIO2/BIO7)*100 and BIO6 Min temperature of coldest month. In the LIG model, the two variables with the highest gain were BIO1 Annual Mean Temperature and BIO16 Precipitation of Wettest Quarter. For the LGM model, the two variables with the highest gain were BIO4 Temperature seasonality (standard deviation*100), and BIO6 Min Temperature of coldest month. The modelling of habitat suitability showed that in the LGM, the habitat suitability for P. infans was better in the lowlands, whereas for the LIG, the habitat suitability increased in the highlands (Fig. 7).

Species distribution modelling for: Current period, Last Inter Glacial (LIG: 0.15–0.10 Myr) and Last Glacial Maximum (LGM: 0.025–0.020 Myr), the probability of presence of the species is in scale of colors
Discussion
Biogeographic and evolutionary history of P. infans
The recovery of two well-differentiated clades within P. infans (Fig 2 and Additional file 8) indicates a long history of isolation, with subsequent genetic differentiation, which seems to be linked to the intense volcanic and tectonic activity in central Mexico during the Pliocene (Figs. 2 and 3). This pattern previously has been reported, in which the genetic differentiation has been linked with the past configuration of the rivers more than the current hydrology of this region of central Mexico, as is the case for the goodeid, Zoogoneticus quitzeoensis (Bean, 1898), and for species of the cyprinid genus Algansea [30, 33].
Populations of P. infans are largely differentiated by the isolation of watersheds as have occurred in other freshwater fishes with low dispersal ability; however, some geographical features within a river or basin have been shown to be sufficient to differentiate populations of P. infans [26].
We recovered the samples from the Rio Grande de Santiago Basin grouped in the two main clades for phylogenetic analyses and in the two haplogroups for haplotype networks (A, B). This pattern, in which the samples from the Santiago River were grouped in two clades, previously was found in another study [26]. Specimens from the Rio Grande de Santiago Basin sampled upstream of the falls, el Salto de Juanacatlan, belong to clade B, whereas populations sampled downstream of the falls were grouped within clade A, suggesting that the Salto de Juanacatlan formation, a waterfall 20 m high, has been an important and ancient barrier promoting differentiation between populations. This geologic feature previously has been shown to represent a barrier for fish faunal interchange between the Santiago and Chapala Lake [26, 38].
Ancestral area reconstructions recovered very low marginal probabilities for biogeographical routes from the ancestral areas of all populations of P. infans (DEC = 0.01; S-DIVA = 0.08; Figs. 3 and 4). Therefore, both ancestral area reconstructions were unable to resolve the state of the node separating clade A from clade B, but were able to reconstruct the ancestral areas and biogeographical routes within each clade.
Despite these low marginal probabilities, the most plausible event that separated the Ameca River and Lake Zacapu of its ancestral area of distribution was a vicariant event. Our date estimation for the diversification of the two main clades (A and B) was ca. 2.83 Myr (1.25–4.41 Myr), between the middle Pliocene and the early Pleistocene periods (Figs. 3 and 4).
The geological activity during the middle Pliocene and early Pleistocene in central Mexico promoted the cladogenesis of clade A from clade B. This deep divergence of the two clades is coincident with the interruption of the ancient connection of the upper Ameca River with drainages in central Mexico by tectonic and volcanic activity at ca. 3–1 Myr [70], (Fig. 3). As a result, this dispersal route could have been blocked at the end of the Pliocene and during the Pleistocene. This occurred when the hydrological systems that shaped the complex Chapala-Lerma Paleosystem (Ameca River, Magdalena, Chapala, Lerma River and the lakes distributed along the Colima graben) became isolated due to volcanic and tectonic activity in the triple junction area, the Ameca and San Marcos faults at ca. 3.5–1.5 Myr (Fig. 3) [71].
The isolation of the region where clade A is distributed during the Pliocene, has also been reported in other freshwater species including the cyprinids Yuriria amatlana Domínguez-Domínguez et al., 2007 and Algansea amecae Pérez-Rodríguez et al., 2009, as well as the goodeid Allotoca goslinei Smith & Miller, 1987 [29, 30, 72, 73]. In addition, other freshwater organisms shown a similar pattern, as Cambarellus chapalanus (Faxon, 1898), which has two divergent genetic groups, one distributed in Chapala Lake and the other in the Ameca River Basin separated at ca. 2.6 Myr [32]. A similar pattern was mentioned before for Poeciliopsis [26], who suggested that the ancestors of the strictly northern clade of Poeciliopsis must have been distributed across the region presently occupied by our clade B, which includes Zacapu.
The discrepancies between the mitochondrial and nuclear genes are related to the mixture of some individuals of haplogroup A with haplogroup B. In this case, considering the high genetic distances with mitochondrial genes, the divergence times, and the results from the AMOVAs, we suggested that it could be the result of retention of ancestral polymorphisms, as has been shown for other freshwater fishes of central Mexico [74].
Biogeography within clade A
The biogeographic analyses showed a dispersal event between Sayula and the Ameca basin, suggesting an early connection more than a million years ago. This connection was previously found for P. infans between the Ameca River and Atotonilco Lake of the Sayula region [26].
The isolation of the Ameca, Etzatlan-San Marcos, Magdalena and Lakes of the Sayula region, could be due to the formation of the current watersheds during the Pleistocene epoch ca. 0.95 Myr (95% HPD: 0.39–1.5 Myr; Figs. 2 and 3), when the connections of the Ameca River and Atotonilco-Sayula Lakes were disrupted by Pleistocene volcanism and the intense tectonic activity of the so called triple junction [71, 75]. This is also congruent with the presence of Ameca splendens Miller & Fitzsimons, 1971, in the Ameca River and Sayula regions, with a divergence time between the two populations calculated in less than a million of years [29].
Other dispersal events from the Ameca to Etzatlan-San Marcos and Magdalena regions were recovered. The climatic changes during this pluvial-interpluvial period, beginning ca. 0.90 Myr [4, 76], could have promoted this dispersal event when Magdalena Lake was considerably larger and extended to Etzatlan-San Marcos area [77]. After that time, the Ameca was isolated from the Etzatlan-San Marcos and Magdalena regions by a vicariant event ca. 0.72 Myr (DEC = 0.209; S-DIVA = 1.0), which could be associated with the reactivation of the Plan de Barrancas fault during the Quaternary (ca. 1.0 Myr), and with the Amatlán de Cañas half graben (Fig. 3) that was formed ca. 3.4 Myr [13, 70, 78].
Finally, a dispersal event from the Ameca River to the Verde and Santiago Rivers also was found (ca. 0.14 Myr), and this is supported by previous findings suggesting that these populations have been connected until very recently through stream capture of the Ameca and Verde Rivers, which was facilitated by the volcanism in the Tepic-Zacoalco graben [26, 79], (Fig. 3).
The biogeographic events that isolated the three recovered sub-clades are supported by the AMOVA analysis, maximizing the ΦCT when samples were grouped into four groups, included the three sub-clades within clade A (Tables 5 and 6), but not when they were grouped according to biogeographic regions as have been proposed for other freshwater fishes, including goodeids and cyprinids [33, 36].
Since goodeids, cyprinids, and P. infans have evolved in spatiotemporal congruence, the differences found in the evolutionary history of P. infans could be related to the biogeographic origin of each group. Poeciliopsis infans has a Neotropical origin, whereas goodeids and cyprinids are of Neartic origin [18]. As a result, we expect that Quaternary climate changes have influenced genetic variation and the distributional patterns of P. infans. Moreover, the genetic diversity was higher for regions clustered in the clade A (lowlands) than for regions grouped in clade B (highlands). This could also be linked to more stable high temperatures in lowland areas, which could also promote the diversification of populations found for this lowland clade (clade A). This pattern has been reported in plants that shown that the habitat and environment changes affect the genetic diversity [80,81,82,83], as is the case of Caragana microphylla, a species distributed in two different habitats that shown that populations from the high temperature region had lower genetic diversity than those from medium and low temperature regions [81]. The only exception of this pattern is the Magdalena Lake population, which has the lowest genetic diversity within this clade, a pattern that is explained due to the history of instability of this hydrological basin, with extreme and intermittent periods of flooding and drying [84], promoting recurrent events of bottlenecks and loss of genetic diversity.
Also, other factors, in addition to the environment, such as life-history traits, breeding systems, dispersal mechanism, geographic variation, range and life span and the histories of populations have affected genetic variation between populations [81,82,83].
Biogeography within clade B
Our results are in accordance with the proposed connections and isolation between the Cuitzeo and Zacapu regions, which has occurred several times during the Pleistocene [85]. The connection between both regions has been postulated to occur through the Chucandiro-Huaniqueo paleo-river, a connection disrupted less than 1 Myr, due to the volcanism of the Tarasco corridor and the activity of the Northeast-Southeast fault system ca. 0.7 to 0.5 Myr [86], (Fig. 3). This disruption has been proposed as the cause of the isolation of different fish species between Cuitzeo and Zacapu including the goodeids Skiffia lermae (Meek, 1902), Goodea atripinnis (Jordan, 1880), Alloophorus robustus (Bean, 1892) and Hubbsina turneri (de Buen, 1940) [29, 85], and the cyprinids Algansea tincella (Valenciennes, 1844) and Yuriria alta (Jordan, 1880) [30, 72].
After the isolation of Zacapu and Cuitzeo, the DEC and S-DIVA showed a dispersal event from Cuitzeo towards the middle Lerma River, an event that is in accordance with recent isolation of this hydrological basin with respect to Zacapu and Cuitzeo lakes. This event was previously proposed for the goodeids Hubbsina turneri [85], and Zoogoneticus quitzeoensis (Bean, 1898), that are distributed in Cuitzeo, Zacapu, and the middle Lerma [33], as well as Neotoca bilineata (Bean, 1887) for which a low level of genetic differentiation (mtDNA) was found for populations from Cuitzeo and the middle Lerma [87].
Regarding the second biogeographic route for this clade, which is different for the DEC and S-DIVA analyses, the most plausible route is a dispersal event from Zacapu toward the lower Lerma Basin, followed by a dispersal event toward Chapala Lake and the Santiago River respectively, as is found in DEC. Zacapu is currently connected with the lower Lerma through the Angulo River, for which P. infans could have dispersed toward Chapala Lake and the Santiago River via the lower Lerma. This information is congruent with the presence of Notropis calientis (Jordan & Snyder, 1899), Yuriria alta, Algansea tincella, Xenotoca variata (Bean, 1887), Chapalichthys encaustus (Jordan & Snyder, 1899), Allotoca dugesii (Bean, 1887) and Moxostoma austrinum Bean, 1880, in the Middle Lerma, Lower Lerma and Grande de Santiago basins, as well as in Chapala Lake region [35, 36].
Human mediated dispersion
In general, we found biogeographic correspondence in the distribution of clades, but incongruences were also found for some populations and areas, as is the case of Balsas, Cotija and Patzcuaro, populations that shown a null genetic and haplotype diversities, with all sampled individuals belong to the most common haplotype of clade B.
These results have two possible explanations: 1) a secondary dispersal and colonization event, which is unlikely due to the historical isolation of all of those drainages with respect to contiguous drainages [29, 88], or 2) a founder effect due a dispersal event mediated by humans. Human-based introductions represent the most probable explanation according to the geographic distribution of related haplotypes and the null genetic diversity of Balsas, Cotija and Patzcuaro regions, since for Patzcuaro this species has been reported as a human-mediate introduction [89, 90] (Tables 3 and 4).
Several species in the family Poeciliidae have been introduced for mosquito control worldwide and have spread successfully to over 40 countries [91]. Other possible ways of introduction of poeciliids is the release of the organisms by aquarists, through the use of this fish as food source for commercial introduced fish, or accidentally transported with commercially important fishes stocked into water bodies, such a species of tilapia (Oreochromis and Tilapia), which have been widely introduced throughout Mexico [89, 92].
Historical demography and distribution modeling
Fluctuations in population size shown by BSP in populations of both clades of P. infans agree with the continuous fluctuations of the climate and water levels of hydrological systems in central Mexico due to glacial and interglacial cycles [93].
For clade A, the analysis of historical demography showed a demographic decline at ca 0.15–0.1 Myr, followed by a recent population expansion, estimated to start around 0.025 Myr in most of the populations analyzed (Fig. 5). These genetic based analyses are congruent with the distribution modeling results, in which drainages where clade A is distributed, show restricted areas with high probabilities (≥0.77) to support populations of P. infans during the LIG (0.15–0.10 Myr), localized mainly to a small area within the Ameca region (Fig. 7). Whereas for the LGM (0.025–0.020 Myr), an increase in the areas with high probabilities (≥0.77) to support populations of P. infans is observed, covering most of the present day distribution of clade A populations. This recent population expansion for almost all populations of clade A could explain why the genetic diversity is highest in this clade rather than in the clade B. It is well known that stable populations that persisted from the LGM to the present harbor disproportionately large amounts of unique genetic diversity [94].
The decline in the clade B population seems to starts after 0.075 Myr, followed by a population expansion at ca. ≤0.018 Myr in all the populations analyzed, however, we take with caution the population expansion of some biogeographic regions for both clades, because the size increase is out of the HPD limits (Fig. 6).
These results are also congruent with the distribution modeling results, since the distribution modeling during LIG (0.15–0.10 Myr) also showed a high proportion of areas where clade B is distributed with high probabilities (≥0.77) to support populations of P. infans. Whereas for the LGM (0.025–0.020 Myr), a decrease in the area with high probability of presence (≥ 0.77) is observed for areas were clade B is distributed. These results are expected for a species with tropical preferences inhabiting highlands, where temperatures declines of 8.5 °C during LGM have been postulated [95], followed by a expansion of the distribution range when the last ice age ended and an increase of the temperature and water level of hydrological systems have been recorded [93]. It has been shown that the climatic fluctuations were accompanied by a loss of genetic diversity and even extinction of populations that were unable to adapt to these changes and find suitable conditions [96], as could be the case for P. infans that is restricted to basin drainages. This is congruent with the distribution modeling of clade B distributed in highlands of central Mexico, and explains the low genetic diversity found in almost all populations within this clade.
Finally, in the present day modeling that represents an Inter-glacial period, it showed an extended distributional area with a probability of presence ≥0.77 in the upper parts of the distributional range for both clades A and B. These results of population expansion in lower areas during Glacial Maximum and in upper areas during Inter Glacial periods are also congruent with the Neotropical origin of P. infans, suggesting that areas that maintain high temperatures are more suitable for P. infans. This is also congruent with reports suggesting the displacement of plant communities as a response to the climate cooling, resulting in the migration to low altitudes [97], for which the climate change in glacial cycles could be responsible of the modifications in the composition of the communities favoring more resistant species to the new environmental conditions [93, 97].
Conservation implications
The regions occupied by P. infans have been heavily impacted by habitat loss due to overexploitation, pollution, habitat degradation, and the introduction of non-native species [98,99,100]. The negative impacts of these anthropogenic activities on native freshwater fishes of central Mexico are well known [33, 101,102,103]. These activities can exacerbate the loss of genetic diversity, which is especially harmful for a species with a long history of instability due to the natural climatic fluctuations, as is the case of P. infans. This is especially true for those populations distributed in clade B (Tables 3 and 4). For this reason, the establishment of each recovered clade and sub-clade as Operational Conservation Unit (OCU) [104] is necessary in order to conserve the unique genetic pool that each clade represents.
Conclusions
The results of this study indicate that P. infans has had a long history of isolation and subsequent genetic differentiation, which appears to be linked to the intense volcanic and tectonic activity in central Mexico. The separation of the two recovered clades appears to have been promoted by the geological activity during the middle Pliocene, and early Pleistocene in central Mexico, ca. 2.83 Myr. Poeciliopsis infans is a co-distributed species with other group of fishes as goodeids and cyprinids, evolving in spatiotemporal congruence. The differences in the recovered biogeographic patterns of P. infans is likely related to the biogeographic origin of each group, since P. infans has Neotropical origin, and goodeids and cyprinids are of Neartic origin. Populations of P. infans distributed in lowlands showed a higher level of genetic diversity than populations distributed in highlands, which could be linked to more stable and higher temperatures in lowland areas. Finally, fluctuations in population size are supported by the continuous fluctuations of the climate and water levels of hydrological systems in central Mexico due to glacial and interglacial cycles.
Abbreviations
- mtDNA:
-
Mitochondrial Deoxyribonucleic Acid
- nDNA:
-
Nuclear Deoxyribonucleic Acid;
- SEMARNAT:
-
Secretaria de Medio Ambiente y Recursos Naturales
References
Castoe TA, Daza JM, Smith EN, Sasa MM, Kuch U, Campbell JA, et al. Comparative phylogeography of pitvipers suggests a consensus of ancient middle American highland biogeography. J Biogeogr. 2009;36(1):88–103.
Ruíz-Sánchez E, ChD S. Influence of the geological history of the trans-Mexican Volcanic Belt on the diversification of Nolina parviflora (Asparagaceae: Nolinoideae). J Biogeogr. 2013;40(7):1336–47.
Hewitt GM. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc. 1996;58(3):247–76.
Hewitt GM. The genetic legacy of the quaternary ice ages. Nature. 2000;405(6789):907–13.
Hewitt GM. Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc B. 2004;359(1442):183–95.
Bermingham E, Martin AP. Comparative mtDNA phylogeography of neotropical freshwater fishes: testing shared history to infer the evolutionary landscape of lower central America. Mol Ecol. 1998;7(4):499–517.
Bryson RW, García-Vázquez UO, Riddle BR. Phylogeography of middle American Gophersnakes: mixed responses to biogeographical barriers across the Mexican transition zone. J Biogeogr. 2011;38:1570–84.
Bryson RW, García-Vázquez UO, Riddle BR. Diversification in the Mexican horned lizard Phrynosoma orbiculare across a dynamic landscape. Mol Phylogenet Evol. 2012;62(1):87–96.
Bryson RW, Chaves J, Smith BT, Miller MJ, Winker K, Pérez-Emán JL, et al. Diversification across the new world within the ‘blue’ Cardinalids (Aves: Cardinalidae). J Biogeogr. 2014;41(3):587–99.
Ghanavi HR, González EG, Doadrio I. Phylogenetic relationships of freshwater fishes of the genus Capoeta (Actinopterygii, Cyprinidae) in Iran. Ecol Evol. 2016;6(22):8205–22.
Perea S, Cobo-Simo M, Doadrio I. Cenozoic tectonic and climatic events in southern Iberian peninsula: implications for the evolutionary history of freshwater fish of the genus Squalius (Actinopterygii, Cyprinidae). Mol Phylogenet Evol. 2016;97:155–69.
Leprieur F, Tedesco PA, Hugueny B, Beauchard O, Dürr HH, Brosse S, et al. Partitioning global patterns of freshwater fish beta diversity reveals contrasting signatures of past climate changes. Ecol Lett. 2011;14(4):325–34.
Ferrari L, Orozco-Esquivel T, Manea V, Manea M. The dynamic history of the trans-Mexican Volcanic Belt and the Mexico subduction zone. Tectonophysics. 2012;522-523:122–49.
Halffter G. Biogeography of the montane entomofauna of Mexico and central America. Annu Rev Entomol. 1987;32:95–114.
Morrone JJ. Halffter’s Mexican transition zone (1962-2014), cenocrons and evolutionary biogeography. J Zool Syst Evol Res. 2015;53(3):249–57.
Marshall CJ, Liebherr JK. Cladistic biogeography of the Mexican transition zone. J Biogeogr. 2000;27(1):203–16.
Halas D, Zamparo D, Brooks DRA. Protocol for studying biotic diversification by taxon pulses. J Biogeogr. 2005;32(2):249–60.
Domínguez-Domínguez O, Pérez-Ponce de León G. ¿La mesa central de México es una provincia biogeográfica? Análisis descriptivo basado en componentes bióticos dulceacuícolas. Rev Mex Biodivers. 2009;80(3):835–52.
West RC. Surface configuration and asociated geology of middle America. In: Wanchope R, West RC, editors. Handbook of middle American Indians, University of Texas Press, Austin; 1964. p. 33–82.
Zaldívar-Riverón A. Nieto-Montes de Oca a, Laclette JP. Phylogeny and evolution of dorsal pattern in the Mexican endemic lizard genus Barisia (Anguidae: Gerrhonotinae). J Zool Syst Evol Res. 2005;43(3):243–57.
Bryson RW, García-Vázquez UO, Riddle BR. Relative roles of Neogene vicariance and quaternary climate change on the historical diversification of bunchgrass lizards (Sceloporus scalaris group) in Mexico. Mol Phylogenet Evol. 2012;62(1):447–57.
Arellano E, González-Cozátl FX, Rogers DS. Molecular systematics of middle American harvest mice Reithrodontomys (Muridae), estimated from mitochondrial cytochrome b gene sequences. Mol Phylogenet Evol. 2005;37(2):529–40.
Puebla-Olivares F, Bonaccorso E, Espinosa de los Monteros A, Omland KE, Llorente-Bousquets JE, Peterson AT, et al. Speciation in the emerald Toucanet (Aulacorhynchus prasinus) complex. Auk. 2008;125(1):39–50.
Castañeda-Rico S, León-Paniagua L, Vázquez-Domínguez E, Navarro-Sigüenza G. Evolutionary diversification and speciation in rodents of the Mexican lowlands: the Peromyscus melanophrys species group. Mol Phylogenet Evol. 2014;70(1):454–63.
Miller RR, Origin SML. Geography of the fishes of Central Mexico. In: Hocutt CH, Wiley EO, editors. The zoogeography of north American freshwater fishes. Wiley-Intersciences publication; 1986. p. 487–519.
Mateos M, Sanjur OI, Vrijenhoek RC. Historical biogeography of the livebearing fish genus Poeciliopsis (Poeciliidae: Cyprinodontiformes). Evolution. 2002;56(5):972–84.
Huidobro L, Morrone JJ, Villalobos JL, Álvarez F. Distributional patterns of freshwater taxa (fishes, crustaceans and plants) from the Mexican transition zone. J Biogeogr. 2006;33(4):731–41.
Domínguez-Domínguez O, Doadrio I, Pérez-ponce de León G. Historical biogeography of some river basins in Central Mexico evidenced by their goodeine freshwater fishes: a preliminary hypothesis using secondary brooks parsimony analysis (PBA). J Biogeogr. 2006;33(8):1437–47.
Domínguez-Domínguez O, Pedraza-Lara C, Gurrola-Sánchez N, Perea S, Pérez-Rodríguez R, Israde-Alcántara I, et al. Historical biogeography of the Goodeinae (Cyprinodontiforms). In: Uribe MC, Grier HJ, editors. Viviparous Fishes II. New life publications; 2010. p. 34–69.
Pérez-Rodríguez R, Domínguez-Domínguez O. Pérez-ponce de León G, Doadrio I. Phylogenetic relationships and biogeography of the genus Algansea Girard (Cypriniformes: Cyprinidae) of Central Mexico inferred from molecular data. BMC Evol Biol. 2009;9:223.
Parra-Olea G, Windfield JC, Velo-Antón G, Zamudio KR. Isolation in habitat refugia promotes rapid diversification in a montane tropical salamander. J Biogeogr. 2012;39(2):353–70.
Pedraza-Lara C, Doadrio I, Breinholt JW, Crandall KA. Phylogeny and evolutionary patterns in the dwarf crayfish subfamily (Decapoda: Cambarellinae). PLoS One. 2012;7(11):e48233.
Domínguez-Domínguez O, Alda F, Pérez-ponce de León G, García-Garitagoitia JL, Doadrio I. Evolutionary history of the endangered fish Zoogoneticus quitzeoensis (bean, 1898) (Cyprinodontiformes: Goodeidae) using a sequential approach to phylogeography base don mitochondrial and nuclear DNA data. BMC Evol Biol. 2008;8:161.
Domínguez-Domínguez O, Pérez-Rodríguez R, Doadrio I. Morphological and genetic comparative analyses of populations of Zoogoneticus quitzeoensis (Cyprinodontiformes: Goodeidae) from Central Mexico, with description of a new species. Rev Mex Biodivers. 2008;79(2):373–83.
Pérez-Rodríguez R, Domínguez-Domínguez O, Mar-Silva F, Doadrio I. Pérez-ponce de León G. The historical biogeography of the southern group of the sucker genus Moxostoma (Teleostei: Catostomidae) and the colonization of Central Mexico. Zool J Linnean Soc. 2016;177(3):633–47.
Pérez-Rodríguez R, Domínguez-Domínguez O, Doadrio I, Cuevas-García E, Pérez-ponce de León G. Comparative historical biogeography of three groups of Neartic freshwater fishes across Central Mexico. J Fish Biol. 2015;86(3):993–1015.
Family Poeciliidae LPHF. In: Reis RE, Kullander SO, CJJr F, editors. Check list of the freshwater fishes of south and central America. Porto Alegre: EDIPUCRS; 2003. p. 555–81.
Miller R, Minckley W, Mark, S. Freshwater Fishes of México. University of Chicago. Estados Unidos de América. 2005.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2.
Stephens M, Donelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73(5):1162–9.
Sousa-Santos C, Robalo JI, Collares-Pereira MJ, Almada VC. Heterozygous indels as useful tools in the reconstruction of DNA sequences and in the assessment of ploidy level and genomic constitution of hybrid organisms. DNA Seq. 2005;16(6):462–7.
Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23(2):254–67.
González-Peña D, Gómez-Blanco D, Reboiro-Jato M, Fernández-Riverola F, Posada D. ALTER: program-oriented conversion of DNA and protein alignments. Nucleic Acids Res. 2010;38(2):14–8.
Kass RE, Raftery AE. Bayes factors. J Amer Statist Assoc. 1995;90(430):773–95.
Lanfear R, Calcott B, Ho SY, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyzes. Mol Biol Evol. 2012;29(6):1695–701.
Silvestro D, Michalak I. RAxMLGUI: a graphical front-end for RAxML. Org Divers Evol. 2012;12(4):335–7.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Alfaro ME, Zooler S, Lutzoni F. Bayes or bootstrap? A simulation study comparing the performance of Bayesian Markov chain Monte Carlo sampling and bootstrapping in assessing phylogenetic confidence. Mol Biol Evol. 2003;20(2):255–66.
Ronquist F, Teslenko M, Van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42.
Rambaut A, Drummond A. Tracer v1.5. 2007. Available: http://tree.bio.ed.ac.uk/software/tracer.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–73.
Zardoya R, Doadrio I. Molecular evidence on the evolutionary and biogeographical patterns of European cyprinids. J Mol Evol. 1999;49(2):227–37.
Machordom A, Doadrio I. Evidence of a cenozoic Betic-Kabilian connection based on freshwater fish phylogeography (Luciobarbus, Cyprinidae). Mol Phylogenet Evol. 2001;18(2):252–63.
Near TJ, Benard MF. Rapid allopatric speciation in logperch darters (Percidae: Percina). Evolution. 2004;58(12):2798–808.
Drummond AJ, Ho SYW, Phillips J, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;5:e88.
Heled J, Drummond AJ. Bayesian inference of population size history from multiple loci. BMC Evol Bio. 2008;8:289.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Res. 2010;10(3):564–7.
Nylander JAA, Olsson U, Alström P, Sanmartín I, Baker A. Accounting for phylogenetic uncertainty in biogeography: a Bayesian approach to dispersal–Vicariance analysis of the thrushes (Aves: Turdus). Syst Biol. 2008;57(2):257–68.
Ree RH, Moore BR, Webb CO, Donoghue MJ. A likelihood framework for inferring the evolution of geographic range on phylogenetic trees. Evolution. 2005;59(11):2299–311.
Ree RH, Smith SA. Maximum-likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst Biol. 2008;57(1):4–14.
Yu Y, Harris AJ, Blair C, He J. RASP (reconstruct ancestral state in phylogenies): a tool for historical biogeography. Mol Phylogenet Evol. 2015;87:46–9.
Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22(5):1185–92.
Elith J, Graham CH, Anderson RP, Dudík M, Ferrier S, Guisan A, et al. Novel methods improve prediction of species’ distributions from occurrence data. Ecography. 2006;29(2):129–51.
Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecol Model. 2006;190(3–4):231–59.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. Very high resolution interpolated climate surfaces for the global land areas. Int J Climatol. 2005;25(15):1965–78.
Pearson RG, Raxworthy CJ, Nakamura M, Peterson AT. Predicting species distributions from small numbers of occurrence records: a test case using cryptic geckos in Madagascar. J Biogeogr. 2007;34(1):102–17.
Graham MH. Confronting multicollinearity in ecological multiple regression. Ecology. 2003;84(11):2809–15.
Araujo MB, Pearson RG, Thuiller W, Erhard M. Validation of species-climate impact models under climate change. Glob Change Biol. 2005;11(9):1504–13.
Ferrari L, Rosas-Elguera J. Late Miocene to quaternary extension and the northern boundary of the Jalisco block, western Mexico. The Tepic-Zacoalco rift revised. Geol. Soc. am. Bull. Special Paper. 1999;334:1–23.
Rosas-Elguera J, Urrutia-Fucugauchi J. Tectonic control on the volcano-sedimentary sequence of the Chapala graben, western Mexico. Int Geol Rev. 1998;40(4):350–62.
Domínguez-Domínguez O, Pompa-Domínguez A, Doadrio I. A new species of the genus Yuriria Jordan & Evermann, 1896 (Actinopterygii, Cyprinidae) from the Ameca basin of the central Mexican plateau. Graellsia. 2007;63(2):259–71.
Pérez-Rodríguez R, Pérez ponce de León G, Domínguez-Domínguez O, Doadrio I. A new species of Algansea (Actinopterygii: Cyprinidae) from the Ameca River basin, in Central Mexico. Rev Mex Biodivers. 2009;80(2):483–90.
Alda F, Reina RG, Doadrio I, Bermingham E. Phylogeny and biogeography of the Poecilia sphenops species complex (Actinopterygii, Poeciliidae) in central America. Mol Phylogenet Evol. 2013;66:1011–26.
Garduño V, Tibaldi A. Kinematic evolution of the continental active triple junction of the western Mexican Volcanic Belt. C R Acad Sci. 1991;2:135–42.
Webb T, Bartlein PJ. Global changes during the last 3 million years: climatic controls and biotic responses. Annu Rev Ecol Evol Syst. 1992;23:141–73.
Conant R. Observations on garter snakes of the Thamnophis eques Complex in the lakes of Mexico’s Transvolcanic Belt, with descriptions of new taxa. American museum Novitates. Bull Am Mus Nat Hist. 2003;3406:64.
Pacheco J, Mortera-Gutiérrez C, Delgado-Granados H, Singh S, Valenzuela R, Shapiro N, et al. Tectonic significance of an earthquake sequence in the Zacoalco half-graben, Jalisco, Mexico. J S Am Earth Sci. 1999;12(6):557–65.
Nelson SA, Carmichael ISE. Pleistocene to recent alkalic volcanism in the región of Sanganguey volcano, Nayarit, Mexico. Contrib Mineral Petrol. 1984;85(4):321–35.
Huang WD, Zhao XY, Zhao X, Zhao HL, Wang SK, Lian J. A combined approach using ISSR and ITS analysis for the characterization of Artemisia halodendron from Horqin sandy land, northern China. Biochem Syst Ecol. 2011;39(4–6):346–51.
Huang WD, Zhao XY, Zhao X, Li Y, Lian J. Effects of environmental factors on genetic diversity of Caragana microphylla in Horqin Sandy land, Northeast China. Ecol. Evol. 2016;6(22):8256–66.
Su ZH, Zhang ML. A range wide geographic pattern of genetic diversity and population structure of Hexinia polydichotoma (Asteraceae) in Tarim Basin and adjacent areas. Biochem Syst Ecol. 2014;56:49–59.
Faye A, Deblauwe V, Mariac C, Damien R, Sonké B, Vigouroux Y, Couvreur TLP. Phylogeography of the genus Podococcus (Palmae/Arecaceae) in central African rain forests: climate stability predicts unique genetic diversity. Mol Phylogenet Evol. 2016;105:126–38.
Perea S, Doadrio I. Phylogeography, historical demography and hábitat suitability modelling of freshwater fishes inhabiting seasonally fluctuating Mediterranean river systems: a case study using the Iberian cyprinid Squalis valentinus. Mol Ecol. 2015;24(14):3706–22.
Moncayo-Estrada R, Israde-Alcántara I, Garduño-Monroy VH. La cherehuita Hubbsina turneri De Buen (1941) (Pisces, Goodeidae). Origen, distribución y su uso en la regionalización de la cuenca del Lerma. Hidrobiologica. 2001;11(1):1–13.
Israde-Alcántara I. Lagos Volcánicos y Tectónicos de Michoacán. In: Garduno-Monroy VH, Corona-Chavez P, Israde-Alcántara I, MennellaL, Arreygue E, Bigioggero B, Chiesa, S, editors. Carta Geológica de Michoacán Escala 1:250000. Universidad Michoacana de San Nicolás de Hidalgo. 1999. p. 45–72.
Ornelas-García CP, Alda F, Díaz-Pardo E, Gutiérrez-Hernández A, Doadrio I. Genetic diversity shaped by historical and recent factors in the live-bearing twoline skiffia Neotoca bilineata. J Fish Biol. 2012;81(6):1963–84.
Corona-Santiago DK, Doadrio I, Domínguez-Domínguez O. Evolutionary history of the live-bearing endemic Allotoca diazi species complex (Actinopterygii, Goodeinae): evidence of founder effect events in the Mexican Prehispanic period. PLoS One. 2015;10(5):e0124138.
Galindo-Villegas J, Sosa-Lima F. Gonopodial system review and a new fish record of Poeciliopsis infans (Cyprinodontiformes: Poeciliidae) for Lake Patzcuaro, Michoacan, Central Mexico. Rev Biol Trop. 2002;50(3–4):1151–7.
Ramírez-Herrejón JP, Castañeda-Sam LS, Moncayo-Estrada R, Caraveo-Patiño J, Balart EF. Trophic ecology of the exotic Lerma livebearer Poeciliopsis infans (Cyprinodontiformes: Poeciliidae) in the Lago de Pátzcuaro, Central Mexico. Rev Biol Trop. 2013;61(3):1289–300.
Welcomme RA. History of international introductions of inland aquatic species. ICES Mar Sci Symp. 1992;194:3–14.
Contreras-MacBeath T, Mejía-Mojica H, Carrillo-Wilson R. Negative impact on the aquatic ecosystems of the state of Morelos, Mexico from introduced aquarium and other commercial fish. Aquarium Sci Conserv. 1998;2(2):67–78.
Caballero M, Lozano-García MS, Vázquez-Selem L, Ortega B. Evidencias de cambio climático y ambiental en las cuencas de altura del centro de México durante el último máximo glacial. Bol Soc Geol Mex. 2010;62(3):359–77.
Hampe A, Petit R. Conserving biodiversity under climate change: the rear edge matters. Ecol Lett. 2005;8(5):461–7.
Lachniet MS, Vázquez-Selem L. Last glacial maximum equilibrium line altitudes in the circum-Caribbean (Mexico, Guatemala, Costa Rica, Colombia, and Venezuela). Quat Int. 2005;138-139:129–44.
Hofreiter M, Stewart JR. Ecological change, range fluctuations and population dynamics during the Pleistocene. Curr Biol. 2009;19(14):R584–94.
Lozano-García S, Torres-Rodríguez E, Ortega B, Vázquez G, Caballero M. Ecosystem responses to climate and disturbances in western Central Mexico during the late Pleistocene and Holocene. Paleogeogr Palaeoclimatol Palaeoecol. 2013;370:184–95.
Alfaro R, Martínez V, Segovia N, Peña P, López M, Armienta M, et al. Radon behavior in springs and wells around Cuitzeo Lake, Lerma River basin, Mexico. Geofís. Int. 2002;41(4):439–46.
De la Vega-Salazar MY, Avila-Luna E, Macías-García C. Ecological evaluation of local extinction: the case of two genera of endemic Mexican fish. Zoogoneticus and Skiffia Biodivers Conserv. 2003;12(10):2043–56.
Domínguez-Domínguez O, Mercado-Silva N, Lyons J. Conservation status of Mexican goodeids: problems, perspectives, and solutions. In: Uribe MC, Grier HJ, editors. Viviparous Fishes. New life publications; 2005. p. 495–504.
Mercado-Silva N, Lyons JD, Maldonado GS, Nava MM. Validation of a fish-based index of biotic integrity for streams and rivers of Central Mexico. Rev Fish Biol Fish. 2002;12(2):179–91.
Domínguez-Domínguez O. Pérez Ponce de León G. Los goodeidos, peces endémicos del centro de México. CONABIO. Biodiversitas. 2007;75:12–5.
Domínguez-Domínguez O, Boto L, Alda F, Pérez-Ponce de León G, Doadrio I. Human impacts on drainages of the mesa central, Mexico, and its genetic effects on an endangered fish, Zoogoneticus quitzeoensis. Conserv Biol. 2007;21(1):168–80.
Doadrio I, Perdices A, Machordom A. Allozymic variation of the endangered killifish Aphanius iberus and its application to conservation. Env Biol Fish. 1996;45(3):259–71.
Acknowledgements
This work was conducted as part of the doctoral studies of R. G. Beltrán-López, and she was supported with a scholarship from the Consejo Nacional de Ciencia y Tecnología (CONACYT: scolarship number 329883). We thank Isaí Betancourt for his help with sampling, and to Berenice García Andrade and Silvia Perea for their help in laboratory and for their comments to improve this work. We thank three anonymous reviewers for their comments that improved this manuscript.
Funding
Funding for this study was provided by the Ministerio de Economía y Competitividad y FEDER, Spain (CGL2016–75262-P) to ID and PRODEM, CIC-UMSNH, Chester Zoo garden and CONACYT sabbatical grant to ODD, as well as funds from the U.S. National Science Foundation (DEB 1354930) to KRP. These funding body do not had a specific role in the design of the study and collection, analyses, and interpretation of data in writing the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its additional files]. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
RGBL, ODD, conceived the ideas; RGBL, ODD, RPR, conducted the fieldwork and collected the specimens; RGBL, ODD, RPR, and ID, analyzed the data and RGBL, ODD, RPR, KP, and ID write the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research has been carried out within an appropriate ethical framework. All procedures performed, including field sampling techniques and laboratory protocols, as anesthetised and euthanasia techniques used in this study were reviewed and approved by a committee of Mexican Ministry of Environmental and Natural Resources (SEMARNAT), under collection permit number PPF/DGOPA-262/17. This procedures were also assessed and approved by the Institutional Biosecurity and Bioethics committee of the Institute of Chemical and Biological Research, Universidad Michoacana de San Nicolás de Hidalgo, México. The specimens were anesthetised using tricaine mesylate (MS-222) to anesthetize the fishes, according with the Official Mexican Norm NOM-051-ZOO-1995 and NOM-033-SAG/ZOO-2014 for humanitarian treatment in the mobilization of animals. The study organism is neither protected nor endangered.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Tissue voucher number, and access number of GenBank. (DOC 416 kb)
Additional file 2:
Primers, PCR conditions, and References. (DOC 35 kb)
Additional file 3:
Models selected with Akaike information criterion and the parameters of each gene. (DOC 32 kb)
Additional file 4:
Geographical coordinates of the 162 sites registers in Colección de Peces de la Universidad Michoacana de San Nicolás de Hidalgo used as presence data for species distribution modelling. (DOC 161 kb)
Additional file 5:
Ambiguously Aligned Regions for S7. (DOC 49 kb)
Additional file 6:
The Bayesian inference tree of P. infans from cytb mitochondrial gene (1083 bp). Bayesian posterior probability (> 0.9; above the branches) and maximum likelihood bootstrap values (> 80%; below the branches) are indicated. (DOC 943 kb)
Additional file 7:
The Bayesian inference tree of P. infans from coxI mitochondrial gene (631 bp). Bayesian posterior probability (> 0.9; above the branches) and maximum likelihood bootstrap values (> 80%; below the branches) are indicated. (DOC 548 kb)
Additional file 8:
The Bayesian inference tree of P. infans from concatenated sequences of two mitochondrial genes (cytb, coxI; 1771 bp). Bayesian posterior probability (> 0.9; above the branches) and maximum likelihood bootstrap values (> 80%; below the branches) are indicated. (DOC 738 kb)
Additional file 9:
The Bayesian inference tree of P. infans from RHO nuclear gene (845 bp). Bayesian posterior probability (> 0.9; above the branches) and maximum likelihood bootstrap values (> 80%; below the branches) are indicated. (DOC 469 kb)
Additional file 10:
The Bayesian inference tree of P. infans from S7 nuclear gene (859 bp). Bayesian posterior probability (> 0.9; above the branches) and maximum likelihood bootstrap values (> 80%; below the branches) are indicated. (DOC 715 kb)
Additional file 11:
The Bayesian inference tree of P. infans from concatenated sequences of two nuclear genes (S7 and RHO: 1704 bp). Bayesian posterior probability (> 0.9) and maximum likelihood bootstrap values (> 80%) are indicated. (DOC 2382 kb)
Additional file 12:
Haplotype networks for nuclear genes, a) S7 gene, b) RHO gene. The two recovered haplogroups are show with labels A and B. (DOC 1189 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Beltrán-López, R.G., Domínguez-Domínguez, O., Pérez-Rodríguez, R. et al. Evolving in the highlands: the case of the Neotropical Lerma live-bearing Poeciliopsis infans (Woolman, 1894) (Cyprinodontiformes: Poeciliidae) in Central Mexico. BMC Evol Biol 18, 56 (2018). https://doi.org/10.1186/s12862-018-1172-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-018-1172-7