
Abstract
Sediments serve as an indicator of the state of the environment, as they reveal, anthropogenic influences (e.g. industry) over time. The knowledge about the composition of sediments, in particular by the speciation, helps in the assessment of the environmental situation. The speciation of mercury in sediments is still being discussed and continues to pose a great challenge for analytical chemists. Despite a broad number of publications in this area, there is no gold-standard about the speciation of mercury in sediments. The reason for this is the growing interest in new, better methods for the speciation of mercury, which increases the number of publications and the uncertainty among the analysts. Therefore, the methodology of mercury speciation in sediments requires improvement and would benefit from a standardized approach. The goal of this review is to give an overview of the existing methods and to discuss the issues of methodology. Discussed parts in this review article include: (1) available reference material, (2) the methodology of extraction, (3) enrichment procedures, (4) separation and (5) detection.
Graphical Abstract


(adapted from [21])

(adapted from [32])

(adapted from [21])

(adapted from [66])

(adapted from [102])

(adapted from [38])
Similar content being viewed by others
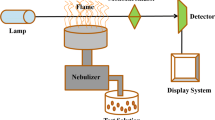
References
Kudo A, Miyahara S (1991) Water Sci Technol 23:283–290
WHO (2010) Public Health and Environment. World Health Organization, Geneva. http://www.who.int/ipcs/features/10chemicals_en.pdf?ua=1
Leermakers M, Baeyens W, Quevauviller P, Horvat M (2005) Trac Trend Anal Chem 24:383–393. https://doi.org/10.1016/j.trac.2004.01.001
Fitzgerald WF, Lamborg CH, Hammerschmidt CR (2007) Chem Rev 107:641–662. https://doi.org/10.1021/cr050353m
Hursh JB, Clarkson TW, Cherian MG, Vostal JJ, Vandermallie R (1976) Arch Environ Health 31:302–309
Syversen T, Kaur P (2012) J Trace Elem Med Bio 26:215–226. https://doi.org/10.1016/j.jtemb.2012.02.004
Gochfeld M (2003) Ecotox Environ Safe 56:174–179. https://doi.org/10.1016/S0147-6513(03)00060-5
Friberg L, Skog E, Wahlberg JE (1961) Acta Derm Venereol 41:40–52
Magos L, Brown AW, Sparrow S, Bailey E, Snowden RT, Skipp WR (1985) Arch Toxicol 57:260–267. https://doi.org/10.1007/Bf00324789
Clarkson TW (2002) Environ Health Persp 110:11–23
Kerper LE, Ballatori N, Clarkson TW (1992) Am J Physiol 262:R761–R765
Aberg B, Ekman L, Falk R, Greitz U, Persson G, Snihs JO (1969) Arch Environ Health 19:478–484
Ullrich SM, Tanton TW, Abdrashitova SA (2001) Crit Rev Environ Sci Technol 31:241–293. https://doi.org/10.1080/20016491089226
Li WC, Tse HF (2015) Environ Sci Pollut Res 22:192–201. https://doi.org/10.1007/s11356-014-3544-x
Issaro N, Abi-Ghanem C, Bermond A (2009) Anal Chim Acta 631:1–12. https://doi.org/10.1016/j.aca.2008.10.020
Aceto M, Foglizzo AM, Mentasti E, Sacchero G, Sarzanini C (1995) Int J Environ Anal Chem 60:1–13. https://doi.org/10.1080/03067319508027222
Mason RP, Reinfelder JR, Morel FMM (1995) Water Air Soil Pollut 80:915–921. https://doi.org/10.1007/Bf01189744
Malm O, Branches FJP, Akagi H, Castro MB, Pfeiffer WC, Harada M, Bastos WR, Kato H (1995) Sci Total Environ 175:141–150. https://doi.org/10.1016/0048-9697(95)04910-X
Weber JH, Puk R (1994) Appl Organomet Chem 8:709–713. https://doi.org/10.1002/aoc.590080723
Hintelmann H, Falter R, Ilgen G, Evans RD (1997) Fresen J Anal Chem 358:363–370. https://doi.org/10.1007/s002160050431
Leopold K, Foulkes M, Worsfold P (2010) Anal Chim Acta 663:127–138. https://doi.org/10.1016/j.aca.2010.01.048
Bowles KC, Apte SC, Maher WA, Bluhdorn DR (2003) Water Air Soil Pollut 147:25–38. https://doi.org/10.1023/A:1024561830113
Bisinoti MC, Junior ES, Jardim WF (2007) J Brazil Chem Soc 18:544–553. https://doi.org/10.1590/S0103-50532007000300008
Araujo BF, Hintelmann H, Dimock B, Sobrinho RD, Bernardes MC, de Almeida MG, Krusche AV, Rangel TP, Thompson F, de Rezende CE (2018) Limnol Oceanogr 63:1134–1145. https://doi.org/10.1002/lno.10758
He TR, Lu J, Yang F, Feng XB (2007) Sci Total Environ 386:53–64. https://doi.org/10.1016/j.scitotenv.2007.07.022
Kannan K, Smith RG, Lee RF, Windom HL, Heitmuller PT, Macauley JM, Summers JK (1998) Arch Environ Contam Toxicol 34:109–118. https://doi.org/10.1007/s002449900294
Mikac N, Niessen S, Ouddane B, Wartel M (1999) Appl Organomet Chem 13:715–725. https://doi.org/10.1002/(Sici)1099-0739(199910)13:10%3C715::Aid-Aoc918%3E3.0.Co;2-4
Kannan K, Falandysz J (1998) Water Air Soil Pollut 103:129–136. https://doi.org/10.1023/A:1004967112178
Wilken RD, Hintelmann H (1991) Water Air Soil Pollut 56:427–437. https://doi.org/10.1007/Bf00342289
Minganti V, Capelli R, Drava G, De Pellegrini R (2007) Chemosphere 67:1018–1024. https://doi.org/10.1016/j.chemosphere.2006.10.053
Gabriel MC, Williamson DG (2004) Environ Geochem Health 26:421–434. https://doi.org/10.1007/s10653-004-1308-0
Ibanez-Palomino C, Lopez-Sanchez JF, Sahuquillo A (2012) Anal Chim Acta 720:9–15. https://doi.org/10.1016/j.aca.2012.01.015
Le Roux S, Baker P, Crouch A (2016) S Afr J Chem S Afr T 69:124–131. https://doi.org/10.17159/0379-4350/2016/v69a15
Templeton DM, Ariese F, Cornelis R, Danielsson LG, Muntau H, Van Leeuwen HP, Lobinski R (2000) Pure Appl Chem 72:1453–1470. https://doi.org/10.1351/pac200072081453
Sarica DY, Turker AR (2012) Clean Soil Air Water 40:523–530. https://doi.org/10.1002/clen.201100535
Braaten HFV, de Wit HA, Harman C, Hagestrom U, Larssen T (2014) Int J Environ Anal Chem 94:381–384. https://doi.org/10.1080/03067319.2013.823489
Martinis EM, Wuilloud RG (2010) J Anal Atom Spectrom 25:1432–1439. https://doi.org/10.1039/c004678g
Amde M, Yin YG, Zhang D, Liu JF (2016) Chem Speciat Bioavailab 28:51–65. https://doi.org/10.1080/09542299.2016.1164019
Yu LP, Yan XP (2003) Trac Trend Anal Chem 22:245–253. https://doi.org/10.1016/S0165-9936(03)00407-2
Rosain RM, Wai CM (1973) Anal Chim Acta 65:279–284. https://doi.org/10.1016/S0003-2670(01)82493-4
Leermakers M, Lansens P, Baeyens W (1990) Fresen J Anal Chem 336:655–662. https://doi.org/10.1007/Bf00331410
Krivan V, Haas HF (1988) Fresen Z Anal Chem 332:1–6. https://doi.org/10.1007/Bf00487020
Stoeppler M, Matthes W (1978) Anal Chim Acta 98:389–392. https://doi.org/10.1016/S0003-2670(01)84069-1
Lansens P, Meuleman C, Baeyens W (1990) Anal Chim Acta 229:281–285. https://doi.org/10.1016/S0003-2670(00)85140-5
Sedlackova L, Kruzikova K, Svobodova Z (2014) Food Chem 150:360–365. https://doi.org/10.1016/j.foodchem.2013.10.041
Li X, Wang Y, Li BH, Feng CH, Chen YX, Shen ZY (2013) Environ Earth Sci 69:1537–1547. https://doi.org/10.1007/s12665-012-1988-1
Kim E, Noh S, Lee YG, Kundu SR, Lee BG, Park K, Han S (2014) Mar Chem 158:59–68. https://doi.org/10.1016/j.marchem.2013.11.004
Zhang T, Kucharzyk KH, Kim B, Deshusses MA, Hsu-Kim H (2014) Environ Sci Technol 48:9133–9141. https://doi.org/10.1021/es500336j
Weiss HV, Shipman WH, Guttman MA (1976) Anal Chim Acta 81:211–217. https://doi.org/10.1016/S0003-2670(00)89480-5
Avramescu ML, Zhu J, Yumvihoze E, Hintelmann H, Fortin D, Lean DRS (2010) Environ Toxicol Chem 29:1256–1262. https://doi.org/10.1002/etc.158
Leermakers M, Nguyen HL, Kurunczi S, Vanneste B, Galletti S, Baeyens W (2003) Anal Bioanal Chem 377:327–333. https://doi.org/10.1007/s00216-003-2116-6
Fabbri D, Felisatti O, Lombardo M, Trombini C, Vassura I (1998) Sci Total Environ 213:121–128. https://doi.org/10.1016/S0048-9697(98)00083-7
Quevauviller P, Fortunati GU, Filippelli M, Bortoli A, Muntau H (1998) Appl Organomet Chem 12:531–539. https://doi.org/10.1002/(Sici)1099-0739(199808/09)12:8/9%3C531::Aid-Aoc758%3E3.0.Co;2-I
Vazquez MJ, Carro AM, Lorenzo RA, Cela R (1997) Anal Chem 69:221–225. https://doi.org/10.1021/ac960513h
Hintelmann H (1999) Chemosphere 39:1093–1105. https://doi.org/10.1016/S0045-6535(99)00180-0
Frohne T, Rinklebe J (2013) Water Air Soil Pollut 224:1591. https://doi.org/10.1007/s11270-013-1591-4
Wallschlager D, Desai MVM, Wilken RD (1996) Water Air Soil Pollut 90:507–520. https://doi.org/10.1007/Bf00282665
Davis A, Bloom NS, Hee SSQ (1997) Risk Anal 17:557–569. https://doi.org/10.1111/j.1539-6924.1997.tb00897.x
Karlsson T, Skyllberg U (2003) Environ Sci Technol 37:4912–4918. https://doi.org/10.1021/es034302n
Ravichandran M (2004) Chemosphere 55:319–331. https://doi.org/10.1016/j.chemosphere.2003.11.011
Khwaja AR, Bloom PR, Brezonik PL (2006) Environ Sci Technol 40:844–849. https://doi.org/10.1021/es051805c
Zhong H, Wang WX (2008) Environ Pollut 151:222–230. https://doi.org/10.1016/j.envpol.2007.01.049
Skyllberg U, Qian J, Frech W, Xia K, Bleam WF (2003) Biogeochemistry 64:53–76. https://doi.org/10.1023/A:1024904502633
Fiorentino JC, Enzweiler J, Angelica RS (2011) Water Air Soil Pollut 221:63–75. https://doi.org/10.1007/s11270-011-0769-x
Manohar DM, Krishnan KA, Anirudhan TS (2002) Water Res 36:1609–1619
Han Y, Kingston HM, Boylan HM, Rahman GMM, Shah S, Richter RC, Link DD, Bhandari S (2003) Anal Bioanal Chem 375:428–436. https://doi.org/10.1007/s00216-002-1701-4
Bloom NS, Preus E, Katon J, Hiltner M (2003) Anal Chim Acta 479:233–248. https://doi.org/10.1016/S0003-2670(02)01550-7
Saniewska D, Beldowska M (2017) Talanta 168:152–161. https://doi.org/10.1016/j.talanta.2017.03.026
Reis AT, Rodrigues SM, Davidson CM, Pereira E, Duarte AC (2010) Chemosphere 81:1369–1377. https://doi.org/10.1016/j.chemosphere.2010.09.030
Bacon JR, Davidson CM (2008) Analyst 133:25–46. https://doi.org/10.1039/b711896a
Andrews JC (2006) Struct Bond 120:1–35. https://doi.org/10.1007/430_011
Kim CS, Bloom NS, Rytuba JJ, Brown GE (2003) Environ Sci Technol 37:5102–5108. https://doi.org/10.1021/es0341485
Reis AT, Coelho JP, Rucandio I, Davidson CM, Duarte AC, Pereira E (2015) Geoderma 237:98–104. https://doi.org/10.1016/j.geoderma.2014.08.019
Barnett MO, Harris LA, Turner RR, Henson TJ, Melton RE, Stevenson RJ (1995) Water Air Soil Pollut 80:1105–1108. https://doi.org/10.1007/Bf01189771
Martin JM, Nirel P, Thomas AJ (1987) Mar Chem 22:313–341. https://doi.org/10.1016/0304-4203(87)90017-X
Gleyzes C, Tellier S, Astruc M (2002) Trac Trend Anal Chem 21:451–467. https://doi.org/10.1016/S0165-9936(02)00603-9
Brombach CC, Gajdosechova Z, Chen B, Brownlow A, Corns WT, Feldmann J, Krupp EM (2015) Anal Bioanal Chem 407:973–981. https://doi.org/10.1007/s00216-014-8254-1
Jagtap R, Krikowa F, Maher W, Foster S, Ellwood M (2011) Talanta 85:49–55. https://doi.org/10.1016/j.talanta.2011.03.022
Bloom NS, Colman JA, Barber L (1997) Fresen J Anal Chem 358:371–377. https://doi.org/10.1007/s002160050432
Rahman GMM, Kingston HM (2005) J Anal Atom Spectrom 20:183–191. https://doi.org/10.1039/b404581e
Tseng CM, deDiego A, Martin FM, Donard OFX (1997) J Anal Atom Spectrom 12:629–635. https://doi.org/10.1039/a700832e
Qian J, Skyllberg U, Tu Q, Bleam WF, Frech W (2000) Fresen J Anal Chem 367:467–473. https://doi.org/10.1007/s002160000364
Xiang WJ, Liu J, Chang M, Zheng CG (2012) Chem Eng J 200:91–96. https://doi.org/10.1016/j.cej.2012.06.025
Roulet M, Guimaraes JRD, Lucotte M (2001) Water Air Soil Pollut 128:41–60. https://doi.org/10.1023/A:1010379103335
Falter R (1999) Chemosphere 39:1051–1073. https://doi.org/10.1016/S0045-6535(99)00178-2
Quevauviller P (1999) Chemosphere 39:1153–1165. https://doi.org/10.1016/S0045-6535(99)00184-8
Carrasco L, Vassileva E (2015) Anal Chim Acta 853:167–178. https://doi.org/10.1016/j.aca.2014.10.026
Canario J, Antunes P, Lavrado J, Vale C (2004) Trac Trend Anal Chem 23:799–806. https://doi.org/10.1016/j.trac.2004.08.009
Dmytriw R, Mucci A, Lucotte M, Pichet P (1995) Water Air Soil Pollut 80:1099–1103. https://doi.org/10.1007/Bf01189770
Liang L, Horvat M, Feng XB, Shang LH, Lil H, Pang P (2004) Appl Organomet Chem 18:264–270. https://doi.org/10.1002/aoc.617
Horvat M, Bloom NS, Liang L (1993) Anal Chim Acta 281:135–152. https://doi.org/10.1016/0003-2670(93)85348-N
Hammerschmidt CR, Fitzgerald WF (2001) Anal Chem 73:5930–5936. https://doi.org/10.1021/ac010721w
Lorenzo RA, Vazquez MJ, Carro AM, Cela R (1999) Trac Trend Anal Chem 18:410–416. https://doi.org/10.1016/S0165-9936(99)00118-1
Tseng CM, DeDiego A, Martin FM, Amouroux D, Donard OFX (1997) J Anal Atom Spectrom 12:743–750. https://doi.org/10.1039/a700956i
Bowles KC, Apte SC (2000) Anal Chim Acta 419:145–151. https://doi.org/10.1016/S0003-2670(00)00997-1
Ramalhosa E, Segade SR, Pereira E, Vale C, Duarte A (2001) Anal Chim Acta 448:135–143. https://doi.org/10.1016/S0003-2670(01)01317-4
Hintelmann H, Wilken RD (1993) Appl Organomet Chem 7:173–180. https://doi.org/10.1002/aoc.590070303
Cattani I, Spalla S, Beone GM, Del Re AAM, Boccelli R, Trevisan M (2008) Talanta 74:1520–1526. https://doi.org/10.1016/j.talanta.2007.09.029
Hintelmann H, Evans RD (1997) Fresen J Anal Chem 358:378–385. https://doi.org/10.1007/s002160050433
Bloom NS (1992) Can J Fish Aquat Sci 49:1010–1017. https://doi.org/10.1139/F92-113
Tseng CM, de Diego A, Pinaly H, Amouraoux D, Donard OFX (1998) J Anal Atom Spectrom 13:755–764. https://doi.org/10.1039/A802344a
Jagtap R, Maher W (2015) Microchem J 121:65–98. https://doi.org/10.1016/j.microc.2015.01.010
Maggi C, Berducci MT, Bianchi J, Giani M, Campanella L (2009) Anal Chim Acta 641:32–36. https://doi.org/10.1016/j.aca.2009.03.033
Rezende PS, Silva NC, Moura WD, Windmoller CC (2018) Microchem J 140:199–206. https://doi.org/10.1016/j.microc.2018.04.006
Kim CS, Brown GE, Rytuba JJ (2000) Sci Total Environ 261:157–168. https://doi.org/10.1016/S0048-9697(00)00640-9
Sladek C, Gustin MS (2003) Appl Geochem 18:567–576. https://doi.org/10.1016/S0883-2927(02)00115-4
Kim CS, Rytuba JJ, Brown GE (2004) J Colloid Interf Sci 271:1–15. https://doi.org/10.1016/S0021-9797(03)00330-8
Biester H, Scholz C (1997) Environ Sci Technol 31:233–239. https://doi.org/10.1021/es960369h
Bollen A, Wenke A, Biester H (2008) Water Res 42:91–100. https://doi.org/10.1016/j.watres.2007.07.011
Higueras P, Oyarzun R, Biester H, Lillo J, Lorenzo S (2003) J Geochem Explor 80:95–104. https://doi.org/10.1016/S0375-6742(03)00185-7
Hojdova M, Navratil T, Rohovec J (2008) Bull Environ Contam Toxicol 80:237–241. https://doi.org/10.1007/s00128-007-9352-y
Piani R, Covelli S, Biester H (2005) Appl Geochem 20:1546–1559. https://doi.org/10.1016/j.apgeochem.2005.04.003
Rallo M, Lopez-Anton MA, Perry R, Maroto-Valer MM (2010) Fuel 89:2157–2159. https://doi.org/10.1016/j.fuel.2010.03.037
Rumayor M, Diaz-Somoano M, Lopez-Anton MA, Martinez-Tarazona MR (2013) Talanta 114:318–322. https://doi.org/10.1016/j.talanta.2013.05.059
Reis AT, Coelho JP, Rodrigues SM, Rocha R, Davidson CM, Duarte AC, Pereira E (2012) Talanta 99:363–368. https://doi.org/10.1016/j.talanta.2012.05.065
Windmoller CC, Silva NC, Andrade PHM, Mendes LA, do Valle CM (2017) Anal Methods UK 9:2159–2167. https://doi.org/10.1039/c6ay03041f
Fernandez-Martinez R, Rucandio I (2013) Anal Methods UK 5:4131–4137. https://doi.org/10.1039/c3ay40566d
Tseng CM, De Diego A, Wasserman JC, Amouroux D, Donard OFX (1999) Chemosphere 39:1119–1136. https://doi.org/10.1016/S0045-6535(99)00182-4
Martin-Doimeadios RCR, Monperrus M, Krupp E, Amouroux D, Donard OFX (2003) Anal Chem 75:3202–3211. https://doi.org/10.1021/ac026411a
Delgado A, Prieto A, Zuloaga O, de Diego A, Madariaga JM (2007) Anal Chim Acta 582:109–115. https://doi.org/10.1016/j.aca.2006.08.051
US EPA SW-846 Update V Mercury species fractionation and quantification by microwave assisted extraction, selective solvent extraction and/or solid phase extraction, method 3200, July 2014. https://www.epa.gov/sites/production/files/2015-12/documents/3200.pdf
Liu Y, Chai XL, Hao YX, Gao XF, Lu ZB, Zhao YC, Zhang J, Cai MH (2015) Environ Sci Pollut R 22:8603–8610. https://doi.org/10.1007/s11356-014-3942-0
He TR, Zhu YZ, Yin DL, Luo GJ, An YL, Yan HY, Qian XL (2015) Environ Sci Pollut R 22:5124–5138. https://doi.org/10.1007/s11356-014-3864-x
Liang P, Lam CL, Chen Z, Wang HS, Shi JB, Wu SC, Wang WX, Zhang J, Wang HL, Wong MH (2013) J Soil Sediment 13:1301–1308. https://doi.org/10.1007/s11368-013-0719-x
Schwartz GE, Redfern LK, Ikuma K, Gunsch CK, Ruhl LS, Vengosh A, Hsu-Kim H (2016) Environ Sci Proc Imp 18:1427–1439. https://doi.org/10.1039/c6em00458j
Nevado JJB, Martin-Doimeadios RCR, Bernardo FJG, Moreno MJ (2008) Anal Chim Acta 608:30–37. https://doi.org/10.1016/j.aca.2007.12.001
Yin YG, Chen M, Peng JF, Liu JF, Jiang GB (2010) Talanta 81:1788–1792. https://doi.org/10.1016/j.talanta.2010.03.039
Turker AR, Cabuk D, Yalcinkaya O (2013) Anal Lett 46:1155–1170. https://doi.org/10.1080/00032719.2012.753608
Yang FF, Li JH, Lu WH, Wen YY, Cai XQ, You JM, Ma JP, Ding YJ, Chen LX (2014) Electrophoresis 35:474–481. https://doi.org/10.1002/elps.201300409
Gao ZB, Ma XG (2011) Anal Chim Acta 702:50–55. https://doi.org/10.1016/j.aca.2011.06.019
Chen BB, Wu YL, Guo XQ, He M, Hu B (2015) J Anal Atom Spectrom 30:875–881. https://doi.org/10.1039/c4ja00312h
Pietila H, Peramaki P, Piispanen J, Majuri L, Starr M, Nieminen T, Kantola M, Ukonmaanaho L (2014) Microchem J 112:113–118. https://doi.org/10.1016/j.microc.2013.10.002
Taylor VF, Carter A, Davies C, Jackson BP (2011) Anal Methods UK 3:1143–1148. https://doi.org/10.1039/c0ay00528b
Margetinova J, Houserova-Pelcova P, Kuban V (2008) Anal Chim Acta 615:115–123. https://doi.org/10.1016/j.aca.2008.03.061
Wilken RD, Falter R (1998) Appl Organomet Chem 12:551–557. https://doi.org/10.1002/(Sici)1099-0739(199808/09)12:8/9%3C551::Aid-Aoc760%3E3.0.Co;2-2
Pietila H, Peramaki P, Piispanen J, Starr M, Nieminen T, Kantola M, Ukonmaanaho L (2015) Chemosphere 124:47–53. https://doi.org/10.1016/j.chemosphere.2014.11.001
Mao YX, Yin YG, Li YB, Liu GL, Feng XB, Jiang GB, Cai Y (2010) Environ Pollut 158:3378–3384. https://doi.org/10.1016/j.envpol.2010.07.031
Leng G, Yin H, Li SB, Chen Y, Dan DZ (2012) Talanta 99:631–636. https://doi.org/10.1016/j.talanta.2012.06.051
Stanisz E, Werner J, Matusiewicz H (2013) Microchem J 110:28–35. https://doi.org/10.1016/j.microc.2013.01.006
Bravo-Sanchez LR, Encinar JR, Martinez JIF, Sanz-Medel A (2004) Spectrochim Acta B 59:59–66. https://doi.org/10.1016/j.sab.2003.10.001
Krystek P, Ritsema R (2004) Appl Organomet Chem 18:640–645. https://doi.org/10.1002/aoc.697
Stoichev T, Martin-Doimeadios RCR, Tessier E, Amouroux D, Donard OFX (2004) Talanta 62:433–438. https://doi.org/10.1016/j.talanta.2003.08.006
Gomez-Ariza JL, Lorenzo F, Garcia-Barrera T, Sanchez-Rodas D (2004) Anal Chim Acta 511:165–173. https://doi.org/10.1016/j.aca.2004.01.051
Centineo G, Gonzalez EB, Sanz-Medel A (2004) J Chromatogr A 1034:191–197. https://doi.org/10.1016/j.chroma.2004.01.051
Munoz J, Gallego M, Valcarcel M (2004) J Chromatogr A 1055:185–190. https://doi.org/10.1016/j.chroma.2004.09.026
Bloxham MJ, Gachanja A, Hill SJ, Worsfold PJ (1996) J Anal Atom Spectrom 11:145–148. https://doi.org/10.1039/ja9961100145
Ho YS, Uden PC (1994) J Chromatogr A 688:107–116. https://doi.org/10.1016/S0021-9673(94)89019-6
Sarzanini C, Sacchero G, Aceto M, Abollino O, Mentasti E (1994) Anal Chim Acta 284:661–667. https://doi.org/10.1016/0003-2670(94)85070-4
Bramanti E, Lomonte C, Onor M, Zamboni R, D’Ulivo A, Raspi G (2005) Talanta 66:762–768. https://doi.org/10.1016/j.talanta.2004.12.031
Wan CC, Chen CS, Jiang SJ (1997) J Anal Atom Spectrom 12:683–687. https://doi.org/10.1039/a605765i
Ackley KL, Sutton KL, Caruso JA (2000) J Anal Atom Spectrom 15:1069–1073. https://doi.org/10.1039/b000986p
Shum SCK, Pang HM, Houk RS (1992) Anal Chem 64:2444–2450. https://doi.org/10.1021/ac00044a025
Blanco RM, Villanueva MT, Uria JES, Sanz-Medel A (2000) Anal Chim Acta 419:137–144. https://doi.org/10.1016/S0003-2670(00)01002-3
Dong LM, Yan XP, Li Y, Jiang Y, Wang SW, Jiang DQ (2004) J Chromatogr A 1036:119–125. https://doi.org/10.1016/j.chroma.2004.02.070
Chen XP, Han C, Cheng HY, Wang YC, Liu JH, Xu ZG, Hu L (2013) J Chromatogr A 1314:86–93. https://doi.org/10.1016/j.chroma.2013.08.104
Tu Q, Qvarnstrom J, Frech W (2000) Analyst 125:705–710. https://doi.org/10.1039/a908880f
Lee TH, Jiang SJ (2000) Anal Chim Acta 413:197–205. https://doi.org/10.1016/S0003-2670(00)00807-2
da Rocha MS, Soldado AB, Blanco-Gonzalez E, Sanz-Medel A (2000) J Anal Atom Spectrom 15:513–518
da Rocha MS, Soldado AB, Blanco-Gonzalez E, Sanz-Medel A (2000) Biomed Chromatogr 14:6–63
Medina I, Rubi E, Mejuto MC, Cela R (1993) Talanta 40:1631–1636. https://doi.org/10.1016/0039-9140(93)80077-5
Mercader-Trejo F, de San Miguel ER, de Gyves J (2005) J Anal Atom Spectrom 20:1212–1217. https://doi.org/10.1039/b505000f
Mercader-Trejo F, Herrera-Basurto R, de San Miguel ER, de Gyves J (2011) Int J Environ Anal Chem 91:1062–1076. https://doi.org/10.1080/03067311003782658
Nguyen TH, Boman J, Leermakers M, Baeyens W (1998) X-ray Spectrom 27:277–282. https://doi.org/10.1002/(Sici)1097-4539(199807/08)27:4%3C277::Aid-Xrs297%3E3.0.Co;2-U
Koplik R, Klimesova I, Malisova K, Mestek O (2014) Czech J Food Sci 32:249–259
Bulska E, Baxter DC, Frech W (1991) Anal Chim Acta 249:545–554. https://doi.org/10.1016/S0003-2670(00)83032-9
Tao H, Murakami T, Tominaga M, Miyazaki A (1998) J Anal Atom Spectrom 13:1085–1093. https://doi.org/10.1039/a803369b
Uria JES, Sanz-Medel A (1998) Talanta 47:509–524
Harrington CF (2000) Trac Trend Anal Chem 19:167–179. https://doi.org/10.1016/S0165-9936(99)00190-9
Kadlecova M, Daye M, Ouddane B (2014) Anal Lett 47:697–706. https://doi.org/10.1080/00032719.2013.848364
Lambertsson L, Lundberg E, Nilsson M, Frech W (2001) J Anal Atom Spectrom 16:1296–1301. https://doi.org/10.1039/b106878b
Park JS, Lee JS, Kim GB, Cha JS, Shin SK, Kang HG, Hong EJ, Chung GT, Kim YH (2010) Water Air Soil Pollut 207:391–401. https://doi.org/10.1007/s11270-009-0144-3
Caricchia AM, Minervini G, Soldati P, Chiavarini S, Ubaldi C, Morabito R (1997) Microchem J 55:44–55. https://doi.org/10.1006/mchj.1996.1357
Hintelmann H, Evans RD, Villeneuve JY (1995) J Anal Atom Spectrom 10:619–624. https://doi.org/10.1039/ja9951000619
Lin LY, Chang LF, Jiang SJ (2008) J Agric Food Chem 56:6868–6872. https://doi.org/10.1021/jf801241w
de Souza SS, Rodrigues JL, Souza VCD, Barbosa F (2010) J Anal Atom Spectrom 25:79–83. https://doi.org/10.1039/b911696f
Houserova P, Matejicek D, Kuban V (2007) Anal Chim Acta 596:242–250. https://doi.org/10.1016/j.aca.2007.06.020
Rahman GMM, Kingston HM (2004) Anal Chem 76:3548–3555. https://doi.org/10.1021/Ac030407x
Rai R, Maher W, Kirkowa F (2002) J Anal Atom Spectrom 17:1560–1563. https://doi.org/10.1039/b208041a
Tu Q, Johnson W, Buckley B (2003) J Anal Atom Spectrom 18:696–701. https://doi.org/10.1039/b300992k
Kuban P, Houserova P, Kuban P, Hauser PC, Kuban V (2007) Electrophoresis 28:58–68. https://doi.org/10.1002/elps.200600457
Soliman EM, Saleh MB, Ahmed SA (2006) Talanta 69:55–60. https://doi.org/10.1016/j.talanta.2005.08.070
Jiang HM, Hu B, Jiang ZC, Qin YC (2006) Talanta 70:7–13. https://doi.org/10.1016/j.talanta.2006.02.047
Landi S, Fagioli F, Locatelli C (1992) J AOAC Int 75:1023–1028
Oda CE, Ingle JD (1981) Anal Chem 53:2305–2309. https://doi.org/10.1021/Ac00237a040
Leopold K, Foulkes M, Worsfold PJ (2009) Trac Trend Anal Chem 28:426–435. https://doi.org/10.1016/j.trac.2009.02.004
Logar M, Horvat M, Akagi H, Pihlar B (2002) Anal Bioanal Chem 374:1015–1021. https://doi.org/10.1007/s00216-002-1501-x
Labatzke T, Schlemmer G (2004) Anal Bioanal Chem 378:1075–1082. https://doi.org/10.1007/s00216-003-2416-x
Campbell MJ, Vermeir G, Dams R, Quevauviller P (1992) J Anal Atom Spectrom 7:617–621. https://doi.org/10.1039/ja9920700617
Seibert EL, Dressler VL, Pozebon D, Curtius AJ (2001) Spectrochim Acta B 56:1963–1971. https://doi.org/10.1016/S0584-8547(01)00334-2
Monperrus M, Tessier E, Veschambre S, Amouroux D, Donard O (2005) Anal Bioanal Chem 381:854–862. https://doi.org/10.1007/s00216-004-2973-7
Jia XY, Han Y, Liu XL, Duan TC, Chen HT (2011) Spectrochim Acta B 66:88–92. https://doi.org/10.1016/j.sab.2010.12.003
Jia XY, Gong DR, Han Y, Wei C, Duan TC, Chen HT (2012) Talanta 88:724–729. https://doi.org/10.1016/j.talanta.2011.10.026
Cheng HY, Wu CL, Shen LH, Liu JH, Xu ZG (2014) Anal Chim Acta 828:9–16. https://doi.org/10.1016/j.aca.2014.04.042
Nevado JJB, Martin-Doimeadios RCR, Krupp EM, Bernardo FJG, Farinas NR, Moreno MJ, Wallace D, Roper MJP (2011) J Chromatogr A 1218:4545–4551. https://doi.org/10.1016/j.chroma.2011.05.036
Zhao YQ, Zheng JP, Fang L, Lin Q, Wu YN, Xue ZM, Fu FF (2012) Talanta 89:280–285. https://doi.org/10.1016/j.talanta.2011.12.029
Li BH (2011) Anal Methods UK 3:116–121. https://doi.org/10.1039/c0ay00480d
Trujillo IS, Alonso EV, Pavon JMC, de Torres AG (2015) J Anal Atom Spectrom 30:2429–2440. https://doi.org/10.1039/c5ja00335k
Garcia-Ordiales E, Covelli S, Rico JM, Roqueni N, Fontolan G, Flor-Blanco G, Cienfuegos P, Loredo J (2018) Chemosphere 198:281–289. https://doi.org/10.1016/j.chemosphere.2018.01.146
Nevado JJB, Martin-Doimeadios RCR, Bernardo FJG, Moreno MJ (2005) J Chromatogr A 1093:21–28. https://doi.org/10.1016/j.chroma.2005.07.054
Cai Y, Monsalud S, Furton KG, Jaffe R, Jones RD (1998) Appl Organomet Chem 12:565–569. https://doi.org/10.1002/(Sici)1099-0739(199808/09)12:8/9%3C565::Aid-Aoc762%3E3.0.Co;2-K
Nevado JJB, Martin-Doimeadios RCR, Moreno MJ (2009) Sci Total Environ 407:2372–2382. https://doi.org/10.1016/j.scitotenv.2008.12.006
Martin-Doimeadios RCR, Krupp E, Amouroux D, Donard OFX (2002) Anal Chem 74:2505–2512. https://doi.org/10.1021/ac011157s
Rodrigues JL, Alvarez CR, Farinas NR, Nevado JJB, Barbosa F, Martin-Doimeadios RCR (2011) J Anal Atom Spectrom 26:436–442. https://doi.org/10.1039/c004931j
Moreno MJ, Pacheco-Arjona J, Rodriguez-Gonzalez P, Preud’Homme H, Amouroux D, Donard OFX (2006) J Mass Spectrom 41:1491–1497. https://doi.org/10.1002/jms.1120
Sanchez-Rodas D, Corns WT, Chen B, Stockwell PB (2010) J Anal Atom Spectrom 25:933–946. https://doi.org/10.1039/b917755h
Cano-Pavon JM, De Torres AG, Sanchez-Rojas F, Canada-Rudner P (1999) Int J Environ Anal Chem 75:93–106. https://doi.org/10.1080/03067319908047303
Heumann KG, Gallus SM, Radlinger G, Vogl J (1998) Spectrochim Acta B 53:273–287. https://doi.org/10.1016/S0584-8547(97)00134-1
Castillo A, Roig-Navarro AF, Pozo OJ (2006) Anal Chim Acta 577:18–25. https://doi.org/10.1016/j.aca.2006.06.024
Guo W, Hu SH, Wang XJ, Zhang JY, Jin LL, Zhu ZL, Zhang HF (2011) J Anal Atom Spectrom 26:1198–1203. https://doi.org/10.1039/c1ja00005e
Jian L, Goessler W, Irgolic KJ (2000) Fresen J Anal Chem 366:48–53. https://doi.org/10.1007/s002160050010
Brombach CC, Chen B, Corns WT, Feldmann J, Krupp EM (2015) Spectrochim Acta B 105:103–108. https://doi.org/10.1016/j.sab.2014.09.014
Guzman-Mar JL, Hinojosa-Reyes L, Serra AM, Hernandez-Ramirez A, Cerda V (2011) Anal Chim Acta 708:11–18. https://doi.org/10.1016/j.aca.2011.09.037
Ai X, Wang Y, Hou XD, Yang L, Zheng CB, Wu L (2013) Analyst 138:3494–3501. https://doi.org/10.1039/c3an00010a
Harrington CF, Romeril J, Catterick T (1998) Rapid Commun Mass Spectrom 12:911–916. https://doi.org/10.1002/(Sici)1097-0231(19980731)12:14%3C911::Aid-Rcm254%3E3.0.Co;2-X
Pena-Pereira F, Lavilla I, Bendicho C, Vidal L, Canals A (2009) Talanta 78:537–541. https://doi.org/10.1016/j.talanta.2008.12.003
Li PJ, He M, Chen BB, Hu B (2015) J Chromatogr A 1415:48–56. https://doi.org/10.1016/j.chroma.2015.08.062
Chen C, Peng MT, Hou XD, Zheng CB, Long Z (2013) Anal Methods UK 5:1185–1191. https://doi.org/10.1039/c2ay26214b
Li PJ, Zhang X, Hu B (2011) J Chromatogr A 1218:9414–9421. https://doi.org/10.1016/j.chroma.2011.10.071
Ting Y, Chen C, Ch’ng BL, Wang YL, Hsi HC (2018) J Hazard Mater 354:116–124. https://doi.org/10.1016/j.jhazmat.2018.04.074
Gilmour C, Bell JT, Soren AB, Riedel G, Riedel G, Kopec AD, Bodaly RA (2018) Sci Total Environ 640:555–569. https://doi.org/10.1016/j.scitotenv.2018.05.276
Blum PW, Hershey AE, Tsui MTK, Hammerschmidt CR, Agather AM (2018) Biogeochemistry 137:181–195. https://doi.org/10.1007/s10533-017-0408-8
Cesario R, Hintelmann H, Mendes R, Eckey K, Dimock B, Araujo B, Mota AM, Canario J (2017) Environ Pollut 226:297–307. https://doi.org/10.1016/j.envpol.2017.03.075
Valdes C, Black FJ, Stringham B, Collins JN, Goodman JR, Saxton HJ, Mansfield CR, Schmidt JN, Yang S, Johnson WP (2017) Environ Sci Technol 51:4887–4896. https://doi.org/10.1021/acs.est.6b05790
Kodamatani H, Maeda C, Balogh SJ, Nollet YH, Kanzaki R, Tomiyasu T (2017) Chemosphere 173:380–386. https://doi.org/10.1016/j.chemosphere.2017.01.053
Cesario R, Monteiro CE, Nogueira M, O’Driscoll NJ, Caetano M, Hintelmann H, Mota AM, Canario J (2016) Water Air Soil Pollut 227:475. https://doi.org/10.1007/s11270-016-3179-2
Mendes LA, de Lena JC, do Valle CM, Fleming PM, Windmoller CC (2016) Appl Geochem 75:32–43. https://doi.org/10.1016/j.apgeochem.2016.10.011
Monteiro CE, Cesario R, O’Driscoll NJ, Nogueira M, Valega M, Caetano M, Canario J (2016) Mar Pollut Bull 104:162–170. https://doi.org/10.1016/j.marpolbul.2016.01.042
Liu B, Schaider LA, Mason RP, Shine JP, Rabalais NN, Senn DB (2015) Estuar Coast Shelf Sci 159:50–59. https://doi.org/10.1016/j.ecss.2015.03.030
Ma X, Yin YG, Shi JB, Liu JF, Jiang GB (2014) Anal Methods UK 6:164–169. https://doi.org/10.1039/c3ay41625a
Kodamatani H, Tomiyasu T (2013) J Chromatogr A 1288:155–159. https://doi.org/10.1016/j.chroma.2013.02.004
Acknowledgements
Financial support from the Federal Institute of Hydrology is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Published in Chromatographia's 50th Anniversary Commemorative Issue.
Rights and permissions
About this article

Cite this article
Hellmann, C., Costa, R.D. & Schmitz, O.J. How to Deal with Mercury in Sediments? A Critical Review About Used Methods for the Speciation of Mercury in Sediments. Chromatographia 82, 125–141 (2019). https://doi.org/10.1007/s10337-018-3625-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-018-3625-y