
Summary
Background The hexapeptide 4A6 (Ac-Thr(tBu)-His(Bzl)-Thr(Bzl)-Nle-Glu(OtBu)-Gly-Bza) was isolated from a peptide library constructed to identify peptide-based transport inhibitors of multidrug resistance (MDR) efflux pumps including P-glycoprotein and Multidrug Resistance-associated Protein 1. 4A6 proved to be a substrate but not an inhibitor of these MDR efflux transporters. In fact, 4A6 and related peptides displayed potent cytotoxic activity via an unknown mechanism. Objective To decipher the mode of cytotoxic activity of 4A6. Methods Screening of 4A6 activity was performed against the NCI60 panel of cancer cell lines. Possible interactions of 4A6 with the 26S proteasome were assessed via proteasome activity and affinity labeling, and cell growth inhibition studies with leukemic cells resistant to the proteasome inhibitor bortezomib (BTZ). Results The NCI60 panel COMPARE analysis revealed that 4A6 had an activity profile overlapping with BTZ. Consistently, 4A6 proved to be a selective and reversible inhibitor of β5 subunit (PSMB5)-associated chymotrypsin-like activity of the 26S proteasome. This conclusion is supported by several lines of evidence: (i) inhibition of chymotrypsin-like proteasome activity by 4A6 and related peptides correlated with their cell growth inhibition potencies; (ii) 4A6 reversibly inhibited functional β5 active site labeling with the affinity probe BodipyFL-Ahx3L3VS; and (iii) human myeloid THP1 cells with acquired BTZ resistance due to mutated PSMB5 were highly (up to 287-fold) cross-resistant to 4A6 and its related peptides. Conclusion 4A6 is a novel specific inhibitor of the β5 subunit-associated chymotrypsin-like proteasome activity. Further exploration of 4A6 as a lead compound for development as a novel proteasome-targeted drug is warranted.
Similar content being viewed by others


Introduction
The central role that the ubiquitin-proteasome system plays in intracellular protein degradation has been exploited as a potential therapeutic target for the treatment of hematological malignancies, solid tumors and cancer and chronic inflammatory diseases [1,2,3,4,5,6,7,8,9,10]. The 26S–proteasome complex is made up of a 20S core unit, consisting of 4 stacked heptameric rings, which form an α7β7β7α7 complex, and is capped by two 19S regulatory units [11,12,13]. The 20S core unit harbors the proteasome’s catalytic domain which is responsible for caspase-like, trypsin-like and chymotrypsin-like activities, associated with the β1, β2 and β5 subunit, respectively [14]. Several types of proteasome inhibitors have been described that reversibly or irreversibly inhibit proteasome activity by targeting one or more of these β subunits [15,16,17,18,19]. Bortezomib (Velcade®, PS341) was the first proteasome inhibitor that was clinically approved and registered for the treatment of refractory multiple myeloma [2, 20].
Bortezomib (BTZ) is a potent reversible proteasome inhibitor (IC50: 3–5 nM) that primarily targets the β5 subunit of the proteasome, although the β1 subunit and its immunoproteasome counterparts are also targeted [15, 21]. While bortezomib is clinically well tolerated, prolonged administration may result in neurotoxicity and drug resistance may emerge [15, 22,23,24]. Thus, alternative proteasome inhibitors are in demand [7, 17, 25,26,27,28,29,30,31].
The hexapeptide 4A6 (Ac-Thr(tBu)-His(Bzl)-Thr(Bzl)-Nle-Glu(OtBu)-Gly-Bza) [32] (Fig. 1) was identified from a peptide library constructed to identify peptide-based inhibitors of multidrug resistance (MDR) efflux transporters including P-glycoprotein (Pgp/ABCB1) and Multidrug Resistance-associated Protein 1 (MRP1/ABCC1) [33]. These ATP-binding cassette transorters extrude a plethora of structurally and mechanistically distinct cytotoxic agents and thus confer multidrug resistance upon various cancer cells [34,35,36]. In recent years, several types of peptides (linear/cyclic, neutral/ hydrophobic) have been identified for their interaction with MDR efflux transporters and/or their potential chemosensitizing capacity; these include cyclosporin A, gramicidin D, valinomycin, ALLN, dolastatin 10, pepstatin A, leupeptin and reversin 121 [37,38,39,40,41,42,43,44,45]. Likewise, 4A6 was found to be a substrate of the MDR efflux transporters ABCB1 and ABCC1, but lacked the ability to reverse efflux pump MDR [32]. In fact, 4A6 and related peptides displayed potent cytotoxic effects via an unknown mechanism [32]. Here we uncovered the mode of action of 4A6 and provide ample evidence that it exerts its pharmacological activity by blocking the chymotrypsin-like activity of the proteasome. This finding warrants the exploration of 4A6 as a lead compound for further development as a novel proteasome-targeted drug.

Chemical structures of 4A6, 4E11 and bortezomib
Materials and methods
Reagents
Bortezomib (Pyrazylcarbonyl-Phe-Leu-boronate) was provided by the VUmc Hospital Pharmacy Department. The cytotoxic peptides 4A6 (Ac-Thr(tBu)-His(Bzl)-Thr(Bzl)-Nle-Glu(OtBu)-Gly-Bza) (monomer and dimer form) and 4E11 (Ac-Thr(OBzl)-Glu(OtBu)-Glu(OBzl)-Asp(OtBu)-Glu(OtBu)-Gly-Bza) were synthesized as described previously [32]. P121/Reversin (Boc-Asp(OBzl)-Lys-(Z)-OtBu) was kindly provided by Prof. Dr. B. Sarkadi (Budapest, Hungary).
Protease Inhibitor Cocktail (PIC) was obtained from Boehringer Mannheim (Ingelheim, Germany). RPMI-1640 medium and fetal calf serum were obtained from Gibco Chem. Co (Grand Isl., NY, U.S.A). All fluorogenic substrates (Suc-Leu-Leu-Val-Tyr-amc, Ac-Arg-Leu-Arg-amc and Z-Leu-Leu-Glu-amc), the proteasome inhibitors Ac-APnLD-H and leupeptin, and all proteasome subunit-related antibodies (β1, β2, and β5) were purchased from Biomol (Plymouth Meeting, PA, U.S.A.). Anti-ubiquitin antibody (sc-8017) was purchased from Santa Cruz (USA). Ruthenium Red was obtained from Sigma Chem Co (USA).
Synthesis of ac-Thr(tBu)-his(Bzl)-Thr(Bzl)-Nle-OH
The tetramer (Ac-Thr(tBu)-His(Bzl)-Thr(Bzl)-Nle-OH), the major cleavage product of 4A6, was synthesized by standard Fmoc-based solid phase peptide synthesis on FmocNorLeu Sasrin resin. The FmocNorLeu resin was prepared by esterification of FmocNorLeu-OH (10 equivalents) with the unloaded resin using N,N′-diisopropylcarbodiimide (DIC, 10 equivalents) in dimethylformamide. The resin was deprotected with 1% trifluoroacetic acid (TFA) in methylenechloride for 2.5 h followed by precipitation of the peptide with diethylether and HPLC purification (Waters 1525 EF HPLC system).
Cell cultures
Human monocytic/macrophage THP1 cells (ATTC, Manassas, USA) were cultured in RPMI-1640 medium supplemented with 5% fetal calf serum, 20 mM HEPES, 2 mM glutamine and 100 μg/ml penicillin/ streptomycin at 5% CO2 and 37 °C. Cell cultures were seeded at a density of 3 × 105 cells/ml and refreshed twice weekly. Bortezomib (BTZ)-resistant THP1 cell lines were obtained by stepwise increasing extracellular concentrations of BTZ over a period of 6 months [46]. In this study, BTZ-resistant THP1 variants were used and grown in the presence of 50 nM (THP1/BTZ50), 100 nM (THP1/BTZ100) and 200 nM bortezomib (THP1/BTZ200) (see Table 1). Some specific experiments also included THP1/BTZ100 cells that were cultured in the absence of BTZ for 6 months (further designated as THP1/BTZ (−100) cells). Mouse thymoma EL4 and human multiple myeloma H929 cells were cultured in RPMI-1640 medium supplemented with 8% fetal calf serum and 100 μg/ml penicillin/streptomycin at 5% CO2 and 37 °C.
Screening of 4A6 using the NCI60 tumor cell line panel
The NCI 60 human tumor cell line screen was used to assess the activity profile of 4A6 against a panel of tumor cell lines of various cell lineage [47]. Concentrations of 4A6 eliciting 50% growth inhibition (GI50) were determined after 48 h drug exposure. 4A6 sensitivity for each individual cell line is depicted relative to the mean GI50 of the total cell line panel.
4A6 cleavage assay
Proteasome was purified from bovine liver as described previously [48]. For digestion assays, 1 μg proteasome was incubated with 1 μg 4A6 in 50 μl of 50 mM Tris-HCl buffer pH 8.5 at 45 °C for 16 h. Subsequently, the reaction mixture was lyophilized and peptides purified using reversed-phase ZipTip®C18 tips (Millipore). The purified peptide mixture was mixed in a 1:1 ratio with 10 mg/ml 2,5-dihydroxybenzoic acid (DHB, Bruker Daltonik) matrix solution in 0.1% TFA and spotted onto a MALDI (matrix assisted laser desorption/ ionization) target plate. MALDI-TOF analysis was performed on an Autoflex, linear MALDI-TOF-MS (Bruker Daltonik GmbH, Bremen, Germany). Spectra were analyzed with flexAnalysis software (Bruker Daltonik).
Growth inhibition assays
Evaluation of drug sensitivity was carried out as described before [49]. Cells were seeded at an initial density of 1.25 × 105 cells/ml in individual wells of a 24-well plate containing up to 50 μl of drug solutions. Inhibition of cell growth was determined after 72 h of incubation at 37 °C by determining the number of viable cells viable cells using trypan blue exclusion. The drug concentration required to inhibit cell growth by 50% compared to untreated controls was defined as the IC50.
Western blot analysis (ubiquitinated proteins/proteasome subunits)
Western blot analysis to determine protein levels of (i) β1, β2 and β5 proteasome subunits and (ii) the accumulation of ubiquitinated proteins after treatment with 4A6 was performed essentially as described previously [46, 49]. Cells were harvested in the mid-log phase of growth and washed 3 times with ice-cold buffered saline pH 7.4. Total cell lysates of 5 × 106 cells were prepared by resuspension in 500 μl lysis buffer containing: 50 mM Tris-HCl (pH 7.6), 5 mM dithiotreitol, 20 μl PIC (Protease Inhibitor Cocktail; 1 tablet/ml H2O), 20% glycerol and 0.5% NP-40. The suspension was sonicated (MSE sonicator, amplitude 7, for 3 × 5 s with 20 s time intervals at 4 °C) and centrifuged in an Eppendorf micro centrifuge (5 min, 12,000 rpm, 4 °C). Protein content of the supernatant was determined by the Bio-Rad protein assay. 20–30 μg of total cell lysates were fractionated on a 10% polyacrylamide gel containing SDS and transferred onto a PVDF membrane. The membranes were pre-incubated overnight at 4 °C in blocking buffer (5% Bio-Rad Blocker in TBS-T; 10 mM Tris-HCl, pH 8.0, 0.15 M NaCl, 0.1% Tween-20) to prevent non-specific antibody binding. After blocking, the membranes were incubated for 1 h at room temperature with primary antibodies for proteasome subunit β1 (1:1000, PW8140), β2 (1:1000, PW8145) and β5 (1:1000, PW8895) or ubiquitin (1:1000, Santa-Cruz, SC-8017). An antibody to α-tubulin was used (1:1000, Santa Cruz, sc-8035) to check and normalize for any loading differences. After 3 washing steps with TBS-T, the membranes were incubated for 1 h with HRP-labelled donkey-anti-rabbit (1:6000, Amersham, UK) or goat-anti-mouse (1:6000, Dako, Glostrup, Denmark) as secondary antibody. Detection of antibody binding was followed by chemoluminescence using Supersignal (Pierce Biotechnology, Rockford, USA) according to the manufacturers’ instructions. Digital Image acquisition was performed using the Versadoc Imaging System (Biorad Lab., Veenendaal, The Netherlands). The signal intensity was determined densitometrically using Quantity One software (Bio-Rad) and was expressed relative to the intensity of the α-tubulin signal.
Proteasome activity in cell lysates and intact cells
Chymotrypsin-like, trypsin-like and caspase-like proteolytic activities of the proteasome were determined in freshly prepared cell lysates as described previously [21, 46]. Five million untreated or bortezomib-exposed THP1 cells were washed 3 times with ice-cold PBS and pelleted by centrifugation (5 min, 12,000 RPM, 4 °C). Cell pellets were then resuspended in an ATP-containing lysis buffer; 10 mM Tris-HCl buffer (pH 7.8) containing 5 mM ATP, 0.5 mM DTT and 5 mM MgCl2, and kept on ice for 10 min. For complete lysis, cells were sonicated (MSE sonicator, amplitude 7, for 3 × 5 s with 20 s time intervals at 4 °C) followed by centrifugation (5 min, 12,000 RPM, 4 °C) to remove cell debris. The supernatant was collected and protein concentration was determined using the Bio-Rad protein assay. Fluorogenic substrates to measure the chymotrypsin-like, trypsin-like and caspase-like activity were Suc-Leu-Leu-Val-Tyr-amc, Ac-Arg-Leu-Arg-amc and Z-Leu-Leu-Glu-amc, respectively, all at a final concentrations of 100 μM. The substrates were incubated with 20 μg of total cell protein extract in the presence or absence of specific inhibitors (bortezomib for chymotrypsin-like activity, Ac-APnLD-H for caspase-like activity and leupeptin- for trypsin-like activity) in a total assay volume of 200 μl. The release of amc (7-amino-4-methyl-coumarin) was monitored online over a 2-h time period at 37 °C with 5 min intervals. Fluorescence was measured on a Tecan SpectraFluor apparatus (Giessen, The Netherlands) using excitation and emission wavelengths of 360 and 465 nm, respectively. Proteolytic activity was calculated from the slopes of the linear portion of the curves. All results were expressed as percentage relative to untreated THP1/WT cells (100%). Inhibition of chymotrypsin-like activity in intact cells was measured by the Proteasome-Glo™ cell-based assay (Promega, Leiden, The Netherlands), using Suc-LLVY-aminoluciferin as a substrate, according to the manufacturer’s instructions.
Proteasome affinity labelling
Proteasome activity profiling assays were performed as described [50, 51] using a close analog of the BodipyFL probe. Briefly, mouse EL4 thymoma cells were incubated at 37 °C for 2 or 24 h with increasing concentrations of 4A6, followed by a 1 h chase with 500 nM probe. In other experiments, human H929 myeloma cells were incubated at 37 °C with 1 μM 4A6 (2 h), 5 μM MG132 (1 h) or 20 nM bortezomib (1 h) and subsequently probed with 500 nM probe (1 h), either directly or after a washing and recovery step. Cells were harvested and lysed for 30 min in NP40 lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP40) at 4°C. The Bradford assay was used to measure protein content. Proteins were denatured by boiling in reducing sample buffer and analyzed by 12% SDS-PAGE using NuPAGE pre-cast gels (Invitrogen). Gels were then scanned for fluorescence emission using a ProXPRESS 2D Proteomic imaging system (Perkin Elmer). Images were analyzed using Totallab analysis software (Nonlinear Dynamics, Newcastle upon Tyne, UK). Sypro staining served as a loading control.
Apoptosis assay
Induction of apoptosis was analyzed by flow cytometry using APOPTEST™-FITC A700 (VPS Diagnostics, Hoeven, the Netherlands) according to the instructions of the manufacturer. In short, induction of apoptosis was determined after 24 h’ drug exposure. One million cells were harvested and washed 3 times with ice-cold PBS. The cell pellet was incubated for 30 min with 7-Amino-actinomycin D (7-AAD) on ice followed by incubation with Annexin-V according to the instructions of the manufacturer. Annexin-V (early apoptosis) and 7-AAD (late apoptosis) staining was measured by flow cytometry (Beckton & Dickinson, FACScalibur) and analysed using FCSexpress V3 software (Denovo software, Thornhill, Canada).
Statistics
Statistical analysis was performed using Analysis of Variance between groups (ANOVA) in Graphpad prism version 6.0. P values <0.05 were considered to be statistically significant.
Results
4A6 vs bortezomib activity against NCI60 panel of tumor cell lines
In order to get an initial insight regarding the cytotoxic activity of 4A6, we first tested 4A6 in the NCI60 tumor cell line panel that is composed of 60 malignant cell lines of distinct tissue lineage [47]. 4A6 showed remarkable activity towards a panel of leukemia, breast cancer, melanoma, and to some extent colon cancer cells (Fig. 2). In contrast, 4A6 proved rather inactive towards a panel of renal cancer cells and lung cancer cells. Moreover, cells with high levels expression of the multidrug efflux transporter Pgp, including HCT-15, ACHN, UO-31 and NCI/ADR-RES [52], displayed marked resistance to 4A6. COMPARE analysis of GI50 values for 4A6 in the NCI-panel of 60 cell lines showed a correlation coefficient (r) of 0.37 with bortezomib (BTZ), an established proteasome inhibitor drug. A side by side comparison of the activity profile of 4A6 and BTZ in the NCI60 panel of tumor cell lines showed overlapping sensitivities (Fig. 2), albeit based on mean log10GI50 concentrations obtained after 2 days of drug exposure, BTZ was 2–3 orders of magnitude more potent than 4A6. These results demonstrate that 4A6 has an overlapping activity profile with BTZ against the NCI60 panel of tumor cell lines; however, in contrast to BTZ, 4A6 activity was compromised by the presence of a Pgp-dependent MDR phenotype.

Cytotoxic activity profiles of bortezomib vs 4A6 against the NCI-panel of 60 malignant cell lines. Data are based on 48 h’ drug exposure and presented as log GI50 for each individual tumor cell line and as GI50 relative to the mean GI50 of all cell lines tested
Cells with acquired resistance to peptide-based proteasome inhibitor bortezomib are cross-resistant to the cytotoxic peptides 4A6 and 4E11
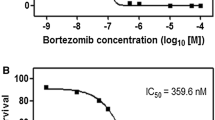
Because of the overlapping activity profile of 4A6 and BTZ in the NCI60 panel, we tested 4A6 in human THP1 cell lines with acquired resistance to BTZ. These cell lines displayed cross-resistance to other known peptide-based proteasome inhibitors (e.g. ALLN, MG132), but also to the linear cytotoxic hexapeptide 4A6, the latter of which has an unknown mechanism of action [46]. To further explore the molecular basis of this observation, THP1 cells with various levels of BTZ-resistance were screened for their sensitivity to 4A6, a dimer form of 4A6, another linear cytotoxic hexapeptide 4E11 (Fig. 1), and the cyclic cytotoxic decapeptide cyclosporin A (Table 1). Within this panel of cytotoxic peptides, 4A6 was the most potent inhibitor of THP1 cell growth (IC50: 0.26 μM), followed by a 3-fold lower potency for the 4A6 dimer and a 15-fold lower potency for 4E11 and cyclosporin A (Table 1). These bortezomib-resistant cell lines displayed the highest levels (up to 287-fold) of cross-resistance to 4A6 (Table 1, Fig. 3) and >60-fold cross resistance to the 4A6-dimer. With respect to the peptide 4E11, a consistently higher IC50 value compared to 4A6 (Table 1) along with limitations in solubility of peptides above a concentration of 50 μM, allowed for the assessment of relatively low level (>13-fold) cross-resistance to 4E11. No cross-resistance of bortezomib-resistant cells was observed for cyclosporin A. Collectively, these results indicate that the peptides 4A6 and 4E11 share properties with known inhibitors of the ubiquitin-proteasome system, including BTZ.

Cross-resistance to 4A6 in bortezomib-resistant cells. Dose response curve for 4A6-induced growth inhibition of wild type (WT) human myelomonocytic THP1 cells and proteasome (bortezomib, BTZ)-resistance selected variants; THP1/BTZ50, THP1/BTZ100 and THP1/BTZ200, selected for growth in extracellular concentrations of 50 nM, 100 nM and 200 nM BTZ, respectively. Results depicted are the means of 3 experiments ± S.D. 4A6 exposure time: 72 h
4A6 is a potent inhibitor of chymotrypsin-like proteasome activity
An intact cell-based luminogenic assay that monitors chymotrypsin-like proteasome activity was used to investigate whether the cytotoxic peptides 4A6 and 4E11 could exert their cytotoxic effect via inhibition of proteasome activity (Fig. 4). Indeed, 4A6 displayed a marked inhibition of chymotrypsin-like proteasome activity (IC50: 0.21 ± 0.05 μM) with a potency 28-fold lower than BTZ (IC50: 0.0074 ± 0.002 μM (Fig. 4a). Likewise, the 4A6-dimer and 4E11 were found to inhibit chymotrypsin-like proteasome activity, though with a lower potency than 4A6 (IC50: 0.49 ± 0.12 μM and 2.4 ± 0.5 μM, respectively). A control peptide Reversin 121, a transport inhibitor of the MDR efflux transporter P-gp [38], had no effect on proteasome activity (IC50: > > 25 μM). Hence, the potency ranking of proteasome inhibitory activity (bortezomib >4A6 > 4A6-dimer >4E11) tightly correlated with their capacity to inhibit cell growth of THP1/WT cells (Table 1).

a Potent inhibition of proteasome activity by the hexameric 4A6 peptide. Luminescent cell-based proteasome assay measuring inhibition of chymotrypsin-like proteasome activity in intact THP1 cells after 1 h exposure to BTZ, the hexameric peptide 4A6, 4A6-dimer, the hexameric peptide 4E11 and, as control, the tripeptide P121/Reversin, a peptide-based transport inhibitor of the MDR protein P-glycoprotein. Results represent the mean of 3 experiments ± S.D. b Inhibition of chymotrypsin-like but not caspase-like and trypsin-like proteasomal activity by 4A6. Chymotrypsin-like, caspase-like and trypsin-like proteasomal activities were determined with specific fluorogenic peptide substrates in cell extracts of THP1 cells after 1 h exposure to the indicated concentrations of 4A6. Controls for selective inhibition of chymotrypsin-like, caspase-like and trypsin-like activity included BTZ (10 nM), Ac-APnLP (25 μM) and leupeptin (20 μM), respectively. Results represent the mean of 3 separate experiments ± S.D
To address whether or not 4A6 is also capable of inhibiting one or both of the other protease activities harbored by the proteasome, chymotrypsin-, caspase- and trypsin-like activities were measured in THP1 cell extracts in the absence or presence of 4A6. Consistent with results shown in Fig. 4a, 4A6 elicited potent inhibitory effects (84–93%) on chymotrypsin-like proteasome activity, but had no inhibitory effect on caspase- and trypsin-like activity over a wide concentration range of 1–100 μM (Fig. 4b). These results demonstrate that 4A6 is a potent and selective inhibitor of chymotrypsin-like proteasome activity.
4A6 is a reversible inhibitor of chymotrypsin-like proteasome activity
Activity probing of constitutive and immunoproteasome β-subunits in EL4 cells that were pre-exposed to 4A6 for 2–24 h revealed a marked and specific inhibition of the β5 subunit with half maximal inhibition at 4A6 concentrations between 0.1 and 0.5 μM and complete inhibition at concentrations >5 μM (Fig. 5a). We next assessed whether this inhibition of β5-subunit could be recovered after removal of 4A6. Data shown in Fig. 5b illustrate that 4A6 is a reversible inhibitor of β5-subunit activity as initial recovery of activity could be observed already after 15 min of 4A6 drug removal and almost complete recovery after 2 h of 4A6 withdrawal. For comparison, the proteasome inhibitor MG132 blocked activity probing of all β-subunits, with a recovery 2 h after drug withdrawal (Fig. 5b). BTZ predominantly inhibited β5-subunit probing but affinity labeling was fully recovered within 2 h after drug withdrawal (Fig. 5b). We finally explored whether 4A6 remained intact as a peptide or could be subject to proteolytic cleavage when exposed to purified proteasomes. Comparison of mass spectra of the intact peptide (Fig. 5c) and the peptide after proteasomal digestion (Fig. 5d) showed that next to 4A6 (m/z 1080.6), one main additional peak appeared after digestion at m/z 749.5, corresponding to the 4-mer peptide Ac-Thr(tBu)-His(Bzl)-Thr(Bzl)-Nle-OH. A smaller peak appeared at m/z 934.6, corresponding to 5-mer peptide Ac-Thr(tBu)-His(Bzl)-Thr(Bzl)-Nle-Glu(OtBu)-OH. This indicates that 4A6 is predominantly cleaved at the P4-P5 position and to a lesser extent at the P5-P6 position. The main 4A6 proteasomal cleavage product, Ac-Thr(tBu)-His(Bzl)-Thr(Bzl)-Nle-OH was synthesized, but did not show any proteasome inhibitory effect or cell growth inhibitory potential (data not shown). Hence, these results suggest that 4A6 is a dual substrate and reversible inhibitor of proteasome subunit β5.

4A6 is a reversible inhibitor of proteasome subunit β5. a EL4 cells were incubated with the indicated concentrations of 4A6 for 2 or 24 h and then probed with a proteasome affinity probe as described in the Materials & Methods section. b H929 cells were incubated with 1 μM 4A6 (2 h), 20 nM BTZ (1 h) or 5 μM MG132 (1 h). Subsequently, cells were either probed directly with the affinity probe (0′) or resuspended in fresh medium without inhibitor and left to recover for the indicated times. As a control, non-treated cells were probed directly. A representative of 2 separate experiments is shown. c MALDI spectrum of 4A6 (m/z 1080.6). d MALDI spectrum of 4A6 after proteasomal digestion, showing the appearance one major cleavage product at m/z 749.5, which corresponds to Ac-Thr(tBu)-His(Bzl)-Thr(Bzl)-Nle-OH
Cellular exposure to 4A6 induces accumulation of ubiquitinated proteins and apoptosis but displays properties distinct of bortezomib
One hallmark of proteasome inhibition is the accumulation of ubiquitinated proteins, which are toxic to cells and induce apoptosis [53, 54]. Exposure of THP1/WT cells to 4A6 and 4E11 for 24 h resulted, just as for the known proteasome inhibitor BTZ, in a marked accumulation of ubiquitinated proteins, illustrated by a characteristic smear upon Western blot probed with an anti-ubiquitin antibody (Fig. 6a). In contrast, the same concentrations of 4A6 and 4E11 did not provoke any accumulation of ubiquitinated proteins in bortezomib-resistant cells. Consistent with these observations was the efficient induction of apoptosis by 4A6 in parental THP1/WT cells but none by 4A6 (over a concentration range of 0–25 μM) in THP1/BTZ200 cells (Fig. 6b and c). For comparison, the anti-cancer drug and topoisomerase II inhibitor etoposide (VP16) was equally effective in inducing apoptosis in THP1/WT and THP1/BTZ200 cells (not shown).

4A6-induced accumulation of ubiquitinated proteins and induction of apoptosis in THP1/WT cells but not in BTZ-resistant cells. a Accumulation of ubiquitinated proteins in THP1/WT cells and BTZ-resistant THP1/BTZ200 cells after 24 h’ exposure to the indicated concentrations of BTZ and 4A6. THP1/BTZ200 cells were allowed a 4 day drug washout period (control) before exposure to BTZ or 4A6. b Induction of apoptosis (Annexin-V positive cells) in THP1/WT cells and THP1/BTZ200 cells after 24 h’ exposure to a concentration range of 4A6. c A representative flow cytometric tracing of apoptosis induction (Annexin-V/7-AAD staining) following 24 h incubation of THP1/WT cells and THP1/BTZ200 cells with 25 μM 4A6. d Ruthenium Red protects from BTZ-induced but not from 4A6-induced cell growth inhibition. THP1 cells were incubated for 72 h with a concentration range of BTZ or 4A6 in the absence (−) or presence (+) of 25 μM Ruthenium Red. The ratio of IC50 values for BTZ and 4A6 in the presence or absence of Ruthenium Red is depicted as fold protection. Mean of 3 separate experiments ± S.D
To explore whether 4A6 shares properties with the known proteasome inhibitor BTZ, we investigated the ability of 4A6 to mimic a reported feature of BTZ, the disregulation of intracellular calcium homeostasis that triggers caspase activation and apoptosis [55]. This process could be counteracted by inhibitors of the mitochondrial calcium uniporter (e.g. Ruthenium Red), thereby providing a protective effect against BTZ [55]. While a marked abrogation of BTZ activity could be obtained by Ruthenium Red, no effect of this compound was observed with respect to 4A6 activity (Fig. 6d). These results suggest that 4A6 has no apparent impact on mitochondrial calcium homeostasis.
4A6 provokes proteasome β5 subunit induction
Given the specific targeting of 4A6 of the β5 subunit of the proteasome, we explored whether exposure to 4A6 had an effect on the expression of the β5 subunit as compared to the other catalytic subunits β1 and β2. To this end, THP1/WT cells and the bortezomib-resistant cell lines THP1/BTZ 100 and THP1/BTZ (−100), the latter being a subline of THP1/BTZ 100 that was grown in the absence of BTZ for 6 months, were exposed to a concentration range of 4A6 (0.1–10 μM) for 24 h (Fig. 7a). No significant effects of 4A6 exposure were observed regarding expression of the β1 and β2 proteasome subunits. In contrast, a dose-dependent increase in proteasome β5 subunit expression was noted in both THP1/WT sublines with relatively low basal levels of β5 expression and the two BTZ-resistant cell lines, including THP1/BTZ (−100) cells that retained a level of cross-resistance to 4A6 similar as THP1/BTZ 100 cells (Fig. 7a). Densitometric analysis showed a 3–5 fold increase in β5 subunit induction in all three cell lines upon exposure to 10 μM 4A6 exposure as compared to drug-free controls (Fig. 7b). This result implies that induction of proteasome β5 subunit expression constitutes a rapid adaptive response upon targeting of this subunit by the inhibitor 4A6.

4A6 induces proteasome β5 subunit overexpression. a Protein expression of β1, β2 and β5 proteasome subunits in THP1/WT cells, the BTZ-resistant cell line THP1/BTZ100 grown in the presence of 100 nM BTZ, and THP1/BTZ (−100) cells, a subline of THP1/BTZ100 that was grown in the absence of BTZ for 6 months. Before incubation with 4A6, THP1/BTZ100 cells were allowed a 4 days BTZ washout period (control/0), after which cells were exposed for 24 h to the indicated concentrations of 4A6. Expression of α-tubulin served as an actual loading control. b Results of scanning of protein band intensities in panel (a) are presented as mean ± S.D. of 3 separate experiments
Discussion
Here we have shown that the cytotoxic hexameric 4A6 peptide elicits its pharmacological activity via selective and reversible inhibition of the chymotrypsin-like proteasome activity. The specific targeting of the chymotrypsin-like proteasome activity by 4A6 was further corroborated by upregulation of the expression of the β5 subunit of the proteasome. Moreover, cells harboring mutations in the β5 subunit which confer resistance to BTZ [21], displayed a marked cross-resistance to 4A6.
Most peptide-based proteasome inhibitors contain tri- or tetrapeptide moieties that dock into one or more of the active site pockets of the proteasome [16, 56]. However, peptides extended with N-terminally linked spacers and specific caps can also retain their proteasome inhibitory potential [50, 57]. Notwithstanding this fact, 4A6, as well as another hexameric peptide (4E11) exhibited a motif and mode of action distinct from known peptide-based proteasome inhibitors. The linear hydrophobic nature of 4A6 likely facilitates its interactions with the β5-subunit of the proteasome that preferentially cleaves after hydrophobic amino acid residues [12, 14]. To this end, we explored whether or not the interaction of 4A6 with the proteasome involves mere steric occlusion of the β5 active site or alternatively, that the 4A6 peptide serves as a cleavage substrate of the proteasome. Consistent with this notion may be the fact that the dimeric form of 4A6, which contains the same amino acid sequence as 4A6 and is therefore also likely to be cleaved by the proteasome, is almost equally effective in inhibiting β5-associated proteasome activity (Fig. 4a and Table 1). In this context, it is important to note that replacement of Thr(Bzl) at the P3 position by Lys(Z) or Ala in 4A6 abolished the cytotoxic effect by 4A6 [32], suggesting that this residue is essential for effective proteasome binding and inhibition.
The marked level of cross-resistance to 4A6 of cells resistant to the proteasome inhibitor BTZ (Table 1, Fig. 3) supports the conclusion that 4A6 and BTZ share a common mode of interaction with the β5 active site. In fact, studies from our laboratory revealed that the molecular basis of BTZ resistance in these cells involved a point mutation in the PSMB5 gene that introduced a single amino acid change (Ala → Thr) at position 49 of the PSMB5 protein [46]. Since the Ala49 position resides in the BTZ binding pocket of PSMB5 and is involved in the interaction with BTZ [16, 30, 56, 58, 59], the Ala49Thr mutation is likely to underlie loss of BTZ binding and acquisition of bortezomib resistance [46]. The even higher levels of cross-resistance to 4A6 than resistance levels to BTZ suggest that Ala49 is even more critical in binding the 4A6 peptide than BTZ. In this respect, it was interesting to note that exposure of BTZ-resistant cells to 4A6 provoked a marked upregulation of mutant PSMB5 protein (Fig. 7), presumably as a compensatory mechanism to counteract loss of proteasome activity due to inhibition by 4A6.
Although the indicated PSMB5 mutation may be the dominant factor in conferring drug resistance to 4A6, it was previously reported that cellular extrusion by the MDR efflux transporters P-gp (ABCB1) and MRP1 (ABCC1) could also confer resistance to 4A6 [32]. This was further illustrated herein in the activity profile of 4A6 in the NCI60 panel of tumor cell lines where cells with a consistent MDR phenotype (mainly P-gp) were markedly less sensitive to 4A6 (Fig. 2). In contrast, such a MDR phenotype had relatively a marginal impact on the activity of BTZ. The presence of the boron group in BTZ most likely abolishes the ability of this compound to serve as a proficient substrate for MDR transporters as compared to other small peptides [60, 61]. Although Fig. 2 demonstrated an overlap in activities against some tumor types (leukemia/breast cancer), the current study indicates (Fig. 6d) that at least one mode of action of 4A6 was distinct from BTZ by not inducing apoptosis/growth inhibition via disregulation of mitochondrial calcium homeostasis [55]. Consistent with this study we showed here that inhibition of the mitochondrial uniporter with ruthenium red abrogated the growth inhibitory effects of BTZ, but had no effect on 4A6 activity.
Collectively, this study reported on 2 novel hexameric peptide-based proteasome inhibitors with several properties distinct from currently identified proteasome inhibitors, including BTZ. One of these peptides, 4A6, may serve as a lead compound for drug development by further optimization of its selective proteasome β5 subunit targeting against leukemia and breast cancer cells. The notion that 4A6 is a bona fide P-gp and MRP1 substrate may on the one hand compromise some of its activity against tumor cell expressing this drug efflux transporter, but on the other hand it may underlie a different, possibly more favorable toxicity profile than BTZ.
Abbreviations
- BTZ :
-
bortezomib
- Reversin-P121 :
-
Boc-Asp(OBzl)-Lys-(Z)-OtBu
- ALLN :
-
N-acetyl-Leu-Leu-norleucinal
- MG132 :
-
Z-Leu-Leu-Leucinal
- MG262 :
-
Z-Leu-Leu-Leu-boronate
- 4A6 :
-
Ac-Thr(tBu)-His(Bzl)-Thr(Bzl)-Nle-Glu(OtBu)-Gly-Bza
- 4E11 :
-
Ac-Thr(OBzl)-Glu(OtBu)-Glu(OBzl)-Asp(OtBu)-Glu(OtBu)-Gly-Bza
- Suc-LLVY-amc :
-
Suc-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin
- AAF-cmk :
-
Ala-Ala-Phe-chloromethylketone
- OtBu :
-
(O-)tert-butyl
- Bzl :
-
Benzyl
- Bza :
-
Benzylamine
- Ac :
-
Acetyl
- Ub :
-
Ubiquitin
- P-gp :
-
P-glycoprotein
- MRP1 :
-
Multidrug resistance associated protein 1
- BCRP :
-
Breast cancer resistance protein
References
Adams J (2004) The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5:417–421
Richardson PG, Mitsiades C, Hideshima T, Anderson KC (2006) Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu Rev Med 57:33–47
Voorhees PM, Orlowski RZ (2006) The proteasome and proteasome inhibitors in cancer therapy. Annu Rev Pharmacol Toxicol 46:189–213
Gatti L, Zuco V, Zaffaroni N, Perego P (2013) Drug combinations with proteasome inhibitors in antitumor therapy. Curr Pharm Des 19:4094–4114
Van der Heijden JW, Oerlemans R, Lems WF, Scheper RJ, Dijkmans BA, Jansen G (2009) The proteasome inhibitor bortezomib inhibits the release of NFkappaB-inducible cytokines and induces apoptosis of activated T cells from rheumatoid arthritis patients. Clin Exp Rheumatol 27:92–98
Dick LR, Fleming PE (2010) Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today 15:243–249
Anderson KC (2012) The 39th David A. Karnofsky lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol 30:445–452
Verbrugge SE, Scheper RJ, Lems WF, de Gruijl TD, Jansen G (2015) Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res Ther 17:17
Manasanch EE, Orlowski RZ (2017) Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol 14:417–433
Cloos J, Roeten MS, Franke NE, van Meerloo J, Zweegman S, Kaspers GJ, Jansen G (2017) (Immuno) proteasomes as therapeutic target in acute leukemia. Cancer Metastasis Rev 36:599–615
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479
Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82:373–428
Lub S, Maes K, Menu E, De Bruyne E, Vanderkerken K, Van Valckenborgh E (2016) Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget 7:6521–6537
Kisselev AF, Goldberg AL (2001) Proteasome inhibitors: from research tools to drug candidates. Chem Biol 8:739–758
Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, Ovaa H, Berkers C, Nicholson B, Chao TH, Neuteboom ST, Richardson P, Palladino MA, Anderson KC (2005) A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 8:407–419
Borissenko L, Groll M (2007) 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev 107:687–717
Kisselev AF, van der Linden WA, Overkleeft HS (2012) Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol 19:99–115
Niewerth D, Dingjan I, Cloos J, Jansen G, Kaspers G (2013) Proteasome inhibitors in acute leukemia. Expert Rev Anticancer Ther 13:327–337
Mitsiades CS (2015) Therapeutic landscape of carfilzomib and other modulators of the ubiquitin-proteasome pathway. J Clin Oncol 33:782–785
Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A, Harousseau JL (2012) Proteasome inhibitors in multiple myeloma: 10 years later. Blood 120:947–959
Kisselev AF, Callard A, Goldberg AL (2006) Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem 281:8582–8590
Argyriou AA, Iconomou G, Kalofonos HP (2008) Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood 112:1593–1599
McConkey DJ, Zhu K (2008) Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat 11:164–179
Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, Cloos J (2015) Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat 18:18–35
Nencioni A, Grunebach F, Patrone F, Ballestrero A, Brossart P (2007) Proteasome inhibitors: antitumor effects and beyond. Leukemia 21:30–36
Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, Bennett MK (2007) Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 67:6383–6391
Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ (2007) Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 110:3281–3290
Orlowski RZ, Kuhn DJ (2008) Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res 14:1649–1657
Kuhn DJ, Orlowski RZ (2012) The immunoproteasome as a target in hematologic malignancies. Semin Hematol 49:258–262
Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, Groll M (2012) Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148:727–738
Huber EM, Groll M (2012) Inhibitors for the immuno- and constitutive proteasome: current and future trends in drug development. Angew Chem Int Ed Eng 51:8708–8720
de Jong MC, Slootstra JW, Scheffer GL, Schroeijers AB, Puijk WC, Dinkelberg R, Kool M, Broxterman HJ, Meloen RH, Scheper RJ (2001) Peptide transport by the multidrug resistance protein MRP1. Cancer Res 61:2552–2557
Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234
Caetano-Pinto P, Jansen J, Assaraf YG, Masereeuw R (2017) The importance of breast cancer resistance protein to the kidneys excretory function and chemotherapeutic resistance. Drug Resist Updat 30:15–27
Li W, Zhang H, Assaraf YG, Zhao K, Xu X, Xie J, Yang DH, Chen ZS (2016) Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat 27:14–29
Shapira A, Livney YD, Broxterman HJ, Assaraf YG (2011) Nanomedicine for targeted cancer therapy: towards the overcoming of drug resistance. Drug Resist Updat 14:150–163
Sharom FJ, Yu X, Diodato G, Chu JW (1996) Synthetic hydrophobic peptides are substrates for P-glycoprotein and stimulate drug transport. Biochem J 320(Pt 2):421–428
Sharom FJ, Yu X, Lu P, Liu R, Chu JW, Szabo K, Muller M, Hose CD, Monks A, Varadi A, Seprodi J, Sarkadi B (1999) Interaction of the P-glycoprotein multidrug transporter (MDR1) with high affinity peptide chemosensitizers in isolated membranes, reconstituted systems, and intact cells. Biochem Pharmacol 58:571–586
Daoud SS, Juliano RL (1989) Modulation of doxorubicin resistance by valinomycin (NSC 122023) and liposomal valinomycin in Chinese hamster ovary cells. Cancer Res 49:2661–2667
Assaraf YG, Borgnia MJ (1994) Probing the interaction of the multidrug-resistance phenotype with the polypeptide ionophore gramicidin D via functional channel formation. Eur J Biochem 222:813–824
Toppmeyer DL, Slapak CA, Croop J, Kufe DW (1994) Role of P-glycoprotein in dolastatin 10 resistance. Biochem Pharmacol 48:609–612
Sarkadi B, Muller M, Homolya L, Hollo Z, Seprodi J, Germann UA, Gottesman MM, Price EM, Boucher RC (1994) Interaction of bioactive hydrophobic peptides with the human multidrug transporter. FASEB J 8:766–770
Eytan GD, Borgnia MJ, Regev R, Assaraf YG (1994) Transport of polypeptide ionophores into proteoliposomes reconstituted with rat liver P-glycoprotein. J Biol Chem 269:26058–26065
Borgnia MJ, Eytan GD, Assaraf YG (1996) Competition of hydrophobic peptides, cytotoxic drugs, and chemosensitizers on a common P-glycoprotein pharmacophore as revealed by its ATPase activity. J Biol Chem 271:3163–3171
Stark M, Assaraf YG (2017) Structural recognition of tubulysin B derivatives by multidrug resistance efflux transporters in human cancer cells. Oncotarget 8:49973–49987
Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zanten I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, Van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G (2008) Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 112:2489–2499.
Shoemaker RH (2006) The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 6:813–823
Raijmakers R, Berkers CR, de Jong A, Ovaa H, Heck AJ, Mohammed S (2008) Automated online sequential isotope labeling for protein quantitation applied to proteasome tissue-specific diversity. Mol Cell Proteomics 7:1755–1762
Oerlemans R, van der Heijden J, Vink J, Dijkmans BA, Kaspers GJ, Lems WF, Scheffer GL, Ifergan I, Scheper RJ, Cloos J, Assaraf YG, Jansen G (2006) Acquired resistance to chloroquine in human CEM T cells is mediated by multidrug resistance-associated protein 1 and provokes high levels of cross-resistance to glucocorticoids. Arthritis Rheum 54:557–568
Berkers CR, Verdoes M, Lichtman E, Fiebiger E, Kessler BM, Anderson KC, Ploegh HL, Ovaa H, Galardy PJ (2005) Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat Methods 2:357–362
Berkers CR, van Leeuwen FW, Groothuis TA, Peperzak V, van Tilburg EW, Borst J, Neefjes JJ, Ovaa H (2007) Profiling proteasome activity in tissue with fluorescent probes. Mol Pharm 4:739–748
Szakacs G, Annereau JP, Lababidi S, Shankavaram U, Arciello A, Bussey KJ, Reinhold W, Guo Y, Kruh GD, Reimers M, Weinstein JN, Gottesman MM (2004) Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell 6:129–137
Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, Van Zantwijk CH, Zweegman S, Chan ET, Kirk CJ, Geerke DP, Schimmer AD, Kaspers GJ, Jansen G, Cloos J (2012) Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 26:757–768
Franke NE, Kaspers GL, Assaraf YG, van Meerloo J, Niewerth D, Kessler FL, Poddighe PJ, Kole J, Smeets SJ, Ylstra B, Bi C, Chng WJ, Horton TM, Menezes RX, Musters RJ, Zweegman S, Jansen G, Cloos J (2016) Exocytosis of polyubiquitinated proteins in bortezomib-resistant leukemia cells: a role for MARCKS in acquired resistance to proteasome inhibitors. Oncotarget 7:74779–74796
Landowski TH, Megli CJ, Nullmeyer KD, Lynch RM, Dorr RT (2005) Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res 65:3828–3836
Groll M, Berkers CR, Ploegh HL, Ovaa H (2006) Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 14:451–456
Kessler BM, Tortorella D, Altun M, Kisselev AF, Fiebiger E, Hekking BG, Ploegh HL, Overkleeft HS (2001) Extended peptide-based inhibitors efficiently target the proteasome and reveal overlapping specificities of the catalytic beta-subunits. Chem Biol 8:913–929
Unno M, Mizushima T, Morimoto Y, Tomisugi Y, Tanaka K, Yasuoka N, Tsukihara T (2002) The structure of the mammalian 20S proteasome at 2.75 a resolution. Structure 10:609–618
Huber EM, Heinemeyer W, Groll M (2015) Bortezomib-resistant mutant proteasomes: structural and biochemical evaluation with carfilzomib and ONX 0914. Structure 23:407–417
Minderman H, Zhou Y, O'loughlin KL, Baer MR (2007) Bortezomib activity and in vitro interactions with anthracyclines and cytarabine in acute myeloid leukemia cells are independent of multidrug resistance mechanisms and p53 status. Cancer Chemother Pharmacol 60:245–255
Verbrugge SE, Assaraf YG, Dijkmans BA, Scheffer GL, Al M, den Uyl D, Oerlemans R, Chan ET, Kirk CJ, Peters GJ, Van der Heijden JW, de Gruijl TD, Scheper RJ, Jansen G (2012) Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther 341:174–182
Acknowledgements
This study is dedicated to the memory of Mariska C. de Jong, who passed away July 2006. We will remember Jerry Slootstra, who sadly passed away in 2014, for his devoted contribution to this study.
The authors thank H. Hilkmann for peptide synthesis and A. de Jong (NKI) for expert technical assistance with proteasome purification and mass spectrometry.
Funding
This study was supported by grants from the Dutch Arthritis Foundation / Reumafonds (grant NRF03-I-40 to GJ) and the Dutch Cancer Society (grant VU96–1256 to RJS and grant NKI 2005–3368 to HO).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Oerlemans, R., Berkers, C.R., Assaraf, Y.G. et al. Proteasome inhibition and mechanism of resistance to a synthetic, library-based hexapeptide. Invest New Drugs 36, 797–809 (2018). https://doi.org/10.1007/s10637-018-0569-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-018-0569-x