
Abstract
Solvent-accessibility change plays a critical role in protein misfolding and aggregation, the culprit for several neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS). Mass spectrometry-based hydroxyl radical (·OH) protein footprinting has evolved as a powerful and fast tool in elucidating protein solvent accessibility. In this work, we used fast photochemical oxidation of protein (FPOP) hydroxyl radical (·OH) footprinting to investigate solvent accessibility in human copper-zinc superoxide dismutase (SOD1), misfolded or aggregated forms of which underlie a portion of ALS cases. ·OH-mediated modifications to 56 residues were detected with locations largely as predicted based on X-ray crystallography data, while the interior of SOD1 β-barrel is hydrophobic and solvent-inaccessible and thus protected from modification. There were, however, two notable exceptions—two closely located residues inside the β-barrel, predicted to have minimal or no solvent accessibility, that were found modified by FPOP (Phe20 and Ile112). Molecular dynamics (MD) simulations were consistent with differential access of peroxide versus quencher to SOD1’s interior complicating surface accessibility considerations. Modification of these two residues could potentially be explained either by local motions of the β-barrel that increased peroxide/solvent accessibility to the interior or by oxidative events within the interior that might include long-distance radical transfer to buried sites. Overall, comparison of modification patterns for the metal-free apoprotein versus zinc-bound forms demonstrated that binding of zinc protected the electrostatic loop and organized the copper-binding site. Our study highlights SOD1 hydrophobic groups that may contribute to early events in aggregation and discusses caveats to surface accessibility conclusions.

Graphical Abstract
Similar content being viewed by others


Introduction
Protein misfolding and aggregation are associated with many human diseases, especially those with neurodegenerative phenotypes, such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS) [1]. The aggregation propensity of proteins and peptides relevant to these diseases is dependent on their intrinsic physico-chemical properties [2]. What causes these proteins and peptides to aggregate and how the aggregation is linked to the disease remain the subject of intensive investigation.
Solvent-accessibility change is an important factor in protein aggregation. The exposure of hydrophobic groups of a protein often precedes its aggregation [3, 4] as the driving force for aggregation is similar to that for protein folding, i.e., the reduction of non-polar solvent-accessible area and the maximization of van der Waals interactions, leading to lower free energy. The aggregation propensity of a protein increases as its non-polar solvent-accessible area expands. Solvent accessibility is thus widely used in predicting the tendency of proteins and peptides to aggregate [5, 6]. The species with exposed hydrophobic groups are believed to represent partially unfolded intermediates that may proceed to form aggregates via misfolding pathways.
Mass spectrometry-based methods, such as hydrogen-deuterium exchange (HDX), are used to probe the accessibility and dynamics of a protein’s polypeptide backbone. Conversely, there has been rapid development of hydroxyl radical (·OH) as a modification reagent for protein footprinting and its use in studying protein side-chain solvent accessibility. The size of ·OH is similar to that of water molecules, and the reaction of ·OH with amino acid side chains is 10 to 2000 times faster than that with the polypeptide backbone [7]. In the presence of quenchers, labeling occurs rapidly in milli-/micro-second time scales [8, 9] faster than protein unfolding [10]. Hydroxyl radicals are generated either by synchrotron radiolysis of water [8, 9] or by laser photolysis of hydrogen peroxide (fast photochemical oxidation of proteins; FPOP) [11]. The fast and irreversible labeling reactions allow various applications of ·OH footprinting in answering important questions like protein folding/unfolding [12, 13], fast biological processes such as metal transport [14], conformational changes upon ligand binding [15], and epitope mapping [16, 17]. Recently, addition of a small molecule dosimeter has been used to improve the accuracy of surface accessibility measurements (SASA) [18,19,20].
From bacteria to humans, copper-zinc superoxide dismutase (SOD1) participates in antioxidant defense by catalyzing the disproportionation of superoxide radicals to dioxygen and H2O2. There has been intense interest in SOD1 since linkage between mutations in the human SOD1 (hSOD1) gene and ALS, a fatal neurodegenerative disease characterized by the death of motor neurons, was discovered in 1993 [21]. To date, over 160 ALS-causing mutations have been identified [22, 23]. As in other neurodegenerative diseases, hSOD1-associated ALS results from a “gain-of-function” mechanism thought to involve the formation of toxic aggregates [22, 23].
Since it is likely that exposure of hydrophobic protein interior often precedes protein aggregation, identification of SOD1 species with exposed hydrophobic groups will thus provide insights into the early events of hSOD1 aggregation in ALS. The solvent accessibility of hSOD1 has been investigated by HDX, revealing an increased accessibility in an ALS mutant (A4V) [24] and two flexible β-strands, β3 and β4, in metal-free hSOD1 [25]. While HDX provides information at the peptide backbone level, we describe here a study of solvent and H2O2 accessibility of hSOD1 at the peptide and residue levels using FPOP. Since hydrogen peroxide and oxygen are the products of the major catalytic activity of SOD1, it is reasonable to expect channels to conduct H2O2 away from the active site toward the solvent milieu. Consequently, OH can be generated by UV-photodissociation events within the protein’s structure potentially complicating the analysis beyond “surface accessibility.”
Materials and Methods
Materials
Formic acid, 30% hydrogen peroxide, sodium chloride, and HPLC-grade solvents were purchased from Thermo Fisher (Waltham, MA). Catalase, l-glutamine, l-methionine, ammonium bicarbonate, sodium phosphate monobasic/dibasic, iodoacetamide, dithiothreitol (DTT), triethylammonium acetate, formaldehyde solution, formaldehyde-13C, d2 solution, sodium cyanoborohydride, and sodium cyanoborodeuteride were obtained from Sigma Aldrich (St. Louis, MO). Trypsin and chymotrypsin were purchased from Promega Corp. (Madison, WI). C18 zip tips were obtained from Millipore (Billerica, MA).
Expression and Purification of SOD1
Saccharomyces cerevisiae cells carrying YEp351-SOD1 were grown in YPD media (1% yeast extract, 2% peptone, 2% dextrose, pH 6) at 30 °C with shaking at 220 rpm to O.D. > 10. Cells were harvested by centrifugation at 12,000×g for 10 min. Isolation of hSOD1 was performed as previously described [26].
FPOP Labeling
The setup of the FPOP system was based on previous reports with minor changes. A KrF excimer laser (GAM Laser Inc., Orlando, FL) operated at 248 nm was used to irradiate the sample solution. The laser was focused through a 50-mm bi-convex lens (Thorlabs, Newton, NJ) to a beam of 2 mm wide (3.5-mm height) onto 150-μm i.d. fused silica tubing (Polymicro Technologies, Pheonix, AZ), the coating of which was removed using an ethanol burner. The samples were pumped through the silica tubing using a syringe-infusion pump (KD Scientific, Holliston, MA).
The preparation of apo- and Zn-bound hSOD1 was performed as previously described [27]. The metal contents were confirmed by inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7500 Series).
Apo- and Zn-bound hSOD1 was mixed with glutamine and phosphate buffered saline (PBS, 10 mM sodium phosphate, 138 mM NaCl, and 2.7 mM KCl, pH 7.4). A 10× stock solution of hydrogen peroxide (H2O2) was made and added just prior to sample infusion for FPOP labeling to achieve a final concentration of 5–15 mM. The final concentrations of SOD1 and glutamine were 32 μM and 20 mM, respectively. Samples were infused at a flow rate of 15 μL/min into tubes containing 100 nM catalase and 30 mM methionine in PBS buffer, in order to remove excess H2O2 and generated reactive radicals. The laser was tuned to 65 mJ/pulse and was pulsed at a frequency of 6 Hz. Given the flow rate, capillary dimensions, laser repeat rate, and beam width above, we estimate 85% of the sample was illuminated minimizing multiple hits in non-uniform flow [28]. In terms of the control samples, the same amounts of H2O2 were added, but the samples were infused in the same time frame without being irradiated by the laser. Holo-hSOD1 was not included in this study due to excessive oxidation in the control (plus H2O2, minus illumination) presumably due to the presence of Cu. The oxidation seen in the minus irradiation control (Figure S1A) was omitted from the analysis.
Mass Spectrometry Sample Preparation
The samples for determining the global modification level were prepared by acidifying an aliquot of each irradiated or control sample and eluting it from C18 zip tips.
To prepare samples for determining the modification at peptide and residue levels, an aliquot of each irradiated or control sample was dried using a speed vac and then resuspended in 50 mM ammonium bicarbonate and 6 M guanidinium hydrochloride, pH 7.8. The samples were reduced with 10 mM DTT for 30 min and alkylated with 30 mM iodoacetamide in the dark for 1 h. They were then diluted 1:4 with 50 mM ammonium bicarbonate, pH 7.8, and digested overnight at 37 °C with trypsin or chymotrypsin at an enzyme:protein ratio of 1:20.
The digested samples were dried using a speed vac and then resuspended in 100 mM triethylammonium acetate. The reductive dimethylation of the lysine groups was performed as previously described [29, 30]. The irradiated (heavy) and control (light) samples were mixed together at a 1:1 M ratio and were then cleaned up by elution from C18 Zip tips (Millipore).
LC-ESI-MS and MS/MS
The global MS spectra were obtained using LTQ (Thermo Scientific) coupled with a HPLC (Agilent 1100 series). Samples were loaded onto a polymeric reverse-phase column (PLRPS; 2.1 × 150 mm, 300 Å, 5 μm; Agilent). We used a 50-min gradient with a 10-min equilibration phase with 2% solvent B (acetonitrile with 0.1% formic acid), a linear increase to 95% solvent B in 30 min, and a final re-equilibration to 2% solvent B for 10 min. Mass spectra were acquired over an m/z range of 300–2000.
The digested samples were analyzed using LTQ Orbitrap XL (Thermo Scientific) equipped with an Eksigent (Dublin, CA) NanoLiquid chromatography-1D plus system and an Eksigent autosampler. They were loaded onto an Agilent 5 × 0.3 mm, 5 μm ID, ZORBAX 300SB-C18 pre-Trap column (Santa Clara, CA) and washed for 10 min with 100% Buffer A (0.1% formic acid) at a flow rate of 6 μL/min. The peptides were then separated on a precision capillary columns, 0.1 × 150 mm, 3 μm ID, 200 Å, MAGIC C18AQ column (San Clemente, CA), using a flow rate of 400 nL/min. The following elution gradient was used: 0–20 min 4–35% Buffer B (100% acetonitrile), 20–24 min 35–80% Buffer B, and 24–26 min 80% Buffer B. The column was then re- equilibrated for 18 min with Buffer A. Mass spectra (ESI-MS) and tandem mass spectra (ESI-MS/MS) were recorded in positive-ion mode with electrospray ionization voltage of 1800 V, capillary voltage of 49 V, tube lens of 130 V, and capillary temperature of 180 °C. Information-dependent acquisition was performed where the five most intense ions were selected in the m/z range of 300–1600 using a 100,000-resolution FTMS scan and subjecting them to MS-MS using broadband collision-induced disassociation of normalized collision energy of 35% and LTQ detection. Dynamic exclusion was applied for a period of 60 s.
Data Analysis
The raw files were imported into Progenesis LC-MS (version 4.1, Nonlinear Dynamics, Durham, NC). The software aligns the chromatograms of the irradiated and control samples, assigns the mass peaks, and creates a merged set of mascot generic format (mgf) files from all acquisitions. The mgf files were then searched for unmodified and modified SOD1 peptides using an in-house software package. Only precursor ions with abundance higher than 5 × 103 were considered for MS/MS search.
The software package constructs a library containing SOD1 peptides with all possible FPOP-induced oxidative modifications and searches for peptides from the mgf files based on their precursor ion masses with errors of ≤ 100 ppm and their MS/MS spectra. Each peptide can have at most one miscleavage. The acetylation of the SOD1 N-terminus and dimethylation of lysine residues (heavy) were set as static modifications. The FPOP-induced variable modifications that were considered for MS/MS search are listed in Table S1. Up to three variable modifications per peptide were considered. The MS/MS spectra of the matches were then visually examined to ensure that they were valid.
For quantification based on stable isotope tagging, individual MS runs were combined, and the peak intensity of dimethyl labeling pairs was calculated by summing-up the three most intense peaks of each peptide (Figure S2). The extent of modification of each peptide is defined as
where IFPOP and Icontrol are the peak intensities of the same peptide in the irradiated and control samples with light and heavy isotope labeling, respectively.
Peptide quantification was performed using Progenesis LC-MS, which quantifies ion abundance from the survey scan data. The fraction of a peptide with specific modifications was calculated as the ratio of ion abundance of this peptide to the total ion abundances of the same peptide that are unmodified or modified.
Molecular Dynamics Simulations to Access the Differential Penetrance of H2O2 and Glutamine into the SOD1 Protein
The differential access of the substrate H2O2 and the amino acid glutamine from the bulk aqueous solvent to the active-site channel of native superoxide dismutase with metal ligands or with metal-depleted apo-SOD protein was further studied using molecular dynamics simulations with the Gromacs software suite of programs (version 5.1.1). Coordinate files for the monomeric metal ligand structure of the protein (2LU5.pdb) or apo-metal-depleted SOD (1HL4.pdb) were obtained from the Protein Data Bank (http://www.rcsb.org/pdb/) and placed in a 64 × 64 Å simulation box. The solute was then added to the protein with either H2O2 (RHQH.pdb) or glutamine (FBCA.pdb) model structures from the Automated Topography Builder (ATB) and Repository version 2.2 (https://atb.uq.edu.au/) to a solvent box concentration of 5 mM using the Build-Solvent cluster utility of VEGA ZZ Molecular Modeling Toolkit (version 3.1.0; nova.disfarm.unimi.it/cms/index.php). The simulation box was then solvated with SPC water, and sodium counter ions were added to achieve neutrality. Minimization and production dynamics runs were carried out employing the Gromos 54a7_atb united atom force field (ATB).
The SOD structure, solute, and solvent system were then refined using the GROMACS suite of molecular dynamics (MD) programs [31] (http://www.gromacs.org). The system was then minimized using a steepest decent strategy followed by a three-step minimization-equilibration process at 311°K for a total of 200 ps. This included both NVT and NPT equilibration phases to allow water and solute molecules to reorient around any of the exposed parts of the protein. Equilibration protocols employed a PME strategy for Coulombic long-range interactions and Berendsen temperature coupling. A Berendsen strategy was also used for pressure coupling in an isotropic mode to emulate solution motion. After equilibration, the system was subjected to a dynamics production run at the same temperature using the Nose-Hoover protocol and pressure (Parrinello-Rahman) values used in the pre-run steps for a period of 300 ns. The Verlet cut-off scheme was employed for all minimization, equilibration, and production steps.
The output of the production run simulation was analyzed with the Gromacs suite of analysis tools while molecular graphics were rendered using Pymol (version 1.7.4.1; Schrodinger, LLC).
Results and Discussion
Zn Binding Stabilizes the hSOD1 Electrostatic Loop
To elucidate the impact of metal binding on solvent exposure, we performed FPOP labeling on apo- and Zn-loaded SOD1 at the same concentration of H2O2. The mass spectra of the undigested proteins were similar (Figure S1), both containing a distribution of 0, + 16, + 32, … Da states, corresponding to the introduction of oxygen atoms. Although its amount was reduced by over 20-fold with FPOP labeling, the unmodified protein remained the most abundant species, in good agreement with other FPOP studies [10, 11]. Both trypsin and chymotrypsin were used for efficient proteolysis of control and modified samples enabling relative quantification of unmodified peptides. Chymotrypsin is typically avoided for quantification of modified residues since it targets residues that are oxidized frequently potentially interfering with enzyme specificity and thus peptide yield.
To achieve fast and accurate analysis of the FPOP data, we used duplex stable isotope dimethyl labeling to quickly identify peptides whose level of unmodified form changed significantly [30, 32] (Figure S2), a similar tactic to the 15N labeling strategy used previously [33]. We found that the region encompassing residues 123–143 became less modified in Zn-hSOD1 (P value < 0.05) (Figure 1A). This region belongs to one of the functional loops, the electrostatic loop (residues 121–144; Figure 2), which acts as an active-site lid, limiting access of solvent to the metal-binding sites. The effect of Zn binding was consistently observed at increased H2O2 concentrations (Figure S3). Our findings are in agreement with a flexible and disordered electrostatic loop as has previously been found in crystallographic [34, 35] and NMR [36] studies of apo-hSOD1. The electrostatic loop thus results in an open clamshell conformation in the metal-free protein with closure induced by binding of Zn.

The electrostatic loop becomes less solvent accessible upon zinc binding. The figure shows the extent of modification of peptides from hSOD1 proteins irradiated in the presence of 5 mM H2O2. The proteins were digested by trypsin (a) and chymotrypsin (b). The extent of modification was determined by comparing the intensities of unmodified peptides between irradiated and control samples (see Experimental Section, Figure S2) (c). The trypsin difference data was superimposed upon the hSOD1 monomer structure with blue representing the region less solvent accessible in the Zn-bound form (residues 123–143) while a short region that became more accessible is shaded in red (116–122). The graphic was generated from PDB 1HL4 using PyMOL software and shows a ribbon diagram of the monomer polypeptide backbone with Zn bound (gray disc). The structure 1HL4 was obtained by X-ray crystallography of the Zn-bound apo-hSOD1 [35]

The ribbon diagram of metal-deficient (20% occupancy of Zn) hSOD1 (PDB: 1HL4) shows that Trp32 is highly solvent accessible while Val118 is shielded by the copper-binding site. Peptide 123–143 (the electrostatic loop is regarded as 121–144) is highlighted in blue. Trp32 and Val118 are shown in orange, and the copper ligands are shown in magenta. The graphic was generated using PyMOL software
Previous NMR data demonstrated that other regions, such as the zinc loop and four β-strands (β4, β5, β7, β8), also showed increased disorder in apo-hSOD1 [36]; in our study, however, these regions displayed similar solvent accessibility in apo- and Zn-hSOD1 (Figure 1). One explanation is that the extent of a region’s exposure to solvent does not always correlate with the motion of the region. For example, β3 (Q16-E25) was almost completely modified by FPOP (see below), although NMR data showed that it was well-ordered in metal-free SOD1 [36].
Site-Specific Oxidation of hSOD1
From trypsin and chymotrypsin digestion, the peptides that carried the highest degree of modification corresponded to residues 24–36 and 21–32, respectively (Figure 1). Noticeably, they both contain Trp32, a surface residue belonging to β3 (Figure 2). Trp32 was found to carry a mass shift of + 32 Da, with respect to the net addition of two oxygen atoms (Table S2). As tryptophan is the second most reactive residue toward ·OH (k = 1.3 × 1010 M−1 s−1) [9], Trp32 is expected to be highly modified. Indeed, compared to the unmodified residues 24–36, the ion abundance of the same peptide carrying a + 32-Da modification on Trp32 was higher by an order of magnitude (Table S3).
Trp32 oxidation occurs naturally in both wild-type and ALS-mutant hSOD1 [37]. Bicarbonate-mediated peroxidase activity of hSOD1 resulting in oxidation of Trp32 alters the copper active-site geometry [38] leading to aggregation of the protein [39]. Conversely, substitution of Trp32 reduces aggregation propensity of ALS-mutant hSOD1 and mitigates its cytotoxicity to motor neurons [37]. As ·OH radicals are continuously produced by Fenton reactions [40] and peroxynitrite (ONOO–) decomposition in vivo, Trp32 oxidation has been suggested to contribute to aggregation of hSOD1 in ALS [41]. Despite the sensitivity of Trp32 to oxidation, when hSOD1 was isolated as detergent-resistant aggregates from spinal cords of ailing ALS-model mice, the protein was predominantly unmodified with its Trp32 residue intact [42].
Using an in-house MS/MS search engine, we identified the modifications of 56 residues in apo-hSOD1 (Tables S1 and S2). They spread throughout the 153-residue SOD1 polypeptide chain and include all but one of the copper and zinc ligands (Figure 3, Table S2). We found 33 unique types of modifications on 16 unique types of amino acids, including not only aromatic residues but also aliphatic ones like alanine, even if the latter are less susceptible toward ·OH-mediated oxidation by 1–3 orders of magnitude [9, 43] consistent with reaction of ·OH with side chains being efficient and unspecific.

Location and mass change of FPOP-induced modifications on Apo-hSOD1. The mass shift(s) are shown below each modified residue. The copper and zinc ligands are highlighted in red and blue, respectively. Cysteine residues are highlighted in yellow
Folding of a β-barrel protein involves the exclusion of water as the interior of the β-barrel forms a hydrophobic cavity. From the X-ray structural data, 24 residues are predicted to have limited solvent accessibility in metal-deficient hSOD1 (Table S4), of which 16 (Ala4, Cys6, Leu8, Ile18, Phe20, Val29, Val31, Ile35, Phe45, Val47, Ala89, Ala95, Leu117, Val119, Ala145, Ile149) are located at the inner wall of the β-barrel. Our results largely agree with the X-ray data, as the majority (15 out of 16) of the residues at the β-barrel inner wall remained unmodified, indicating that the β-barrel was largely folded when the labeling occurred.
Oxidation of Inaccessible Residues
The oxidized products of several completely inaccessible residues, such as Val118, were found (Figure 3, Table S2). It resides at the exterior of β-strand β7 and is deeply buried under the copper active site formed by four histidines, His46, His48, His63, and His120 (Figure 2). As a defined conformation at the copper site would protect Val118 from oxidation, its oxidation reveals an unstructured state of the apoprotein copper site. This finding is consistent with the NMR data, demonstrating that β-strands β4 and β7, while holding Val118 and three copper-binding histidines, are significantly disordered as a consequence of the lack of both of the metal ions [36]. In our study, none of the peptides with oxidized Val118 was found in Zn-loaded hSOD1 (Table S5), in agreement with the previous finding that Zn binding organizes all the β-strands [36, 44].
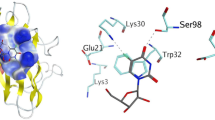
Finally, we were surprised to find two modification sites inside the β-barrel. Phe20, a residue at the β-barrel inner wall, is spatially close to Ile112, a residue at the β-barrel plug. The distance between Phe20 C3 and Ile112 Cδ is 3.7 Å (Figure 4). Despite minimal or zero solvent accessibility (ASA = 0.37 and 0 Å2, respectively), Phe20 and Ile112 of apo-hSOD1 both carried oxidative modifications (Figure 3, Table S2) with their MS/MS spectra shown in Figure S4. Based on label-free quantification, the percentages of peptides with oxidized Phe20 and Ile112 were ~ 6% and ~ 0.06%, respectively (Table S6 and S7). The oxidized products were consistently detected from irradiated apo-hSOD1, although the extents of modification were only marginal. Oxidation of Phe20 but not Ile112 was observed after FPOP of Zn-hSOD1 (Table S6 and S7). The role of these residues in the aggregation of hSOD1 is under investigation.

The modifications of residues, Phe20 and Ile112, with limited solvent accessibility (PDB: 1HL4). The numbers are the measured distances(Å). The graphic was generated using PyMOL software
The observation by FPOP of oxidation in the hydrophobic core of a folded β-barrel protein surprised us since it challenges the “surface accessibility” paradigm. Firstly, oxidation could result from transient local unfolding of the β-barrel that exposes otherwise inaccessible residue side chains. Oxidation of Met residues (Serpin Met374 and 385) with crystal-structure-derived zero SASA was observed by Wang and Chance who used MD simulations to observe “dynamic excursions” of the S atoms that apparently enabled their hydroxyl radical oxidation in radiolysis experiments [45]. Analysis of metal-free hSOD1 solution structure has suggested that the β-barrel exhibits “breathing” motions [36] and that two β-strands, β7 and β8, adopt an open conformation [36], which enlarges the β-barrel mouth close to the N- and C-termini (Figure S5). Our previous finding that the third and fourth β-strands, β3 and β4, are prone to unfolding provides another pathway that enhances H2O2, O2, and water molecule access to interior residues [25]. The modifications of Phe20 and Ile112, if resulting from the partial unfolding of the hSOD1 β-barrel, could implicate a transition state with an increased non-polar solvent-accessible area that predisposes aggregate formation. As mentioned previously, SOD1 presents a special case where both H2O2 and O2 require internal pathways to leave the active-site region and travel to the aqueous milieu giving opportunity for UV photodissociation to directly generate ·OH within the interior “solvent-inaccessible” domain of the protein or for direct oxidation of internal residues to occur prior to reaction with O2. If small molecule quenchers (or dosimeters) do not have access to the interior then ·OH would have a lifetime reflective of diffusion prior to reaction with protein, which would be expected to be very short compared to the lifetime in quencher solution given the immediate proximity of protein. With available X-ray structures of both holo- and apo-hSOD1, we used molecular simulations to further investigate differential small molecule access to the interior of apo-hSOD1 (Figures S6–S9). While H2O2 freely penetrated both structures regardless of metal occupancy, the free amino acid glutamine remained excluded in both cases for the duration of the simulation. Differential access of peroxide versus small molecule quencher/dosimeter complicates SASA for the small SOD1 protein.
One further possibility is that long-range electron/hole transfer could move oxidation sites thereby contributing to the modification of Phe20. Multi-step tunneling/hopping of electrons/holes between reactants separated by several nanometers has been observed in a variety of metalloproteins by Gray and Winkler [46, 47]. The higher occurrence frequencies of aromatic residues in oxidoreductases with respect to other protein classes are believed to originate from their roles in mediating these long-range electron/hole transfer events, as the reactions are faster between aromatic amino acids [46, 48]. Phe20 is spaced at 12.7 Å from a surface histidine, His110 (Figure 4). The Phe20 radical (Phe20·) could form via a hole transfer from oxidized His43 (His43·) with a predicted tunneling time of nanoseconds, much shorter than the time required for the unfolding of β-barrel proteins [49]. Oxygen atoms would then be added rapidly to the Phe20 aromatic ring, given proximity to O2. Given that previous studies show addition of ·OH resulting in a transient histidyl radical [50], this explanation may be less likely. The lifetime of secondary oxidants in FPOP has been discussed with respect to the aqueous phase [51].
Conclusions
In this study, we have demonstrated the successful application of FPOP in identifying solvent-exposed groups of hSOD1. The combination of peptide- and residue-level analysis gave detailed information about the solution structure of hSOD1 that was in substantial agreement with widely available structural information from the literature. The electrostatic loop and the copper-binding site were both disordered in the apoprotein and became organized when Zn was bound. Adding complexity, we found evidence for oxidative modifications of two residues (Phe20 and Ile112) that should be inaccessible inside the β-barrel, potentially resulting from either partial unfolding of the β-barrel or from long-distance electron/hole tunneling from His43 radicals formed on the protein surface. Additionally, our molecular simulation analysis revealed differential access of peroxide reagent over larger quencher molecules. Differential access of reagent, quencher, and dosimeter molecules challenges the assumptions that underlie the FPOP-SASA relationship, at least for the small hSOD1. Despite caveats, the FPOP analysis described promises to be a powerful tool for identification of intermediates that are produced in the early stage of hSOD1 aggregation.
References
Chiti, F., Dobson, C.M.: Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 (2006)
Luheshi, L.M., Crowther, D.C., Dobson, C.M.: Protein misfolding and disease: from the test tube to the organism. Curr. Opin. Chem. Biol. 12, 25–31 (2008)
Tartaglia, G.G., Cavalli, A., Pellarin, R., Caflisch, A.: The role of aromaticity, exposed surface, and dipole moment in determining protein aggregation rates. Protein Sci. 13, 1939–1941 (2004)
Linding, R., Schymkowitz, J., Rousseau, F., Diella, F., Serrano, L.: A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 342, 345–353 (2004)
Tartaglia, G.G., Pawar, A.P., Campioni, S., Dobson, C.M., Chiti, F., Vendruscolo, M.: Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol. 380, 425–436 (2008)
Goldschmidt, L., Teng, P.K., Riek, R., Eisenberg, D.: Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. U. S. A. 107, 3487–3492 (2010)
Buxton, G.V., Greenstock, C.L., Helman, W.P., Ross, A.B.: Critical-review of rate constants for reactions of hydrated electrons, hydrogen-atoms and hydroxyl radicals (·Oh/·O–) in aqueous-solution. J. Phys. Chem. Ref. Data. 17, 513–886 (1988)
Takamoto, K., Chance, M.R.: Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu. Rev. Biophys. Biomol. Struct. 35, 251–276 (2006)
Xu, G.H., Chance, M.R.: Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 107, 3514–3543 (2007)
Gau, B.C., Sharp, J.S., Rempel, D.L., Gross, M.L.: Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 81, 6563–6571 (2009)
Hambly, D.M., Gross, M.L.: Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 16, 2057–2063 (2005)
Chen, J., Rempel, D.L., Gross, M.L.: Temperature jump and fast photochemical oxidation probe submillisecond protein folding. J. Am. Chem. Soc. 132, 15502–15504 (2010)
Gau, B.C., Chen, J., Gross, M.L.: Fast photochemical oxidation of proteins for comparing solvent-accessibility changes accompanying protein folding: data processing and application to barstar. Biochim. Biophys. Acta. 1834, 1230–1238 (2013)
Gupta, S., Chai, J., Cheng, J., D'Mello, R., Chance, M.R., Fu, D.: Visualizing the kinetic power stroke that drives proton-coupled zinc(II) transport. Nature. 512, 101–104 (2014)
Zhang, H., Gau, B.C., Jones, L.M., Vidavsky, I., Gross, M.L.: Fast photochemical oxidation of proteins for comparing structures of protein-ligand complexes: the calmodulin-peptide model system. Anal. Chem. 83, 311–318 (2011)
Yan, Y., Chen, G., Wei, H., Huang, R.Y., Mo, J., Rempel, D.L., Tymiak, A.A., Gross, M.L.: Fast photochemical oxidation of proteins (FPOP) maps the epitope of EGFR binding to adnectin. J. Am. Soc. Mass Spectrom. 25, 2084–2092 (2014)
Jones, L.M., Sperry, J., Carroll, J., Gross, M.L.: Fast photochemical oxidation of proteins for epitope mapping. Anal. Chem. 83, 7657–7661 (2011)
Xie, B., Sharp, J.S.: Hydroxyl radical dosimetry for high flux hydroxyl radical protein footprinting applications using a simple optical detection method. Anal. Chem. 87, 10719–10723 (2015)
Xie, B., Sood, A., Woods, R.J., Sharp, J.S.: Quantitative protein topography measurements by high resolution hydroxyl radical protein footprinting enable accurate molecular model selection. Sci. Rep. 7, 4552 (2017)
Niu, B., Zhang, H., Giblin, D., Rempel, D.L., Gross, M.L.: Dosimetry determines the initial OH radical concentration in fast photochemical oxidation of proteins (FPOP). J. Am. Soc. Mass Spectrom. 26, 843–846 (2015)
Rosen, D.R., Siddique, T., Patterson, D., Figlewicz, D.A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O'Regan, J.P., Deng, H.X., et al.: Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 362, 59–62 (1993)
Sheng, Y., Chattopadhyay, M., Whitelegge, J., Valentine, J.S.: SOD1 aggregation and ALS: role of metallation states and disulfide status. Curr. Top. Med. Chem. 12, 2560–2572 (2012)
Sheng, Y., Abreu, I.A., Cabelli, D.E., Maroney, M.J., Miller, A.F., Teixeira, M., Valentine, J.S.: Superoxide dismutases and superoxide reductases. Chem. Rev. 114, 3854–3918 (2014)
Shaw, B.F., Durazo, A., Nersissian, A.M., Whitelegge, J.P., Faull, K.F., Valentine, J.S.: Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J. Biol. Chem. 281, 18167–18176 (2006)
Durazo, A., Shaw, B.F., Chattopadhyay, M., Faull, K.F., Nersissian, A.M., Valentine, J.S., Whitelegge, J.P.: Metal-free superoxide dismutase-1 and three different amyotrophic lateral sclerosis variants share a similar partially unfolded beta-barrel at physiological temperature. J. Biol. Chem. 284, 34382–34389 (2009)
Chan, P.K., Chattopadhyay, M., Sharma, S., Souda, P., Gralla, E.B., Borchelt, D.R., Whitelegge, J.P., Valentine, J.S.: Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc. Natl. Acad. Sci. U. S. A. 110, 10934–10939 (2013)
Potter, S.Z., Zhu, H., Shaw, B.F., Rodriguez, J.A., Doucette, P.A., Sohn, S.H., Durazo, A., Faull, K.F., Gralla, E.B., Nersissian, A.M., Valentine, J.S.: Binding of a single zinc ion to one subunit of copper-zinc superoxide dismutase apoprotein substantially influences the structure and stability of the entire homodimeric protein. J. Am. Chem. Soc. 129, 4575–4583 (2007)
Konermann, L., Stocks, B.B., Czarny, T.: Laminar flow effects during laser-induced oxidative labeling for protein structural studies by mass spectrometry. Anal. Chem. 82, 6667–6674 (2010)
Boersema, P.J., Raijmakers, R., Lemeer, S., Mohammed, S., Heck, A.J.: Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 4, 484–494 (2009)
Capri, J., Whitelegge, J.P.: Full membrane protein coverage digestion and quantitative bottom-up mass spectrometry proteomics. Methods Mol. Biol. 1550, 61–67 (2017)
Pronk, S., Pall, S., Schulz, R., Larsson, P., Bjelkmar, P., Apostolov, R., Shirts, M.R., Smith, J.C., Kasson, P.M., van der Spoel, D., Hess, B., Lindahl, E.: GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 29, 845–854 (2013)
Hsu, J.L., Chen, S.H.: Stable isotope dimethyl labelling for quantitative proteomics and beyond. Philos. Trans. A Math Phys. Eng. Sci. 374 (2016). https://doi.org/10.1098/rsta.2015.0364
Wang, X., Watson, C., Sharp, J.S., Handel, T.M., Prestegard, J.H.: Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure. 19, 1138–1148 (2011)
Banci, L., Bertini, I., Boca, M., Calderone, V., Cantini, F., Girotto, S., Vieru, M.: Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc. Natl. Acad. Sci. U. S. A. 106, 6980–6985 (2009)
Strange, R.W., Antonyuk, S., Hough, M.A., Doucette, P.A., Rodriguez, J.A., Hart, P.J., Hayward, L.J., Valentine, J.S., Hasnain, S.S.: The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J Mol. Biol. 328, 877–891 (2003)
Banci, L., Bertini, I., Cramaro, F., Del Conte, R., Viezzoli, M.S.: Solution structure of Apo Cu, Zn superoxide dismutase: role of metal ions in protein folding. Biochemistry. 42, 9543–9553 (2003)
Taylor, D.M., Gibbs, B.F., Kabashi, E., Minotti, S., Durham, H.D., Agar, J.N.: Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 282, 16329–16335 (2007)
Karunakaran, C., Zhang, H., Crow, J.P., Antholine, W.E., Kalyanaraman, B.: Direct probing of copper active site and free radical formed during bicarbonate-dependent peroxidase activity of bovine and human copper, zinc-superoxide dismutases. Low-temperature electron paramagnetic resonance and electron nuclear double resonance studies. J. Biol. Chem. 279, 32534–32540 (2004)
Zhang, H., Andrekopoulos, C., Joseph, J., Chandran, K., Karoui, H., Crow, J.P., Kalyanaraman, B.: Bicarbonate-dependent peroxidase activity of human Cu, Zn-superoxide dismutase induces covalent aggregation of protein: intermediacy of tryptophan-derived oxidation products. J. Biol. Chem. 278, 24078–24089 (2003)
Burkitt, M.J., Mason, R.P.: Direct evidence for in vivo hydroxyl-radical generation in experimental iron overload: an ESR spin-trapping investigation. Proc. Natl. Acad. Sci. U. S. A. 88, 8440–8444 (1991)
Beckman, J.S., Beckman, T.W., Chen, J., Marshall, P.A., Freeman, B.A.: Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U. S. A. 87, 1620–1624 (1990)
Shaw, B.F., Lelie, H.L., Durazo, A., Nersissian, A.M., Xu, G., Chan, P.K., Gralla, E.B., Tiwari, A., Hayward, L.J., Borchelt, D.R., Valentine, J.S., Whitelegge, J.P.: Detergent-insoluble aggregates associated with amyotrophic lateral sclerosis in transgenic mice contain primarily full-length, unmodified superoxide dismutase-1. J. Biol. Chem. 283, 8340–8350 (2008)
Huang, W., Ravikumar, K.M., Chance, M.R., Yang, S.: Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: a protection factor analysis. Biophys. J. 108, 107–115 (2015)
Banci, L., Bertini, I., Cantini, F., D'Onofrio, M., Viezzoli, M.S.: Structure and dynamics of copper-free SOD: the protein before binding copper. Protein Sci. 11, 2479–2492 (2002)
Wang, L., Chance, M.R.: Structural mass spectrometry of proteins using hydroxyl radical based protein footprinting. Anal. Chem. 83, 7234–7241 (2011)
Winkler, J.R., Gray, H.B.: Long-range electron tunneling. J. Am. Chem. Soc. 136, 2930–2939 (2014)
Winkler, J.R., Gray, H.B.: Electron flow through metalloproteins. Chem. Rev. 114, 3369–3380 (2014)
Gray, H.B., Winkler, J.R.: Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc. Natl. Acad. Sci. U. S. A. 112, 10920–10925 (2015)
Kubelka, J., Hofrichter, J., Eaton, W.A.: The protein folding ‘speed limit’. Curr. Opin. Struct. Biol. 14, 76–88 (2004)
Lassmann, G., Eriksson, L.A., Lendzian, F., Lubitz, W.: Structure of a transient neutral histidine radical in solution: EPR continuous-flow studies in a Ti3+/EDTA-Fenton system and density functional calculations. J. Phys. Chem. A. 104, 9144–9152 (2000)
Vahidi, S., Konermann, L.: Probing the time scale of FPOP (fast photochemical oxidation of proteins): radical reactions extend over tens of milliseconds. J. Am. Soc. Mass Spectrom. 27, 1156–1164 (2016)
Acknowledgements
This work was supported by NINDS grant P01-NS49134. We thank Dr. Melissa Sondej and Puneet Souda for their aid in mass spectrometry and Dr. Bon-Kyung Koo for her aid in protein expression and purification. The authors thank Prof. Michael Gross and his co-workers for introducing us to the practicalities of setting up FPOP.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
ESM 1
(PDF 2388 kb)
Rights and permissions
About this article

Cite this article
Sheng, Y., Capri, J., Waring, A. et al. Exposure of Solvent-Inaccessible Regions in the Amyloidogenic Protein Human SOD1 Determined by Hydroxyl Radical Footprinting. J. Am. Soc. Mass Spectrom. 30, 218–226 (2019). https://doi.org/10.1007/s13361-018-2075-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-018-2075-y