
Abstract
Purpose of Review
Post-prandial lipemia (PPL), characterized by elevated levels of triglyceride (TG) following a meal, is an independent risk factor for cardiovascular disease. This review summarizes current knowledge on the genetic and epigenetic determinants of the PPL TG response and provides perspectives on future directions.
Recent Findings
Recent studies suggested that PPL-related traits have heritability between 38 and 80%. Genomics studies identified genetic variants in or near APOA1/C3/A4/A5 cluster region affecting PPL TG levels. Epigenomics studies found DNA methylation levels of many genes known to be related to lipid metabolism including CPT1A gene are associated with fasting TG and PPL TG.
Summary
Both genetic polymorphisms and epigenetic modifications are important determinants of PPL variation. Epigenetics may have even more significant impact than genetic variants on PPL. Further studies with multi-omics system biology approach are needed to fully elucidate the mechanisms of PPL regulation to combat the atherogenic effect of PPL.
Similar content being viewed by others


Introduction
Cardiovascular disease (CVD) is the leading cause of death in the USA with prevention and treatment efforts largely targeted at traditional CVD factors (e.g., fasting LDL, blood pressure, adiposity, and smoking). Identifying novel CVD risk factors and understanding more completely the mechanisms by which the established risk factors exert their effects remain a priority of CVD risk prevention, so that these risks can be more fully mitigated.
The association of hypertriglyceridemia (HTG) with CHD has long been recognized. HTG has been consistently shown through longitudinal [1,2,3] to increase CVD risk independently of other known risk factors, and genetic variants that are associated specifically with elevated triglycerides are also associated with CVD [4,5,6]. The importance of HTG (triglyceride (TG) > 150 mg/dL) as a CVD risk factor is highlighted by its high prevalence (i.e., frequency of 25% in the USA [7]). While elevated levels of fasting TG have long been known to predict CVD, fasting levels do not reflect the wide fluctuations and sustained high levels that can occur throughout the day. Unlike the relatively constant plasma LDL-C level, circulating TG increases after a meal, peaking at ~ 4 h and slowly returning to the fasting level after 6–8 h following a meal [8,9,10,11,12]. Consequently, the significance of TG as a CVD risk factor should be recognized in the context that most of the day is spent in the post-prandial state (i.e., post-prandial lipemia, PPL).
A systematic assessment of PPL requires an oral fat tolerance test (OFTT) whereby the fasting subject ingests a standardized fat meal and TG levels are measured at regular intervals for up to 6 h to determine the rate of TG increase and clearance. Multiple cross-sectional studies have now been revealed an association between an elevated TG response after a standardized OFTT and delayed clearance of TGs with presence of CVD [13,14,15,16,17]. Moreover, non-fasting TG levels have been significantly associated with incident CVD events, and this association is stronger than that for fasting TG and is independent of LDL and other known CVD risk factors [18,19,20,21].
The atherogenic mechanisms underlying PPL are only broadly understood. Initially, there is a rapid and prolonged change in circulating lipid profiles after ingesting a high-fat meal, characterized by production of TG-rich remnant particles from lipolysis of chylomicrons and generation of very low density lipoproteins from liver, as well as a reduction in high density lipoprotein (HDL) [22,23,24,25]. These changes are pro-atherogenic and occur in the context of post-prandial inflammation with ensuing endothelial dysfunction [26] and pro-thrombotic activity [24]. Post-prandial inflammation appears to be the central event triggering these atherogenic changes that follow prolonged/intensified PPL [27, 28]. Consistent with the post-prandial inflammation state, a single high-fat meal induces increased secretion of the inflammatory biomarker interleukin-6 (IL-6) during the post-meal period and activation of white blood cells in terms of both the cell count and individual cell activation markers (e.g., granulocyte and mononuclear cells) [29, 30•]. Exactly how this sequence of events unfolds is unclear.
Behavioral and Clinical Determinants of PPL
Variability in the PPL response at the population level is influenced by a range of factors, including sex, age, baseline TG level, aerobic capacity, adiposity, diet, and disease status (e.g., diabetes, insulin resistance, periphery artery disease, hypertension, coronary artery diseases, and inflammation) [2, 8, 13,14,15,16,17, 31]. As an example, there has been much interest in studying the effect of exercise on PPL [32], with numerous studies showing that acute exercise prior to a high-fat meal can effectively attenuate the PPL response (e.g., see review by Teeman et al. [33]). Much current research has been directed into characterizing the optimal duration, intensity, and types of physical exercise that most efficiently reduce the PPL response.
Genetic Determinants of PPL
The genetic basis of HTG has been well recognized from early studies of monogenetic forms of this disorder. The most common monogenetic forms of HTG are due to loss-of-function mutations in genes related to metabolism of triglyceride-rich lipoproteins, such as LPL, APOC2, APOA5, LMF1, GPIHBP1, and GPD1 [34]. Of these, a loss of function mutation in the lipoprotein lipase gene (LPL) is the most common etiology of monogenic HTG. The main function of LPL is to hydrolyze TG in chylomicrons and VLDL particles. In contrast to these monogenic forms, the majority of HTG is due to a polygenic form of the condition. Heritability analyses have revealed that genetic effects can account for 54–59% of the variation of fasting TG in population [35, 36]. In fact, the most recent large-scale genome-wide association studies (GWAS) have identified at least 224 common and low-frequency SNPs from 137 genes/regions that are associated with triglyceride levels, mostly measured in the fasting state [37]. Among these genomic loci are many with known functions in lipid metabolism, such as LPL, APOA5, APOB, APOC1, APOE, and GCKR. However, it is currently unknown how most of other associated loci outside of these known lipid metabolism genes are involved in the regulation of TG metabolism.
The heritability of the PPL TG, assessed as the integrated area under the curve of TG across 6 h, was estimated at 38% in Amish subjects from the Heredity and Phenotype Intervention (HAPI) Heart Study [38]; the slope of the TG increase at 3.5 h after OFTT had a heritability of ~ 80% in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study [39•]. Multiple genetic association studies of PPL TG have been carried out, most utilizing the candidate gene approach and focusing on genes known to be related to lipid absorption, apolipoproteins, and lipid clearance pathways, including the APOA1/C3/A4/A5 gene cluster, ABCA1, CETP, GCKR, IL6, LPL, PLIN1, andTCF7L2 [40, 41]. Unlike candidate gene studies, the GWAS approach has the appeal of offering an agnostic survey of the whole genome and could discover novel candidates.
To date, GWAS of PPL TG have been conducted in only the Amish HAPI Heart [11] and GOLDN studies [42••]. In 2008, the Amish HAPI Heart Study identified a null mutation (R19X) in APOC3 that is carried by ~ 5% of Amish subjects in Lancaster County, PA, USA. The R19X carriers had approximately one half of the levels of apoC-III protein compared to non-carriers and demonstrated a significantly reduced PPL TG excursion during the OFTT. These subjects also had a relatively cardioprotective risk profile that included lower coronary calcification scores [11]. This cardioprotective phenotype occurs because apoC-III inhibits lipoprotein lipase (LPL), the primary hydrolyzer of TG-rich chylomicrons. The mutation leads to increased LPL activity, thereby promoting chylomicron hydrolysis and reducing PPL.
The GWAS of PPL conducted in the GOLDN Study, which was based on 872 subjects of European ancestry, identified two SNPs significantly associated with PPL TG excursion at genome-wide thresholds of significance level (i.e., p < 5E−08), one of which, rs964184, was replicated in the Amish HAPI Study [42••]. rs964184 is located near ZPR1 and close to the APOA1/C3/A4/A5 cluster. rs964184 falls in a linkage disequilibrium block that includes APOA5, the gene-encoding apolipoprotein A5, a component of HDL and which plays a major role in regulating important determinant of plasma TG level. rs964184 has been previously associated with fasting TG [43], TG response to fenofibrate [44], fat intake and TG response interaction [45], metabolic syndrome [46], and coronary artery disease [47].
Epigenetic Determinants of PPL
The two SNPs significantly associated with PPL TG in the GOLDN Study (rs964184 and rs10243693) account for only 4.5% of the variation in PPL TG AUC in the GOLDN population [42••, 48•], and the Amish APOC3 R19X variant (rs76353203) accounts for virtually none of the phenotypic variation in European populations because the frequency of this variant is so rare outside of the Amish. Even for the relatively well-studied fasting TG measure, it is estimated that one half of the variance in this trait cannot be explained by the common and rare variants identified in current GWAS [49]. For example, 86 common SNPs associated with increased TG through meta-analysis account for less than 12% variance of triglyceride levels in the Framingham heart study [43, 50]. The missing heritability of HTG could be due to unstudied rare genetic variants, gene-gene interactions, epigenetics, and/or gene-environment interactions [49].
Epigenetic changes constitute a potential source of variability in PPL TG excursions that is not captured by genome-wide genotype data. Epigenetic changes refer to molecular factors or processes that do not affect the DNA sequence but can influence gene expression [51] via several mechanisms, including the following: (1) methylation of DNA nucleotide base almost exclusively at cytosines of CpG (5’—C—phosphate—G—3’) dinucleotides in human; (2) biochemical modification of the histone molecules that package DNA sequences; and (3) non-coding RNAs that interact with DNA methylation and chromatin modification machinery. Epigenetics has been shown to regulate many important biological processes, including development, diseases, and response to behaviors [52,53,54,55,56]. Epigenetic regulation can be modified by both genetic and environmental factors, for example, genetic polymorphisms can act as quantitative trait loci (QTL) to influence epigenetic modification of DNA sequence; in addition, epigenetic modification of DNA sequence can be dynamic and respond to environmental changes. Thus, epigenetic variation represents the interface of gene and environment interaction, and this provides another source of variation in PPL response across subjects. This review will focus on DNA methylation, the most studied mechanism of epigenetic regulation.
Similar to GWAS, an epigenome-wide association study (EWAS) examines associations between epigenetic variation, mostly DNA methylation variation, and a particular trait. To date, six EWAS have been published of fasting TG (see Table 1). These have revealed associations of fasting TG with DNA methylation sites at 34 unique loci mapping to 17 different genes [48•, 59•, 60•, 61•, 62•, 63•]. The only EWAS of PPL TG published to date is from the GOLDN Study [57••]. This analysis was based on 979 GOLDN subjects who underwent a standard OFTT, and DNA was purified peripheral lymphocytes at baseline before OFTT. The goal of this analysis was to identify DNA methylation sites at baseline that predicted PPL TG response to the OFTT. Eight loci at five genes (LPP, CPT1A, APOA5, SREBF1, and ABCG1) had methylation levels at baseline that were significantly associated with PPL TG at genome-wide significance (P < 1.1 × 10−7) [57••]; interestingly, these eight CpG sites together account for 14.9% of the variance in PPL TG and 16.3% of the phenotypic variance in fasting TG in the GOLDN Study cohort [57••]; in contrast, the two previously identified genetic loci for PPL TG (rs964184 and rs10243693) explained only 4.5% phenotypic variance for both PPL TG AUC and fasting TG. This suggests that epigenetics may be a more significant contributor than DNA sequence variation to PPL TG variability at the population level. Four out of the five identified genes have established roles in lipid metabolism; four CpG loci themselves, mapping to the genes CPT1A, APOA5, SREBF1, and ABCG1, have been directly reported to be associated with fasting TG in European populations (KORA and InCHIANTI) [63•]. The association of PPL TG with CPT1A methylation, which was the most strongly associated locus, was particularly noteworthy because of the function of this gene. CPT1A (Carnitine Palmitoyltransferase 1A) is a key enzyme for the mitochondrial oxidation of long-chain fatty acid, catalyzing the transfer of the acyl group of long-chain fatty acid-CoA conjugates onto carnitine that is essential for the mitochondrial uptake of long-chain fatty acids, which is a metabolism product and component of triglyceride, and their subsequent beta-oxidation. Thus, it is not surprising that CPT1A might play major role in PPL and fasting triglyceride metabolism.
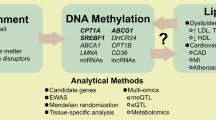
It must be pointed out that the baseline epigenetic profiles correlated with PPL TG response could be inherited inter-generationally or could be the consequence of certain behavior/environmental factors. Besides the heritable epigenetic profiles in epigenome, the aforementioned behavioral/environmental factors that are associated with PPL TG response variation, such as exercise, diet, and smoking, could also modify epigenetic machinery and gene transcription both acutely and chronically, which in turn could alter the PPL TG response after meal intake. For example, both acute and chronic exercise could induce wide-spread DNA methylation and gene expression changes in skeletal muscle and in peripheral blood cells [64,65,66]. One time or long-term dietary interventions also could induce significant DNA methylation and gene expression changes [67, 68]. It was found that dietary polyunsaturated fatty acids can modify the epigenome and some of the methylation changes were correlated with changes in plasma triglyceride [68, 69]. The interplay among genetics, epigenetics, and environmental/behaviors factors and how these factors may collectively lead to an abnormal PPL TG response and increased CVD risk are depicted in a working hypothesis model in Fig. 1. As implied by this model, an integrative multi-omics, multi-system approach is necessary to dissect the pro-atherosclerosis mechanism of HTG.

Theoretical working model on genetic and epigenetic determinants of PPL TG
Conclusions and Future Directions
Genetic and epigenetic association studies are complementary strategies to identify molecular mechanisms underlying adverse PPL TG excursion after meals. However, we are still in the infancy stage with only a handful of genetic variants and DNA methylation loci identified that explain only a small portion of the population variation in PPL TG excursion. Further work is needed on several fronts to advance our understanding of PPL TG as a risk factor for cardiovascular diseases and its management. First, we must increase the sample size for genomic, epigenetic, and epidemiologic studies of PPL TG. Currently, PPL has been understudied in large population studies due to the resource demand in administering a standard fat meal challenge and time course blood sampling. There is also not a solid consensus on the standards for the fat meal challenge. Second, we will need to devote efforts to dissect the dynamic changes in epigenomics, transcriptomic, metabolomics, and proteomics along with PPL TG excursion. Multi-level-omics data has not yet been systematically collected over time for PPL TG. Although Mendelian randomization analysis has demonstrated a causal link between increased TG and cardiovascular risk, the mediating mechanisms remain largely unknown. Natural history studies with extended time course sampling of PPL TG excursions are needed to reveal such mediating signaling pathways and pathological events that are perturbed by the increased PPL TG and which eventually lead to foam cell formation and atherosclerosis plaque development. Third, we will need to design effective behavior intervention to effectively manage PPL TG and prevent CVD. To date, multiple behavioral factors such as diet, exercise, and smoking have been associated with PPL TG variation, but it is not clear what kinds of diet combination or what kind of exercise regime would be optimal to reduce PPL TG exposure. Last but not the least, we will need to design more effective pharmacological treatment for managing PPL TG. Fibrates are the most often used drugs that could specifically target TG and lower TG levels by ∼ 36% [70]; however, the cardiovascular outcome of fibrate therapy varied across different studies so far [71]. Moreover, fibrates could result in adverse effects such as increases in creatinine levels, myopathy, and rhabdomyolysis. Mechanistic genetic and epigenetic study of factors that influence PPL TG response could help the discovery of novel therapy targets.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Toth PP. Triglyceride-rich lipoproteins as a causal factor for cardiovascular disease. Vasc Health Risk Manag. 2016;12:171–83. https://doi.org/10.2147/VHRM.S104369.
Boren J, Matikainen N, Adiels M, Taskinen MR. Postprandial hypertriglyceridemia as a coronary risk factor. Clin Chim Acta. 2014;431:131–42. https://doi.org/10.1016/j.cca.2014.01.015.
Skretteberg PT, Grytten AN, Gjertsen K, Grundvold I, Kjeldsen SE, Erikssen J, et al. Triglycerides-diabetes association in healthy middle-aged men: modified by physical fitness? A long term follow-up of 1962 Norwegian men in the Oslo Ischemia Study. Diabetes Res Clin Pract. 2013;101(2):201–9. https://doi.org/10.1016/j.diabres.2013.06.001.
Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45(11):1345–52. https://doi.org/10.1038/ng.2795.
Holmes MV, Asselbergs FW, Palmer TM, Drenos F, Lanktree MB, Nelson CP, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J. 2015;36(9):539–50. https://doi.org/10.1093/eurheartj/eht571.
Thomsen M, Varbo A, Tybjaerg-Hansen A, Nordestgaard BG. Low nonfasting triglycerides and reduced all-cause mortality: a mendelian randomization study. Clin Chem. 2014;60(5):737–46. https://doi.org/10.1373/clinchem.2013.219881.
Carroll M, Kit B, Lacher D. Trends in elevated triglyceride in adults: United States, 2001–2012. NCHS Data Brief. 2015;198:198.
Alcala-Diaz JF, Delgado-Lista J, Perez-Martinez P, Garcia-Rios A, Marin C, Quintana-Navarro GM, et al. Hypertriglyceridemia influences the degree of postprandial lipemic response in patients with metabolic syndrome and coronary artery disease: from the CORDIOPREV study. PloS One. 2014;9(5):e96297. https://doi.org/10.1371/journal.pone.0096297.
Wojczynski MK, Glasser SP, Oberman A, Kabagambe EK, Hopkins PN, Tsai MY, et al. High-fat meal effect on LDL, HDL, and VLDL particle size and number in the Genetics of Lipid-Lowering Drugs and Diet Network (GOLDN): an interventional study. Lipids Health Dis. 2011;10:181. https://doi.org/10.1186/1476-511X-10-181.
Yunoki K, Nakamura K, Miyoshi T, Enko K, Kohno K, Morita H, et al. Ezetimibe improves postprandial hyperlipemia and its induced endothelial dysfunction. Atherosclerosis. 2011;217(2):486–91. https://doi.org/10.1016/j.atherosclerosis.2011.04.019.
Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322(5908):1702–5. https://doi.org/10.1126/science.1161524.
Cohn JS, McNamara JR, Cohn SD, Ordovas JM, Schaefer EJ. Postprandial plasma lipoprotein changes in human subjects of different ages. J Lipid Res. 1988;29(4):469–79.
Patsch JR, Miesenbock G, Hopferwieser T, Muhlberger V, Knapp E, Dunn JK, et al. Relation of triglyceride metabolism and coronary artery disease. Studies in the postprandial state. J Vasc Res. 1992;12(11):1336–45.
Groot PH, van Stiphout WA, Krauss XH, Jansen H, van Tol A, van Ramshorst E, et al. Postprandial lipoprotein metabolism in normolipidemic men with and without coronary artery disease. J Vasc Res.. 1991;11(3):653–62.
Meyer E, Westerveld HT, de Ruyter-Meijstek FC, van Greevenbroek MM, Rienks R, van Rijn HJ, et al. Abnormal postprandial apolipoprotein B-48 and triglyceride responses in normolipidemic women with greater than 70% stenotic coronary artery disease: a case-control study. Atherosclerosis. 1996;124(2):221–35.
Mero N, Malmstrom R, Steiner G, Taskinen MR, Syvanne M. Postprandial metabolism of apolipoprotein B-48- and B-100-containing particles in type 2 diabetes mellitus: relations to angiographically verified severity of coronary artery disease. Atherosclerosis. 2000;150(1):167–77.
Boquist S, Ruotolo G, Tang R, Bjorkegren J, Bond MG, de Faire U, et al. Alimentary lipemia, postprandial triglyceride-rich lipoproteins, and common carotid intima-media thickness in healthy, middle-aged men. Circulation. 1999;100(7):723–8.
Steiner G. Triglyceride-rich lipoproteins and atherosclerosis, from fast to feast. Ann Med. 1993;25(5):431–5.
Austin MA. Plasma triglyceride as a risk factor for cardiovascular disease. Can J Cardiol. 1998;14(Suppl B):14B–7B.
Cullen P. Evidence that triglycerides are an independent coronary heart disease risk factor. Am J Cardiol. 2000;86(9):943–9.
Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298(3):309–16. https://doi.org/10.1001/jama.298.3.309.
Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O’Cochlain F, Gao F, et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36(4):382–7. https://doi.org/10.1038/ng1329.
Sanders TA. Dietary fat and postprandial lipids. Curr Atheroscler Rep. 2003;5(6):445–51.
Tholstrup T, Miller GJ, Bysted A, Sandstrom B. Effect of individual dietary fatty acids on postprandial activation of blood coagulation factor VII and fibrinolysis in healthy young men. Am J Clin Nutr. 2003;77(5):1125–32.
Duttaroy AK. Postprandial activation of hemostatic factors: role of dietary fatty acids. Prostaglandins Leukot Essent Fatty Acids. 2005;72(6):381–91. https://doi.org/10.1016/j.plefa.2005.03.003.
Vogel RA, Corretti MC, Plotnick GD. Effect of a single high-fat meal on endothelial function in healthy subjects. Am J Cardiol. 1997;79(3):350–4.
Lundman P, Boquist S, Samnegard A, Bennermo M, Held C, Ericsson CG, et al. A high-fat meal is accompanied by increased plasma interleukin-6 concentrations. Nutr Metab Cardiovasc Dis. 2007;17(3):195–202. https://doi.org/10.1016/j.numecd.2005.11.009.
Nappo F, Esposito K, Cioffi M, Giugliano G, Molinari AM, Paolisso G, et al. Postprandial endothelial activation in healthy subjects and in type 2 diabetic patients: role of fat and carbohydrate meals. J Am Coll Cardiol. 2002;39(7):1145–50.
Herieka M, Erridge C. High-fat meal induced postprandial inflammation. Mol Nutr Food Res. 2014;58(1):136–46. https://doi.org/10.1002/mnfr.201300104.
• Emerson SR, Kurti SP, Harms CA, Haub MD, Melgarejo T, Logan C, et al. Magnitude and timing of the postprandial inflammatory response to a high-fat meal in healthy adults: a systematic review. Adv Nutr. 2017;8(2):213–25. https://doi.org/10.3945/an.116.014431. This study conducted a systematic review on the post-prandial changes of five proinflammatory markers and their timing, which provided insights for further studies.
Wang F, Lu H, Liu F, Cai H, Song Z, Guo F, et al. Effects of a liquid high-fat meal on postprandial lipid metabolism in type 2 diabetic patients with abdominal obesity. Nutr Metab (Lond). 2017;14:54. https://doi.org/10.1186/s12986-017-0211-5.
Silva Correa C, Rebolledo Cobos RC, Reischak-Oliveira A. Strength exercise and training in postprandial lipaemia. J Sports Med Phys Fitness. 2015;55(9):1037–45.
Teeman CS, Kurti SP, Cull BJ, Emerson SR, Haub MD, Rosenkranz SK. Postprandial lipemic and inflammatory responses to high-fat meals: a review of the roles of acute and chronic exercise. Nutr Metab (Lond). 2016;13:80. https://doi.org/10.1186/s12986-016-0142-6.
Schwarzova L, Hubacek JA, Vrablik M. Genetic predisposition of human plasma triglyceride concentrations. Physiol Res. 2015;64(Suppl 3):S341–54.
van Dongen J, Willemsen G, Chen WM, de Geus EJ, Boomsma DI. Heritability of metabolic syndrome traits in a large population-based sample. J Lipid Res. 2013;54(10):2914–23. https://doi.org/10.1194/jlr.P041673.
Rahman I, Bennet AM, Pedersen NL, de Faire U, Svensson P, Magnusson PK. Genetic dominance influences blood biomarker levels in a sample of 12,000 Swedish elderly twins. Twin Res Hum Genet. 2009;12(3):286–94. https://doi.org/10.1375/twin.12.3.286.
MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45(D1):D896–901. https://doi.org/10.1093/nar/gkw1133.
Mitchell BD, McArdle PF, Shen H, Rampersaud E, Pollin TI, Bielak LF, et al. The genetic response to short-term interventions affecting cardiovascular function: rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. Am Heart J. 2008;155(5):823–8. https://doi.org/10.1016/j.ahj.2008.01.019.
• Irvin MR, Zhi D, Aslibekyan S, Claas SA, Absher DM, Ordovas JM, et al. Genomics of post-prandial lipidomic phenotypes in the Genetics of Lipid lowering Drugs and Diet Network (GOLDN) study. PloS One. 2014;9(6):e99509. https://doi.org/10.1371/journal.pone.0099509. This paper reported the GWAS and EWAS of 11 post-prandial sterols and 35 post-prandial fatty acids.
Jackson KG, Poppitt SD, Minihane AM. Postprandial lipemia and cardiovascular disease risk: Interrelationships between dietary, physiological and genetic determinants. Atherosclerosis. 2012;220(1):22–33. https://doi.org/10.1016/j.atherosclerosis.2011.08.012.
Perez-Martinez P, Delgado-Lista J, Perez-Jimenez F, Lopez-Miranda J. Update on genetics of postprandial lipemia. Atheroscler Suppl. 2010;11(1):39–43. https://doi.org/10.1016/j.atherosclerosissup.2010.03.002.
•• Wojczynski MK, Parnell LD, Pollin TI, Lai CQ, Feitosa MF, O’Connell JR, et al. Genome-wide association study of triglyceride response to a high-fat meal among participants of the NHLBI Genetics of Lipid Lowering Drugs and Diet Network (GOLDN). Metabolism. 2015;64(10):1359–71. https://doi.org/10.1016/j.metabol.2015.07.001. This study is one of the only two GWAS on PPL TG reported so far.
Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–13. https://doi.org/10.1038/nature09270.
Aslibekyan S, Goodarzi MO, Frazier-Wood AC, Yan X, Irvin MR, Kim E, et al. Variants identified in a GWAS meta-analysis for blood lipids are associated with the lipid response to fenofibrate. PloS One. 2012;7(10):e48663. https://doi.org/10.1371/journal.pone.0048663.
Parnell LD, Blokker BA, Dashti HS, Nesbeth PD, Cooper BE, Ma Y, et al. CardioGxE, a catalog of gene-environment interactions for cardiometabolic traits. BioData Min. 2014;7:21. https://doi.org/10.1186/1756-0381-7-21.
Kristiansson K, Perola M, Tikkanen E, Kettunen J, Surakka I, Havulinna AS, et al. Genome-wide screen for metabolic syndrome susceptibility Loci reveals strong lipid gene contribution but no evidence for common genetic basis for clustering of metabolic syndrome traits. Circ Cardiovasc Genet. 2012;5(2):242–9. https://doi.org/10.1161/CIRCGENETICS.111.961482.
Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43(4):333–8. https://doi.org/10.1038/ng.784.
• Irvin MR, Zhi D, Joehanes R, Mendelson M, Aslibekyan S, Claas SA, et al. Epigenome-wide association study of fasting blood lipids in the Genetics of Lipid-lowering Drugs and Diet Network study. Circulation. 2014;130(7):565–72. https://doi.org/10.1161/CIRCULATIONAHA.114.009158. This is the EWAS of fasting TG reported by the GOLDN study.
Lewis GF, Xiao C, Hegele RA. Hypertriglyceridemia in the genomic era: a new paradigm. Endocr Rev. 2015;36(1):131–47. https://doi.org/10.1210/er.2014-1062.
Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–83. https://doi.org/10.1038/ng.2797.
Wang KC, Chang HY. Epigenomics: technologies and applications. Circ Res. 2018;122(9):1191–9. https://doi.org/10.1161/CIRCRESAHA.118.310998.
Duan L, Liu C, Hu J, Liu Y, Wang J, Chen G et al. Epigenetic mechanisms in coronary artery disease: the current state and prospects. Trends Cardiovasc Med. 2017. https://doi.org/10.1016/j.tcm.2017.12.012.
Fernandez-Sanles A, Sayols-Baixeras S, Subirana I, Degano IR, Elosua R. Association between DNA methylation and coronary heart disease or other atherosclerotic events: a systematic review. Atherosclerosis. 2017;263:325–33. https://doi.org/10.1016/j.atherosclerosis.2017.05.022.
Zheng J, Cheng J, Zhang Q, Xiao X. Novel insights into DNA methylation and its critical implications in diabetic vascular complications. Biosci Rep. 2017;37(2). https://doi.org/10.1042/BSR20160611.
Jia L, Zhu L, Wang JZ, Wang XJ, Chen JZ, Song L, et al. Methylation of FOXP3 in regulatory T cells is related to the severity of coronary artery disease. Atherosclerosis. 2013;228(2):346–52. https://doi.org/10.1016/j.atherosclerosis.2013.01.027.
Zhuang J, Peng W, Li H, Wang W, Wei Y, Li W, et al. Methylation of p15INK4b and expression of ANRIL on chromosome 9p21 are associated with coronary artery disease. PloS One. 2012;7(10):e47193. https://doi.org/10.1371/journal.pone.0047193.
•• Lai CQ, Wojczynski MK, Parnell LD, Hidalgo BA, Irvin MR, Aslibekyan S, et al. Epigenome-wide association study of triglyceride postprandial responses to a high-fat dietary challenge. J Lipid Res. 2016;57(12):2200–7. https://doi.org/10.1194/jlr.M069948. This paper reported the first and the only EWAS of PPL TG performed in GOLDN study.
Truong V, Huang S, Dennis J, Lemire M, Zwingerman N, Aissi D, et al. Blood triglyceride levels are associated with DNA methylation at the serine metabolism gene PHGDH. Sci Rep. 2017;7(1):11207. https://doi.org/10.1038/s41598-017-09552-z.
• Braun KVE, Dhana K, de Vries PS, Voortman T, van Meurs JBJ, Uitterlinden AG, et al. Epigenome-wide association study (EWAS) on lipids: the Rotterdam Study. Clin Epigenetics. 2017;9:15. https://doi.org/10.1186/s13148-016-0304-4. This is a recent EWAS of fasting TG.
• Hedman AK, Mendelson MM, Marioni RE, Gustafsson S, Joehanes R, Irvin MR et al. Epigenetic patterns in blood associated with lipid traits predict incident coronary heart disease events and are enriched for results from genome-wide association studies. Circ Cardiovasc Genet. 2017;10(1). https://doi.org/10.1161/CIRCGENETICS.116.001487. This is a recent EWAS of fasting TG.
• Sayols-Baixeras S, Subirana I, Lluis-Ganella C, Civeira F, Roquer J, Do AN, et al. Identification and validation of seven new loci showing differential DNA methylation related to serum lipid profile: an epigenome-wide approach. The REGICOR study. Hum Mol Genet. 2016;25(20):4556–65. https://doi.org/10.1093/hmg/ddw285. This is a recent EWAS of fasting TG.
• Dekkers KF, van Iterson M, Slieker RC, Moed MH, Bonder MJ, van Galen M, et al. Blood lipids influence DNA methylation in circulating cells. Genome Biol. 2016;17(1):138. https://doi.org/10.1186/s13059-016-1000-6. This is a recent EWAS of fasting TG.
• Pfeiffer L, Wahl S, Pilling LC, Reischl E, Sandling JK, Kunze S, et al. DNA methylation of lipid-related genes affects blood lipid levels. Circ Cardiovasc Genet. 2015;8(2):334–42. https://doi.org/10.1161/CIRCGENETICS.114.000804. This is a recent EWAS of fasting TG.
Barres R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15(3):405–11. https://doi.org/10.1016/j.cmet.2012.01.001.
Robson-Ansley PJ, Saini A, Toms C, Ansley L, Walshe IH, Nimmo MA, et al. Dynamic changes in dna methylation status in peripheral blood Mononuclear cells following an acute bout of exercise: potential impact of exercise-induced elevations in interleukin-6 concentration. J Biol Regul Homeost Agents. 2014;28(3):407–17.
Lindholm ME, Marabita F, Gomez-Cabrero D, Rundqvist H, Ekstrom TJ, Tegner J, et al. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics. 2014;9(12):1557–69. https://doi.org/10.4161/15592294.2014.982445.
de Mello VD, Kolehmanien M, Schwab U, Pulkkinen L, Uusitupa M. Gene expression of peripheral blood mononuclear cells as a tool in dietary intervention studies: what do we know so far? Mol Nutr Food Res. 2012;56(7):1160–72. https://doi.org/10.1002/mnfr.201100685.
Tremblay BL, Guenard F, Rudkowska I, Lemieux S, Couture P, Vohl MC. Epigenetic changes in blood leukocytes following an omega-3 fatty acid supplementation. Clin Epigenetics. 2017;9:43. https://doi.org/10.1186/s13148-017-0345-3.
Burdge GC, Lillycrop KA. Fatty acids and epigenetics. Curr Opin Clin Nutr Metab Care. 2014;17(2):156–61. https://doi.org/10.1097/MCO.0000000000000023.
Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES. Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds: a meta-analysis of randomized controlled trials. J Am Coll Cardiol. 2005;45(2):185–97. https://doi.org/10.1016/j.jacc.2004.10.031.
Kelly MS, Beavers C, Bucheit JD, Sisson EM, Dixon DL. Pharmacologic approaches for the management of patients with moderately elevated triglycerides (150–499 mg/dL). J Clin Lipidol. 2017;11(4):872–9. https://doi.org/10.1016/j.jacl.2017.05.014.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Xu declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal studies performed by any of the authors.
Additional information
This article is part of the Topical Collection on Cardiovascular Genetics
Rights and permissions
About this article

Cite this article
Xu, H. Genetic and Epigenetic Regulations of Post-prandial Lipemia. Curr Genet Med Rep 6, 124–131 (2018). https://doi.org/10.1007/s40142-018-0146-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-018-0146-9