
Abstract
CCAAT/enhancer binding protein α (C/EBPα) dimerizes via its leucine zipper (LZ) domain to bind DNA via its basic region and activate transcription via N-terminal trans-activation domains. The activity of C/EBPα is modulated by several serine/threonine kinases and via sumoylation, its gene is activated by RUNX1 and additional transcription factors, its mRNA stability is modified by miRNAs, and its mRNA is subject to translation control that affects AUG selection. In addition to inducing differentiation, C/EBPα inhibits cell cycle progression and apoptosis. Within hematopoiesis, C/EBPα levels increase as long-term stem cells progress to granulocyte–monocyte progenitors (GMP). Absence of C/EBPα prevents GMP formation, and higher levels are required for granulopoiesis compared to monopoiesis. C/EBPα interacts with AP-1 proteins to bind hybrid DNA elements during monopoiesis, and induction of Gfi-1, C/EBPε, KLF5, and miR-223 by C/EBPα enables granulopoiesis. The CEBPA ORF is mutated in approximately 10 % of acute myeloid leukemias (AML), leading to expression of N-terminally truncated C/EBPαp30 and C-terminal, in-frame C/EBPαLZ variants, which inhibit C/EBPα activities but also play additional roles during myeloid transformation. RUNX1 mutation, CEBPA promoter methylation, Trib1 or Trib2-mediated C/EBPαp42 degradation, and signaling pathways leading to C/EBPα serine 21 phosphorylation reduce C/EBPα expression or activity in additional AML cases.
Similar content being viewed by others


Introduction
CCAAT/enhancer binding protein α (C/EBPα), the prototypical basic region-leucine zipper transcription factor, plays a key role during hematopoiesis. Herein, I first review its molecular properties, protein interactions, protein modifications and other levels of post-transcriptional control, and its ability to affect not only differentiation, but also cell proliferation and survival. The role C/EBPα plays during normal myelopoiesis will then be described, including regulation of its expression and activity by other factors, and finally, the central role that reduced C/EBPα activity or expression plays during myeloid transformation will be delineated.
Dimerization, DNA-binding, and trans-activation
C/EBPα was initially purified from hepatocytes based on its ability to bind the CCAAT box in the herpes simplex virus thymidine kinase promoter and the enhancer core sequences of simian virus 40, polyomavirus, and murine sarcoma virus [1, 2], followed by isolation of its cDNA from the same source [3]. The C-terminus of C/EBPα was noted to lack helix-breaking glycine and proline residues and to contain leucines spaced every seven residues, predicting formation of an amphipathic α-helix with a hydrophobic surface capable of mediating dimerization. This domain was designated the leucine zipper (LZ) [4]. Mutagenic analysis revealed that DNA-binding by C/EBPα requires dimerization via its 36-residue LZ followed by site-specific DNA contact by the adjacent 37-residue basic region (BR), the combination also designated the bZIP domain [5, 6]. bZIP domain:DNA co-crystallization and structural analysis confirmed this model of DNA-binding, identifying specific contacts between the C/EBPα BR and the DNA major groove and phospho-ribose backbone [7, 8]. The BR also contains the C/EBPα nuclear localization signal [9, 10].
C/EBPα was further found capable of transcriptional activation of the serum albumin promoter via a specific binding site, dependent upon integrity of an N-terminal and internal trans-activation domains (TADs) [9, 11, 12], as diagrammed (Fig. 1). Interactions of co-activators or co-repressors with C/EBPα have not been extensively characterized, although the SWI/SNF complex was found to contact a central TAD to facilitate gene activation [13], the TIP60 histone acetyltransferase binds C/EBPα to increase trans-activation [14], and DEK, a protein that interacts with histone modifiers, mediates phosphorylation-dependent activity of the N-terminal C/EBPα TAD [15]. C/EBPα can also impact gene expression independent of DNA-binding; for example, interaction of the non-DNA contact surface of its BR with E2F1 reduces c-Myc transcription [16, 17], and C/EBPα displaces HDAC1 or HDAC3 from chromatin-bound NF-κB p50 to activate Bcl2, Flip, or Nfkb1 gene transcription [18–20].

Diagram of C/EBPα, showing the location of its trans-activation domains, basic region, leucine zipper, initiating AUG residues, protein modifications, and protein interactions
The bZIP family of TFs contains three major sub-families, the C/EBP proteins that in addition to C/EBPα include C/EBPβ, C/EBPδ, C/EBPε, C/EBPγ, and CHOP, the AP-1 proteins that include c-Fos, c-Jun and related proteins, and the CREB/ATF proteins. The C/EBPs readily homo- or hetero-dimerize via their LZ domains to bind to the DNA element 5′-T(T/G)NNGNAA(T/G) with similar affinity [21, 22], Jun and Fos proteins heterodimerize to bind the AP-1 consensus site 5′-TGA(C/G)TCA, and CREB/ATF proteins homo- or hetero-dimerize to bind the DNA element 5′-TCAGCTGA. AP-1 proteins also heterodimerize with small Maf proteins to bind an extended site, 5′-TGA(C/G)TCAGCA [23]. If one designates the repeating α-helical residues in the LZ as abcdefg, the leucines occupy position d, and other hydrophobic residues occupy position a, creating a hydrophobic surface that assists dimerization but with low affinity. Salt bridges between positively or negatively charged e and g residues strengthen the interaction and account for dimerization specificity [24]. C/EBP and AP-1 but not Maf proteins also hetero-dimerize, with reduced affinity compared with C/EBPα homodimers, to bind hybrid DNA elements [25, 26], and C/EBP:ATF hetero-dimerization also occurs [27], further extending the range of cis elements bound by C/EBP proteins.
Translational, protein modification, and miRNA control
In addition to translation of the dominant 42-kd C/EBPα isoform from a canonical N-terminal AUG, use of an internal AUG leads to expression of a 30-kd isoform lacking the N-terminal TAD [28, 29]. In addition, an extended-C/EBPα 46-kd isoform initiating from a non-canonical upstream CUG/GUG contains a nucleolar-localization motif and interacts with nucleophosmin [30]. A conserved upstream open reading frame (uORF) located between this non-canonical translation initiation site and that corresponding to the 42-kd isoform, but with a different reading frame, is thought to control the ratio of p42 vs. p30 translation, dependent upon mTOR activation of eIF-4E and PKR inhibition of eIF-2α, with increased initiation from the uORF due to reduced PKR or increased mTOR activity leading to increased p30 translation [31, 32]. In addition, calreticulin interacts with GCN nucleotide repeats in Cebpa RNA to inhibit its translation [33].
ERK binds an FXFP motif and phosphorylates C/EBPα on S21, near but upstream of the N-terminal TAD, to reduce C/EBPα trans-activation activity, consequent in part to reduced DEK interaction [15, 34]. GSK-3 phosphorylates T222 and T226, dependent on S230 phosphorylation to stimulate C/EBPα activity [35], phosphorylation of S248 via Ras-dependent PKCδ activation increases C/EBPα trans-activation and is required for induction of 32Dcl3 granulocytic differentiation [36], and PKCδ modifies additional residues, with S299 modification capable of attenuating C/EBPα DNA-binding [37].
C/EBPα contains a conserved motif IKQEP, with K159 modification by SUMO-1 reducing C/EBPα activity via increased HDAC3 interaction [8–41]. Known sites of C/EBPα protein modification, along with its protein:protein interactions, are diagrammed (Fig. 1).
MicroRNA-690 directly targets Cebpa RNA to reduce its expression in a myeloid-derived suppressor cell subset [42], and the Trib1 or Trib2 adaptor proteins facilitate COP1 E3 ubiquitin ligase-mediated C/EBPα degradation, preferentially of the 42 kd isoform [43–45].
Regulation of cell proliferation, survival, and quiescence
The finding that mature hepatocytes express higher levels of C/EBPα than hepatoma cells provided the first indication that C/EBPα might negatively control cell proliferation [11]. This author’s finding that wild-type C/EBPα but not variants incapable of DNA-binding suppress 3T3-L1 preadipocyte colony formation prompted further experiments with estradiol-regulated C/EBPα-ER that revealed direct inhibition of 3T3-L1 cell cycle progression [46]. C/EBPα inhibits proliferation via induction of p21, via interaction of residues 175–187 with CDK2 and CDK4, and via interaction of the outer surface of its BR with E2F proteins, the latter mechanism most active in myeloid cells [16, 47–49]. In hepatocytes, PI3K/AKT mediated S193 dephosphorylation reduces Cdk2/Cdk4 interaction [50]. Of note, mice lacking C/EBPα residues 180–194 have no abnormalities and their fetal liver cells display normal proliferative parameters [51]. C/EBPα also induces miR-34a, which targets E2F3 to limit myeloid cell proliferation [52].
C/EBPα slows apoptosis of the Ba/F3, 32Dcl3, and HF-1 hematopoietic cell lines upon cytokine withdrawal, correlated with increased Bcl-2 and FLIP expression, and C/EBPα or an LZ mutant incapable of DNA-binding directly interacts with NF-κB p50 to bind chromatin and induce Bcl-2 expression and promoter activity, whereas a BR mutant does not bind p50 or induce Bcl-2 [18, 19]. C/EBPα or C/EBPβ have much higher affinity for NF-κB p50 than they do for NF-κB p65, allowing these C/EBPs to displace HDAC1 or HDAC3 from chromatin-bound p50 to induce NF-κB target genes even in the absence of canonical NF-κB activation [20, 53].
Finally, indicating a role for C/EBPα in maintaining stem cell quiescence, Mx1-Cre-mediated Cebpa ORF deletion in adult mice draws long-term hematopoietic stem cells (LT-HSC) into cell cycle, induces apoptosis, and leads to stem cell exhaustion in competitive transplantation assays [54, 55].
Regulation of normal myelopoiesis
Expression
C/EBPα is abundant in several cell lineages, including adipocytes, hepatocytes, and type II pneumocytes [56, 57]. Within hematopoiesis, C/EBPα is preferentially expressed in the granulocyte, monocyte, and eosinophil as compared to the lymphoid or megakaryocyte/erythroid lineages [58, 59]. Amongst marrow stem/progenitor cells, low level Cebpa RNA expression is detected in Lin−Sca-1+c-Kit+ (LSK) cells, increases twofold as these progress to the common myeloid progenitor (CMP) and tenfold further as CMP develop into GMP [60]. Cebpa is detectable as well in LT-HSC defined by the LSK/SLAM surface markers [54].
Consequence of reduced expression
Cebpa-/- mice are neonatal lethal due to hepatic dysfunction, but display impaired myelopoiesis [61, 62]. Cebpa(-/-) fetal liver or marrow from Cebpa(f/f);Mx1-Cre adult mice exposed to pIpC to induce Cre expression and biallelic deletion of the Cebpa ORF have markedly reduced GMP and myeloid colony-forming units (CFU), with increased CMP, LSK, and megakaryocyte/erythroid progenitors (MEP), and their peripheral blood has markedly reduced neutrophils and monocytes and absence of eosinophils, with twofold elevated platelets and mild lymphocytosis [60, 63]. Beyond the GMP, threefold Cebpa knockdown prevents granulopoiesis but not monopoiesis, while sixfold knockdown prevents commitment to either lineage and increases erythropoiesis [64].
Contribution to monopoiesis
Reduced levels of C/EBPα may contribute to monopoiesis by hetero-dimerizing with AP-1 proteins such as c-Jun and c-Fos via their respective LZ domains followed by binding to hybrid C/EBP:AP-1 DNA sites, 5′-TGA(C/T)GCAA, commonly found in regulatory elements of genes expressed specifically in monocyte/macrophages and in the FosB gene promoter. These hybrid elements often co-localize with PU.1-binding sites; in contrast, PU.1 sites in B cell-specific genes are not found near C/EBP:AP-1 hybrid sites [26, 65]. Use of artificial acidic and basic LZs to direct specific hetero-dimer formation revealed that C/EBPα:c-Fos or C/EBPα:c-Jun but not C/EBPα:C/EBPα or c-Jun:c-Fos complexes direct monocytic commitment of murine myeloid progenitors [25]. Induction of monopoiesis by exogenous C/EBPα may reflect its interaction with endogenous AP-1 proteins, as a variant harboring the GCN4 LZ was inactive [66–68]. Consistent with fewer but still evident e/g LZ salt bridges, C/EBP:AP-1 affinity is approximately twofold weaker than C/EBP:C/EBP affinity, and semi-quantitative Western blot analysis of C/EBP and AP-1 proteins in myeloid cell lines indicates that hetero-dimers could readily form, as was detected by oligonucleotide pull-down, and may be favored by AP-1 protein induction during monopoiesis [26].
Regulation of granulopoiesis
Formation of active C/EBPα homo-dimers may be a prerequisite for granulopoiesis. C/EBPα induces transcription of several regulatory proteins required for subsequent lineage maturation, including the transcription factors C/EBPε, Gfi-1, and KLF5 [64, 69–72]. Absence of C/EBPε leads to secondary granule deficiency [73], Gfi-1(-/-) mice develop severe neutropenia [74, 75], and KLF5 contributes to 32Dcl3 granulopoiesis [76]. In addition, C/EBPα induces miR-223, leading to degradation of NFI-A mRNA to enhance granulopoiesis [77], and C/EBPα induces miR-30c, which down-regulates Notch1 expression to again favor neutrophilic lineage specification [78]. In addition, C/EBPα cooperates with PU.1, c-Myb, and RUNX1 to activate genes such as myeloperoxidase, neutrophil elastase, lysozyme, lactoferrin, G-CSF receptor (GCSFR), M-CSF receptor (MCSFR), and GM-CSF receptor in immature or mature granulocytic or immature monocytic cells, as previously reviewed [79, 80].
Regulation of Cebpa gene expression during myelopoiesis
By sorting Lin−Sca-1−c-Kit+ marrow cells into GCSFR+MCSFR− vs. GCSFR−MCSFR+ subsets we enriched for CFU-G vs. CFU-M, demonstrating 2.5-fold increased Cebpa RNA in CFU-G [64]. In addition, Runx1 was enriched 1.5-fold, Gfi1 fivefold, Cebpe 14-fold, and Klf5 eightfold in CFU-G, whereas Irf8 was enriched fourfold and Klf4 twofold in CFU-M, and PU.1 levels were similar in both. Thus, induction of Cebpa transcription to favor C/EBPα homodimer over C/EBPα:AP-1 heterodimer formation may contribute to granulocyte lineage specification.
C/EBPα auto-activates its own promoter, and RUNX1 activates the Cebpa promoter modestly, via two conserved non-consensus binding sites, and strongly activates an evolutionarily conserved, 450 bp +37 kb Cebpa enhancer, via four consensus RUNX1 cis elements [81, 82]. ChIP-Seq data demonstrates that the Cebpa enhancer binds RUNX1 as well as SCL, GATA2, LMO2, LYL1, PU.1, Erg, Fli-1, HoxA9, Meis1, Gfi-1b, and C/EBPα [83–85], and notably RUNX1 binding was not evident elsewhere in the Cebpa locus [85]. The enhancer also binds p300 and contains the enhancer-specific H3K4me1 histone modification [82]. In transgenic mice in which the +37 kb Cebpa enhancer and 845 bp promoter directs expression of a cytoplasmically truncated hCD4 reporter, surface marker analysis and CFU assays demonstrate that the Cebpa enhancer/promoter regulatory elements are preferentially active in myeloid compared to lymphoid or erythroid progenitors. In addition, competitive transplantation and FACS analyses demonstrate reporter activity in phenotypic and the large majority of functional LT-HSC [86]. And consistent with these findings, sorting of CMP or LSK/SLAM LT-HSC into hCD4+ and hCD4− subsets revealed that endogenous Cebpa mRNA is highly enriched in hCD4+ CMP [86] or LT-HSC (unpublished). Related results were obtained with another mouse model in which a cDNA encoding Cre recombinase was inserted into the Cebpa locus, followed by breeding to a strain that expresses YFP only in the presence of Cre [87].
LEF-1, reduced in cases of severe congenital neutropenia, activates Cebpa gene expression via a binding site located in its promoter region [88], whereas HIF-1α represses Cebpa transcription via an additional promoter site [89], although HIF-1α may also augment myeloid differentiation via direct interaction with C/EBPα [90]. Finally, a 4.5 kb nuclear, polyA(-), coding-strand RNA encompassing the 2.6 kb Cebpa mRNA interacts with DNMT1 via stem–loop RNA structures to limit Cebpa promoter methylation and increase gene expression [91].
Regulation of C/EBPα activity during myelopoiesis
G-CSF or M-CSF signals direct lineage choice of single, sorted GMP [92]. Stimulation of Lin− marrow myeloid progenitors with G-CSF preferentially activates STAT3 and SHP2, whereas M-CSF more potently activates PLCγ, PKC, and ERK [93]. As noted, ERK phosphorylates C/EBPα S21 to reduce its activity [34], and M-CSF but not G-CSF increases phospho-S21-CEBPα in Lin− marrow cells, dependent on ERK activation [93]. A homo-dimer of unmodified C/EBPα, expressed at increased levels due to Cebpa gene induction by RUNX1 and additional factors, might be required to mediate granulopoiesis, while a phospho-S21-C/EBPα:AP-1 hetero-dimer might still be capable of activating monocyte/macrophage-specific genes in cooperation with PU.1 (Fig. 2). ERK stabilizes c-Fos and induces AP-1 gene induction via Ets:SRF:SRF terniary complex activation to potentially further facilitate monopoiesis while impeding granulocyte lineage development [94]. The p38 MAP kinase can also modify CEBPα S21 to impede neutrophil development [95], and an MKK6-p38MAPK pathway induces C/EBPα proteasomal degradation to facilitate trans-differentiation of inflammatory neutrophils to monocytes [96].

Diagram of a C/EBPα:C/EBPα homodimer and a C/EBPα:AP-1 heterodimer bound to consensus cis elements, with cooperating transcription factors and gene induction events that enable their contribution to granulopoiesis versus monopoiesis
The SHP2 tyrosine phosphatase inactivates IRF8 [97], a transcription factor required for monopoiesis, and acts on RUNX1 to increase its ability to generate megakaryocytes and CD8 T cells [98]. Induction of SHP2 phosphorylation by G-CSF might occur secondary to activation of Src kinases [99], and we find that tyrosine phosphorylation of Runx1 by Src, or potentially additional kinases, increase its trans-activation potency to facilitate granulopoiesis (unpublished). As noted, sumoylation of C/EBPα reduces its activity, and hyposumoylation increases myelopoiesis in zebrafish embryos [100].
Cross-talk with additional transcription factors
PU.1, an Ets transcription factor, is also a key regulator of myeloid development. PU.1-/- mice lack B cells and monocytes and have markedly reduced neutrophils [101]. In contrast to C/EBPα, higher levels of PU.1 are required for monocyte as compared to granulocyte lineage development; Mx1-Cre mediated deletion of the 236 bp −14 kb PU.1 enhancer leads to 80 % reduction of PU.1 with loss of monopoiesis but preservation of granulopoiesis [102]. RUNX1 also contributes to myelopoiesis. The −14 kb PU.1 and +37 kb Cebpa enhancers are each activated by RUNX1, with exposure of Runx1(f/f);Mx1-Cre mice to pIpC reducing Cebpa mRNA twofold and PU.1 mRNA 1.5-fold in CMP and GMP [82, 103]. Notably, from a functional perspective Runx1 deletion impairs granulopoiesis while increasing monopoiesis, leading us to propose that Cebpa is more critical than PU.1 as a RUNX1 target during both normal and malignant myelopoiesis [82]. The PU.1 promoter and −14 kb enhancer are each activated by C/EBPα [104, 105]. The quantitative importance of this regulation during myelopoiesis is uncertain given that higher levels of C/EBPα favor granulopoiesis, whereas higher levels of PU.1 favor monopoiesis. PU.1 induction by C/EBPα could potentially vary during hematopoiesis, e.g. increased at the GMP stage to facilitate myeloid vs. megakaryocyte/erythroid commitment and then reduced to facilitate granulocyte vs. monocyte lineage specification.
By forming a C/EBPα:c-Jun heterodimer via LZ interaction, C/EBPα can divert c-Jun:c-Fos complexes from auto-activating the c-Jun promoter [106], though we have suggested that during myelopoiesis AP-1 complexes are not depleted by C/EBP proteins, but rather that C/EBP:AP-1 heterodimers binds hybrid DNA elements to activate monocytic genes in cooperation with PU.1 and AP-1 [25]. C/EBPα can directly interact with PU.1 to inhibit activation of a model PU.1 reporter [107], although cooperative activation of myeloid genes such as neutrophil elastase by C/EBPα and PU.1 indicates that this mechanism is not always operative [108]. IRF8 directly interacts with C/EBPα to inhibit its interaction with endogenous C/EBPα target genes, thereby preventing granulopoiesis to favor monopoiesis [109]. Perhaps C/EBPα:AP-1 hetero-dimers are less sensitive to IRF8 inhibition, allowing them to contribute to monopoiesis in the presence of IRF8. C/EBPα down-regulates Pax5 RNA through an uncertain mechanism to inhibit lymphopoiesis [110]. Exogenous C/EBPα impairs and reduced C/EBPα enhances erythropoiesis [60, 63, 64, 70]. The effect of reduced C/EBPα on erythropoiesis might reflect decreased PU.1 levels, leading to GATA-1 derepression [111, 112]. However, Cebpa knockdown only reduced PU.1 1.6-fold while markedly enhancing erythropoiesis, leading to the speculation that direct regulation of GATA-1 or a GATA-1 co-factor by C/EBPα might also restrict the CMP to MEP transition [64]. Indeed avian C/EBPβ down-regulates FOG mRNA to enable eosinophil lineage development [113]. Of note, induction of GATA-2 in C/EBPα-expressing GMP leads to eosinophil lineage commitment [59]. Finally, mice lacking the bHLH transcription factor Twist-2 have increased neutrophils, monocytes, and basophils, in part reflecting the ability of Twist-2 to inhibit C/EBPα trans-activation [114].
Role of reduced C/EBPα expression or activity in myeloid transformation
CEBPA ORF mutations
The protein-coding CEBPA ORF is mutated in approximately 10 % of AML cases, most often those with FAB M1 or M2 morphology lacking t(8;21) [115, 116]. Two categories of mutations occur. N-terminal mutations lead to premature translational termination of C/EBPαp42 and increased levels of C/EBPαp30, lacking a TAD. C-terminal mutations occur in the LZ, often in its first α-helix, preventing DNA-binding. About 50 % of AMLs with CEBPA ORF mutations have an N-terminal mutation on one allele and a C-terminal mutation on the other, and patients with double-mutant CEBPA have improved prognosis compared to those with only one mutant allele [117]. Approximately 10 % of AML cases with CEBPA N-terminal mutations acquire the alteration via the germline [117]. Less than 5 % of patients with myelodysplastic syndrome (MDS) harbor CEBPA mutations, and these are most often mono-allelic [116, 118, 119].
The large majority of C-terminal mutations are in-frame, indicating that these C/EBPαLZ variants contribute to transformation not only due to their lack of DNA-binding activity, but also as active oncoproteins. Both C/EBPαp30 and several C/EBPαLZ mutants retain the ability to interact with NF-κB p50 to induce Bcl-2 and inhibit apoptosis [18, 19, 53], a C/EBPαLZ variant interfered with the ability of C/EBPαp42 to activate transcription in cooperation with PU.1 [119], potentially reflecting sequestration of PU.1 or co-activators, and additional protein interactions might further allow C/EBPαLZ variants to contribute to transformation.
The ability of C/EBPαp42 to inhibit cell cycle progression consequent to interaction with E2F proteins is dependent upon integrity of its N-terminus [16]; therefore, C/EBPαp30 likely has reduced ability to inhibit proliferation of leukemic blasts via effects on cell cycle regulator proteins, as is observed [120, 121]. C/EBPαp30 can zipper with C/EBPαp42 and weaken its trans-activation strength, and p30:p30 homodimers compete with p42:42 homodimers for binding to DNA [115]. p30 binds a subset of C/EBP sites with reduced affinity compared with p42 but binds others with equal affinity, retains a TAD and induces multiple genes not affected by p42, including the Ubc9 SUMO ligase, and may further alter progenitor biology via protein interactions [115, 121–124].
Heterozygous WT/p30 knockin mice have no hematologic abnormalities; in contrast p30/p30 mice uniformly develop AML by one year [120]. Preleukemic p30/p30 mice are neutropenic but retain GMP and myeloid CD11blowc-kit+ CFUs that replate indefinitely in IL-3/SCF. Biallelic C/EBPαLZ mice lack GMP and only develop delayed erythroleukemia, whereas LZ/p30 mice develop myeloid transformation with more rapid kinetics than p30/p30 mice [125]. C/EBPαp30 apparently provides C/EBPα activity sufficient to generate GMP but insufficient for further myeloid maturation, and these GMP are then subject to further mutations leading to full myeloid transformation. In contrast, Cebpa(-/-) mice do not develop GMP or AML, but have increased CMP and LSK [60], consistent with the finding that p30/p30 or LZ/p30 leukemia-initiating cells (LIC) reside predominantly in the GMP or LinloSca-1−c-kit+CD11blo populations [120, 121]. Of note, in the absence of C/EBPα, neither Bcr-Abl, MLL-ENL, nor HoxA9/Meis1 induce myeloid transformation, consistent with the idea that a minimal level of C/EBPα activity is required to generate GMP as a substrate for transformation [85, 126, 127]. Consistent with this idea, co-expression of the FLT3ITD-activated tyrosine kinase receptor with C/EBPp30 and a C/EBPαLZ mutant accelerates transformation, with preleukemic GMP expansion [128].
In contrast to C/EBPp30, any one of several C/EBPαLZ variants block 32Dcl3 myeloid differentiation, induce marrow CFU replating upon retroviral transduction, and induce AML upon transplantation of transduced marrow cells, alone and accelerated by FLT3ITD [119, 129]. In addition, C/EBPαLZ variants reduce Mcsfr/Csf1r expression, and ectopic Csf1r cooperates with a C/EBPαLZ variant harboring a C-terminal LZ mutation to induce myeloid transformation [129]. In contrast to these findings with C/EBPαp30, LIP, an N-terminally truncated C/EBPβ isoform, but not full-length C/EBPβ, induces indefinite myeloid CFU replating and AML in vivo, potentially via C/EBPα inhibition [130].
RUNX1 down-modulation
Alterations affecting RUNX1 or its partner CBFβ are common in AML [131]. 12 % of AMLs harbor t(8;21), leading to expression of RUNX1-ETO, which binds RUNX1 cis elements to represses Cebpa transcription [132]. 8 % of AMLs, mainly the M4eo subset, have inv(16), expressing CBFβ-SMMHC, which binds RUNX1 to inhibit its activity [133], and 13 % of cases, most often M0, have inactivating point mutations in the RUNX1 DNA-binding domain or TAD [134]. RUNX1 point mutations are also present in about one-third of cases in which patients with the myeloproliferative diseases chronic myeloid leukemia (CML), polycythemia vera, or essential thrombocytopenia have progressed to AML [135, 136], and RUNX1 point mutations are also found at high frequency in therapy-related MDS and in ~3 % of sporadic MDS cases [137]. RUNX1 point mutation most often occurs in its DNA-binding domain on one allele; these variants do not bind DNA, but dominantly inhibit RUNX1 trans-activation, potentially via interference with RUNX1 dimerization and by competition for CBFβ, a protein required for RUNX1 DNA-binding [137, 138]. CML blast crisis also occasionally manifests t(3;21), expressing RUNX1/MDS1/EVI1 (RME), fusing the RUNX1 DNA-binding domain with MDS1/EVI1 to generate a potent repressor of RUNX1 targets; RME also cooperates with Bcr-Abl to induce AML [139].
Reduced CEBPA transcription consequent to each of these RUNX1 alterations, resulting from reduced activity of the CEBPA promoter and its +41 kb enhancer (homologous to the murine +37 kb enhancer), may be central to their ability to contribute to myeloid transformation. ChIP-Seq data for RUNX1-ETO from two human AML patient samples and from the Kasumi-1 cell line demonstrates exclusive binding at the +41 kb CEBPA enhancer [140]. In contrast, CEBPA enhancer point mutations or small deletions were not seen in AML cases [141]. Of note, Mx1-Cre-mediated Runx1 gene deletion in adult mice does not lead to AML, likely due to modest twofold Cebpa mRNA reduction in GMP, whereas active repression of the Cebpa +37 kb enhancer by RUNX1-ETO or dominant-inhibition of RUNX1, RUNX2, and RUNX3 by CBFβ-SMMHC or RUNX1 mutants likely results in further Cebpa suppression.
Mice lacking the RUNX1-regulated PU.1 −14 kb enhancer uniformly develop AML by 6 months [102]. However, PU.1 gene mutations occur in <1 % of human AML cases [142]. As noted, our study of Runx1-deleted mice led us to suggest that reduction in C/EBPα rather than PU.1 activity is the more critical consequence of RUNX1 oncoprotein expression in human AMLs [82].
Other alterations in AML affecting C/EBPα
Approximately 50 % of AML cases have reduced CEBPA mRNA, with about threefold median reduction amongst these cases, maximum tenfold, vs. CD34+CD38+ (mainly myeloid) progenitors [143]. CEBPA promoter CpG hyper-methylation, evident in 37 % of AML cases [143], may in part reflect reduced activity of RUNX1 or other transcription factors that normally activate the promoter as well as selection of preleukemic blasts with repressive promoter methylation. Approximately 3 % of AML cases harbor dense CEBPA promoter hyper-methylation leading to silencing of CEBPA expression and up-regulation of T cell genes [144]; in these cases the gene encoding C/EBPγ, located just upstream of the CEBPA gene, is markedly induced due to derepression of E2F proteins [145]. The ability of C/EBPγ to zipper with other C/EBPs and reduce their trans-activation potency might contribute to myeloid transformation in these cases. Leukemic blasts from patients with silenced CEBPA or those with biallelic CEBPA ORF mutations have a gene expression profile similar that obtained from the preleukemic LIC population isolated from mice harboring biallelic C- and N-terminal C/EBPα mutant variants or those with reduced Sox4, a gene directly repressed by C/EBPα [146].
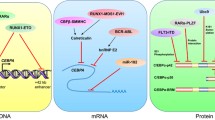
Over-expression of Trib1 or Trib2, which direct C/EBPαp42 but not p30 degradation via the E3 ubiquitin ligase COP1, occurs in 15–20 % of AMLs, and Trib1 or Trib2 are transforming in murine models [43, 147, 148]. FLT3ITD activates ERK and CDK1 to stimulate phosphorylation of C/EBPαS21, reducing C/EBPα trans-activation strength [149, 150], and Bcr-Abl inhibits translation of Cebpa mRNA by inducing hnRNP E2 [151]. Pathways known to down-modulate C/EBPα in AML are summarized, along with additional pathway that might be relevant (Fig. 3).

Pathways, both described and potential, that mediate reduced C/EBPα expression or activity in AML cases via effects on CEBPA transcription, mRNA translation or stability, or protein expression, stability, or activity
Conclusions
The ability of C/EBPα to homo-dimerize and also to hetero-dimerize with other bZIP proteins extends the range of DNA elements it interacts with and provides opportunity for regulation of both C/EBPα and its partners. Future investigations should provide further insight into the role these interactions and C/EBPα protein modifications play during myelopoiesis, including determining whether C/EBPα S21 phosphorylation precluded activation of granulocytic genes but still allow induction of monocytic genes as a heterodimer with AP-1 proteins and assessing the role of additional C/ΕΒPα modifications, e.g., lysine acetylation or arginine methylation. The capacity of C/EBPα to inhibit cell proliferation requires unique control during different stages of hematopoiesis to allow the requisite balance between the proliferative drive and induction of lineage-specific genes. For example, GMP harbor high-levels of C/EBPα and yet have high proliferative potential—do they possess a mechanism to suppress C/EBPα-mediated inhibition of G1 to S cell cycle progression? Further elucidating how the N-terminus of C/EBPα contributes to cell cycle inhibition upon BR interaction with E2F proteins might provide relevant insight. Several C/EBPα protein modifications have been identified, but their role during normal myelopoiesis is poorly understood.
Existence of multiple pathways to C/EBPα inhibition supports the idea that reduction of C/EBPα expression or activity is central to the pathogenesis of AML. Perhaps means can be found to reactivate C/EBPα as “differentiation therapy” for AML or MDS, either by inducing expression of normal CEBPA alleles, targeting signaling pathways, miRNAs, or non-coding RNAs to favor increased C/EBPα expression or activity, or inducing other C/EBP family members that might substitute for C/EBPα. In particular, C/EBPβ induces granulopoiesis in response to cytokine signals in the absence of C/EBPα, and replacement of the Cebpa ORF by Cebpb is well tolerated [152, 153].
Conflict of interest
The author declares that he has no conflict of interest.
References
Graves BJ, Johnson PF, McKnight SL. Homologous recognition of a promoter domain common to the MSV LTR and the HSV tk gene. Cell. 1986;44:565–76.
Johnson PF, Landschulz WH, Graves BJ, McKnight SL. Identification of a rat liver nuclear protein that binds to the enhancer core element of three animal viruses. Genes Dev. 1987;1:133–46.
Landschulz WH, Johnson PF, Adashi EY, Graves BJ, McKnight SL. Isolation of a recombinant copy of the gene encoding C/EBP. Genes Dev. 1988;2:786–800.
Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–64.
Landschulz WH, Johnson PF, McKnight SL. The DNA binding domain of the rat liver nuclear protein C/EBP is bipartite. Science. 1989;243:1681–8.
Agre P, Johnson PF, McKnight SL. Cognate DNA binding specificity retained after leucine zipper exchange between GCN4 and C/EBP. Science. 1989;246:922–6.
Vinson CR, Sigler PB, McKnight SL. Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science. 1989;246:911–6.
Miller M, Shuman JD, Sebastian T, Dauter Z, Johnson PF. Structural basis for DNA recognition by the basic region leucine zipper transcription factor CCAAT/enhancer-binding protein α. J Biol Chem. 2003;278:15178–84.
Friedman AD, McKnight SL. Identification of two polypeptide segments of CCAAT/enhancer-binding protein required for transcriptional activation of the serum albumin gene. Genes Dev. 1990;4:1416–26.
Williams SC, Angerer ND, Johnson PF. C/EBP proteins contain nuclear localization signals imbedded in their basic regions. Gene Expr. 1997;6:371–85.
Friedman AD, Landschulz WH, McKnight SL. CCAAT/enhancer binding protein activates the promoter of the serum albumin gene in cultured hepatoma cells. Genes Dev. 1989;3:1314–22.
Nerlov C, Ziff EB. Three levels of functional interaction determine the activity of CCAAT/enhancer binding protein-α on the serum albumin promoter. Genes Dev. 1994;8:350–62.
Pedersen TA, Kowenz-Leutz E, Leutz A, Nerlov C. Cooperation between C/EBPα TBP/TFIIB and SWI/SNF recruiting domains is required for adipocyte differentiation. Genes Dev. 2001;15:3208–16.
Bararia D, Trivedi AK, Zada AA, Greif PA, Mulaw MA, Christopeit M, et al. Proteomic identification of the MYST domain histone acetyltransferase TIP60 (HTATIP) as a co-activator of the myeloid transcription factor C/EBPα. Leukemia. 2008;22:800–7.
Koleva RI, Ficarro SB, Radomska HS, Carrasco-Alfonso MJ, Alberta JA, Webber JT, et al. C/EBPα and DEK coordinately regulate myeloid differentiation. Blood. 2012;119:4878–88.
Porse BT, Pedersen TA, Xu X, Lindberg B, Wewer UM, Friis-Hansen L, et al. E2F repression by C/EBPα is required for adipogenesis and granulopoiesis in vivo. Cell. 2001;107:247–58.
Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR, et al. G. c-Myc is a critical target for C/EBPα in granulopoiesis. Mol Cell Biol. 2001;21:3789–806.
Paz-Priel I, Cai DH, Wang D, Kowalski J, Blackford A, Liu H, et al. CCAAT/enhancer binding protein α (C/EBPα) and C/EBPα myeloid oncoproteins induce bcl-2 via interaction of their basic regions with nuclear factor-κB p50. Mol Cancer Res. 2005;3:585–96.
Paz-Priel I, Ghosal AK, Kowalski J, Friedman AD. C/EBPα or C/EBPα oncoproteins regulate the intrinsic and extrinsic apoptotic pathways by direct interaction with NF-κB p50 bound to the bcl-2 and FLIP gene promoters. Leukemia. 2009;23:365–74.
Paz-Priel I, Houng S, Dooher J, Friedman AD. C/EBPα and C/EBPα oncoproteins regulate nfkb1 and displace histone deacetylases from NF-κB p50 homodimers to induce NF-κB target genes. Blood. 2011;117:4085–94.
Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5:1538–52.
Williams SC, Cantwell CA, Johnson PF. A family of C/EBP-related proteins capable of forming covalently linked leucine zipper dimers in vitro. Genes Dev. 1991;5:1553–67.
Kataoka K, Noda M, Nishizawa M. Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both Fos and Jun. Mol Cell Biol. 1994;14:700–12.
Vinson CR, Hai T, Boyd SM. Dimerization specificity of the leucine zipper-containing bZIP motif on DNA binding: prediction and rational design. Genes Dev. 1993;7:1047–58.
Cai DH, Wang D, Keefer J, Yeamans C, Hensley K, Friedman AD. C/EBPα:AP-1 leucine zipper heterodimers bind novel DNA elements, activate the PU.1 promoter and direct monocyte lineage commitment more potently than C/EBPα homodimers or AP-1. Oncogene. 2008;27:2772–9.
Hong S, Skaist AM, Wheelan SJ, Friedman AD. AP-1 protein induction during monopoiesis favors C/EBP: AP-1 heterodimers over C/EBP homodimerization and stimulates FosB transcription. J Leukoc Biol. 2011;90:643–51.
Shuman JD, Cheong J, Coligan JE. ATF-2 and C/EBPα can form a heterodimeric DNA binding complex in vitro. Functional implications for transcriptional regulation. J Biol Chem. 1997;272:12793–800.
Descombes P, Schibler U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell. 1991;67:569–79.
Lin FT, MacDougald OA, Diehl AM, Lane MD. A 30-kDa alternative translation product of the CCAAT/enhancer binding protein α message: transcriptional activator lacking antimitotic activity. Proc Natl Acad Sci USA. 1993;90:9606–10.
Müller C, Bremer A, Schreiber S, Eichwald S, Calkhoven CF. Nucleolar retention of a translational C/EBPα isoform stimulates rDNA transcription and cell size. EMBO J. 2010;29:897–909.
Lincoln AJ, Monczak Y, Williams SC, Johnson PF. Inhibition of CCAAT/enhancer-binding protein α and β translation by upstream open reading frames. J Biol Chem. 1998;273:9552–60.
Calkhoven CF, Müller C, Leutz A. Translational control of C/EBPα and C/EBPβ isoform expression. Genes Dev. 2000;14:1920–32.
Timchenko LT, Iakova P, Welm AL, Cai ZJ, Timchenko NA. Calreticulin interacts with C/EBPα and C/EBPβ mRNAs and represses translation of C/EBP proteins. Mol Cell Biol. 2002;22:7242–57.
Ross SE, Radomska HS, Wu B, Zhang P, Winnay JN, Bajnok L, et al. Phosphorylation of C/EBPα inhibits granulopoiesis. Mol Cell Biol. 2004;24:675–86.
Ross SE, Erickson RL, Hemati N, MacDougald OA. Glycogen synthase kinase 3 is an insulin-regulated C/EBPα kinase. Mol Cell Biol. 1999;19:8433–41.
Behre G, Singh SM, Liu H, Bortolin LT, Christopeit M, Radomska HS, et al. Ras signaling enhances the activity of C/EBPα to induce granulocytic differentiation by phosphorylation of serine 248. J Biol Chem. 2002;277:26293–9.
Mahoney CW, Shuman J, McKnight SL, Chen HC, Huang KP. Phosphorylation of CCAAT-enhancer binding protein by protein kinase C attenuates site-selective DNA binding. J Biol Chem. 1992;267:19396–403.
Kim J, Cantwell CA, Johnson PF, Pfarr CM, Williams SC. Transcriptional activity of CCAAT/enhancer-binding proteins is controlled by a conserved inhibitory domain that is a target for sumoylation. J Biol Chem. 2002;277:38037–44.
Subramanian L, Benson MD, Iñiguez-Lluhí JA. A synergy control motif within the attenuator domain of CCAAT/enhancer-binding protein α inhibits transcriptional synergy through its PIASy-enhanced modification by SUMO-1 or SUMO-3. J Biol Chem. 2003;278:9134–41.
Hankey W, Silver M, Sun BS, Zibello T, Berliner N, Khanna-Gupta A. Differential effects of sumoylation on the activities of CCAAT enhancer binding protein α (C/EBPα) p42 versus p30 may contribute in part, to aberrant C/EBPα activity in acute leukemias. Hematol Rep. 2011;3:e5.
Ren J, Li D, Li Y, Lan X, Zheng J, Wang X, et al. HDAC3 interacts with sumoylated C/EBPα to negatively regulate the LXRα expression in rat hepatocytes. Mol Cell Endocrinol. 2013;374:35–45.
Hegde VL, Tomar S, Jackson A, Rao R, Yang X, Singh UP, et al. Distinct microRNA expression profile and targeted biological pathways in functional myeloid-derived suppressor cells induced by Δ9-tetrahydrocannabinol in vivo: regulation of CCAAT/enhancer-binding protein α by microRNA-690. J Biol Chem. 2013;288:36810–26.
Keeshan K, He Y, Wouters BJ, Shestova O, Xu L, Sai H, et al. Tribbles homolog 2 inactivates C/EBPα and causes acute myelogenous leukemia. Cancer Cell. 2006;10:401–11.
Keeshan K, Bailis W, Dedhia PH, Vega ME, Shestova O, Xu L, et al. Transformation by Tribbles homolog 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood. 2010;116:4948–57.
Yoshida A, Kato JY, Nakamae I, Yoneda-Kato N. COP1 targets C/EBPα for degradation and induces acute myeloid leukemia via Trib1. Blood. 2013;122:1750–60.
Umek RM, Friedman AD, McKnight SL. CCAAT-enhancer binding protein: a component of a differentiation switch. Science. 1991;251:288–92.
Timchenko NA, Wilde M, Nakanishi M, Smith JR, Darlington GJ. CCAAT/enhancer-binding protein α (C/EBP α) inhibits cell proliferation through the p21 (WAF-1/CIP-1/SDI-1) protein. Genes Dev. 1996;10:804–15.
Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ, et al. C/EBPα arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell. 2001;8:817–28.
Wang QF, Cleaves R, Kummalue T, Nerlov C, Friedman AD. Cell cycle inhibition mediated by the outer surface of the C/EBPα basic region is required but not sufficient for granulopoiesis. Oncogene. 2003;22:2548–57.
Wang GL, Iakova P, Wilde M, Awad S, Timchenko NA. Liver tumors escape negative control of proliferation via PI3 K/Akt-mediated block of C/EBPα growth inhibitory activity. Genes Dev. 2004;18:912–25.
Porse BT, Pedersen TA, Hasemann MS, Schuster MB, Kirstetter P, Luedde T, et al. The proline-histidine-rich CDK2/CDK4 interaction region of C/EBPα is dispensable for C/EBPα-mediated growth regulation in vivo. Mol Cell Biol. 2006;26:1028–37.
Pulikkan JA, Peramangalam PS, Dengler V, Ho PA, Preudhomme C, Meshinchi S, et al. C/EBPα regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood. 2010;116:5638–49.
Dooher JE, Paz-Priel I, Houng S, Baldwin AS Jr, Friedman AD. C/EBPα, C/EBPα oncoproteins, or C/EBPβ preferentially bind NF-κB p50 compared with p65, focusing therapeutic targeting on the C/EBP:p50 interaction. Mol Cancer Res. 2011;9:1395–405.
Ye M, Zhang H, Amabile G, Yang H, Staber PB, Zhang P, et al. C/EBPα controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat Cell Biol. 2013;15:385–94.
Hasemann MS, Lauridsen FK, Waage J, Jakobsen JS, Frank AK, Schuster MB, et al. C/EBPα is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet. 2014;10:e1004079.
Birkenmeier EH, Gwynn B, Howard S, Jerry J, Gordon JI, Landschulz WH, et al. Tissue-specific expression, developmental regulation, and genetic mapping of the gene encoding CCAAT/enhancer binding protein. Genes Dev. 1989;3:1146–56.
Flodby P, Barlow C, Kylefjord H, Ahrlund-Richter L, Xanthopoulos KG. Increased hepatic cell proliferation and lung abnormalities in mice deficient in CCAAT/enhancer binding protein α. J Biol Chem. 1996;271:24753–60.
Scott LM, Civin CI, Rorth P, Friedman AD. A novel temporal expression pattern of three C/EBP family members in differentiating myelomonocytic cells. Blood. 1992;80:1725–35.
Iwasaki H, Mizuno S, Arinobu Y, Ozawa H, Mori Y, Shigematsu H, et al. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 2006;20:3010–21.
Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBPα. Immunity. 2004;21:853–63.
Wang ND, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV, Wilde MD, Taylor LR, Wilson DR, Darlington GJ. Impaired energy homeostasis in C/EBPα knockout mice. Science. 1995;269:1108–12.
Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein α-deficient mice. Proc Natl Acad Sci USA. 1997;94:569–74.
Heath V, Suh HC, Holman M, Renn K, Gooya JM, Parkin S, et al. C/EBPα deficiency results in hyperproliferation of hematopoietic progenitor cells and disrupts macrophage development in vitro and in vivo. Blood. 2004;104:1639–47.
Ma O, Hong S, Guo H, Ghiaur G, Friedman AD. Granulopoiesis requires increased C/EBPα compared to monopoiesis, correlated with elevated Cebpa in immature G-CSF receptor versus M-CSF receptor expressing cells. PLoS One. 2014;9:e95784.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89.
Wang D, D’Costa J, Civin CI, Friedman AD. C/EBPα directs monocytic commitment of primary myeloid progenitors. Blood. 2006;108:1223–9.
Fukuchi Y, Shibata F, Ito M, Goto-Koshino Y, Sotomaru Y, Ito M, et al. Comprehensive analysis of myeloid lineage conversion using mice expressing an inducible form of C/EBPα. EMBO J. 2006;25:3398–410.
Suh HC, Gooya J, Renn K, Friedman AD, Johnson PF, Keller JR. C/EBPα determines hematopoietic cell fate in multipotential progenitor cells by inhibiting erythroid differentiation and inducing myeloid differentiation. Blood. 2006;107:4308–16.
Wang X, Scott E, Sawyers CL, Friedman AD. C/EBPα bypasses granulocyte colony-stimulating factor signals to rapidly induce PU.1 gene expression, stimulate granulocytic differentiation, and limit proliferation in 32D cl3 myeloblasts. Blood. 1999;94:560–71.
Cammenga J, Mulloy JC, Berguido FJ, MacGrogan D, Viale A, Nimer SD. Induction of C/EBPα activity alters gene expression and differentiation of human CD34+ cells. Blood. 2003;101:2206–14.
Lidonnici MR, Audia A, Soliera AR, Prisco M, Ferrari-Amorotti G, Waldron T, et al. Expression of the transcriptional repressor Gfi-1 is regulated by C/ΕΒPα and is involved in its proliferation and colony formation-inhibitory effects in p210BCR/ABL-expressing cells. Cancer Res. 2010;70:7949–59.
Federzoni EA, Humbert M, Torbett BE, Behre G, Fey MF, Tschan MP. CEBPA-dependent HK3 and KLF5 expression in primary AML and during AML differentiation. Sci Rep. 2014;4:4261.
Lekstrom-Himes JA, Dorman SE, Kopar P, Holland SM, Gallin JI. Neutrophil-specific granule deficiency results from a novel mutation with loss of function of the transcription factor CCAAT/enhancer binding protein ε. J Exp Med. 1999;189:1847–52.
Karsunky H, Zeng H, Schmidt T, Zevnik B, Kluge R, Schmid KW, et al. Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet. 2002;30:295–300.
Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, et al. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity. 2003;18:109–20.
Diakiw SM, Kok CH, To LB, Lewis ID, Brown AL, D’Andrea RJ. The granulocyte-associated transcription factor Krüppel-like factor 5 is silenced by hypermethylation in acute myeloid leukemia. Leuk Res. 2012;36:110–6.
Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPα regulates human granulopoiesis. Cell. 2005;123:819–31.
Katzerke C, Madan V, Gerloff D, Bräuer-Hartmann D, Hartmann JU, Wurm AA, et al. Transcription factor C/EBPα-induced microRNA-30c inactivates Notch1 during granulopoiesis and is downregulated in acute myeloid leukemia. Blood. 2013;122:2433–42.
Friedman AD. Transcriptional regulation of granulocyte and monocyte development. Oncogene. 2002;21:3377–90.
Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene. 2007;26:6816–28.
Christy RJ, Kaestner KH, Geiman DE, Lane MD. CCAAT/enhancer binding protein gene promoter: binding of nuclear factors during differentiation of 3T3-L1 preadipocytes. Proc Natl Acad Sci USA. 1991;88:2593–7.
Guo H, Ma O, Speck NA, Friedman AD. Runx1 deletion or dominant inhibition reduces Cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood. 2012;119:4408–18.
Wilson NK, Foster SD, Wang X, Knezevic K, Schütte J, Kaimakis P, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7:532–44.
Huang Y, Sitwala K, Bronstein J, Sanders D, Dandekar M, Collins C, et al. Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood. 2012;119:388–98.
Collins C, Wang J, Miao H, Bronstein J, Nawer H, Xu T, et al. C/EBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc Natl Acad Sci USA. 2014;111:9899–904.
Guo H, Ma O, Friedman AD. The Cebpa +37-kb enhancer directs transgene expression to myeloid progenitors and to long-term hematopoietic stem cells. J Leukoc Biol. 2014;96:419–26.
Wölfler A, Danen-van Oorschot AA, Haanstra JR, Valkhof M, Bodner C, Vroegindeweij E, et al. Lineage-instructive function of C/EBPα in multipotent hematopoietic cells and early thymic progenitors. Blood. 2010;116:4116–25.
Skokowa J, Cario G, Uenalan M, Schambach A, Germeshausen M, Battmer K, et al. LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat Med. 2006;12:1191–7.
Seifeddine R, Dreiem A, Blanc E, Fulchignoni-Lataud MC, Le Frère Belda MA, Lecuru F, et al. Hypoxia down-regulates CCAAT/enhancer binding protein-α expression in breast cancer cells. Cancer Res. 2008;68:2158–65.
Jiang Y, Xue ZH, Shen WZ, Du KM, Yan H, Yu Y, et al. Desferrioxamine induces leukemic cell differentiation potentially by hypoxia-inducible factor-1 α that augments transcriptional activity of CCAAT/enhancer-binding protein-α. Leukemia. 2005;19:1239–47.
Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. 2013;503:371–6.
Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic cytokines can instruct lineage choice. Science. 2009;325:217–8.
Jack GD, Zhang L, Friedman AD. M-CSF elevates c-Fos and phospho-C/EBPα(S21) via ERK whereas G-CSF stimulates SHP2 phosphorylation in marrow progenitors to contribute to myeloid lineage specification. Blood. 2009;114:2172–80.
Buchwalter G, Gross C, Wasylyk B. Ets ternary complex transcription factors. Gene. 2004;324:1–14.
Geest CR, Buitenhuis M, Laarhoven AG, Bierings MB, Bruin MC, Vellenga E, et al. p38 MAP kinase inhibits neutrophil development through phosphorylation of C/EBPα on serine 21. Stem Cells. 2009;27:2271–82.
Köffel R, Meshcheryakova A, Warszawska J, Hennig A, Wagner K, Jörgl A, et al. Monocytic cell differentiation from band-stage neutrophils under inflammatory conditions via MKK6 activation. Blood. 2014;124:2713–24.
Huang W, Horvath E, Eklund EA. PU.1, interferon regulatory factor (IRF) 2, and the interferon consensus sequence-binding protein (ICSBP/IRF8) cooperate to activate NF1 transcription in differentiating myeloid cells. J Biol Chem. 2007;282:6629–43.
Huang H, Woo AJ, Waldon Z, Schindler Y, Moran TB, Zhu HH, et al. A Src family kinase-Shp2 axis controls RUNX1 activity in megakaryocyte and T-lymphocyte differentiation. Genes Dev. 2012;26:1587–601.
Futami M, Zhu QS, Whichard ZL, Xia L, Ke Y, Neel BG, et al. G-CSF receptor activation of the Src kinase Lyn is mediated by Gab2 recruitment of the Shp2 phosphatase. Blood. 2011;118:1077–86.
Yuan H, Zhou J, Deng M, Zhang Y, Chen Y, Jin Y, et al. Sumoylation of CCAAT/enhancer-binding protein α promotes the biased primitive hematopoiesis of zebrafish. Blood. 2011;117:7014–20.
Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–7.
Rosenbauer F, Wagner K, Kutok JL, Iwasaki H, Le Beau MM, Okuno Y, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat Genet. 2004;36:624–30.
Huang G, Zhang P, Hirai H, Elf S, Yan X, Chen Z, et al. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat Genet. 2008;40:51–60.
Kummalue T, Friedman AD. Cross-talk between regulators of myeloid development: C/EBPα binds and activates the promoter of the PU.1 gene. J Leukoc Biol. 2003;74:464–70.
Yeamans C, Wang D, Paz-Priel I, Torbett BE, Tenen DG, Friedman AD. C/EBPα binds and activates the PU.1 distal enhancer to induce monocyte lineage commitment. Blood. 2007;110:3136–42.
Rangatia J, Vangala RK, Treiber N, Zhang P, Radomska H, Tenen DG, et al. Downregulation of c-Jun expression by transcription factor C/EBPα is critical for granulocytic lineage commitment. Mol Cell Biol. 2002;22:8681–94.
Reddy VA, Iwama A, Iotzova G, Schulz M, Elsasser A, Vangala RK, et al. Granulocyte inducer C/EBPα inactivates the myeloid master regulator PU.1: possible role in lineage commitment decisions. Blood. 2002;100:483–90.
Oelgeschläger M, Nuchprayoon I, Lüscher B, Friedman AD. C/EBP, c-Myb, and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol Cell Biol. 1996;16:4717–25.
Kurotaki D, Yamamoto M, Nishiyama A, Uno K, Ban T, Ichino M, et al. IRF8 inhibits C/EBPα activity to restrain mononuclear phagocyte progenitors from differentiating into neutrophils. Nat Commun. 2014;5:4978.
Hsu CL, King-Fleischman AG, Lai AY, Matsumoto Y, Weissman IL, Kondo M. Antagonistic effect of CCAAT enhancer-binding protein-α and Pax5 in myeloid or lymphoid lineage choice in common lymphoid progenitors. Proc Natl Acad Sci USA. 2006;103:672–7.
Rekhtman N, Radparvar F, Evans T, Skoultchi AI. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev. 1999;13:1398–411.
Zhang P, Behre G, Pan J, Iwama A, Wara-Aswapati N, Radomska HS, et al. Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proc Natl Acad Sci USA. 1999;96:8705–10.
Querfurth E, Schuster M, Kulessa H, Crispino JD, Döderlein G, Orkin SH, et al. Antagonism between C/EBPβ and FOG in eosinophil lineage commitment of multipotent hematopoietic progenitors. Genes Dev. 2000;14:2515–25.
Sharabi AB, Aldrich M, Sosic D, Olson EN, Friedman AD, Lee SH, et al. Twist-2 controls myeloid lineage development and function. PLoS Biol. 2008;6:e316.
Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-α (C/EBPα), in acute myeloid leukemia. Nat Genet. 2001;27:263–70.
Gombart AF, Hofmann WK, Kawano S, Takeuchi S, Krug U, Kwok SH, et al. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein α in myelodysplastic syndromes and acute myeloid leukemias. Blood. 2002;99:1332–40.
Paz-Priel I, Friedman A. C/EBPα dysregulation in AML and ALL. Crit Rev Oncog. 2011;16:93–102.
Fuchs O, Provaznikova D, Kocova M, Kostecka A, Cvekova P, Neuwirtova R, et al. CEBPA polymorphisms and mutations in patients with acute myeloid leukemia, myelodysplastic syndrome, multiple myeloma and non-Hodgkin’s lymphoma. Blood Cells Mol Dis. 2008;40:401–5.
Kato N, Kitaura J, Doki N, Komeno Y, Watanabe-Okochi N, Togami K, et al. Two types of C/EBPα mutations play distinct but collaborative roles in leukemogenesis: lessons from clinical data and BMT models. Blood. 2011;117:221–33.
Kirstetter P, Schuster MB, Bereshchenko O, Moore S, Dvinge H, Kurz E, et al. Modeling of C/EBPα mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13:299–310.
Schuster MB, Frank AK, Bagger FO, Rapin N, Vikesaa J, Porse BT. Lack of the p42 form of C/EBPα leads to spontaneous immortalization and lineage infidelity of committed myeloid progenitors. Exp Hematol. 2013;41:882–93.
Cleaves R, Wang QF, Friedman AD. C/EBPαp30, a myeloid leukemia oncoprotein, limits G-CSF receptor expression but not terminal granulopoiesis via site-selective inhibition of C/EBP DNA binding. Oncogene. 2004;23:716–25.
Wang C, Chen X, Wang Y, Gong J, Hu G. C/EBPα30 plays transcriptional regulatory roles distinct from C/EBPα42. Cell Res. 2007;17:374–83.
Geletu M, Balkhi MY, Peer Zada AA, Christopeit M, Pulikkan JA, Trivedi AK, et al. Target proteins of C/EBPαp30 in AML: C/EBPαp30 enhances sumoylation of C/EBPαp42 via up-regulation of Ubc9. Blood. 2007;110:3301–9.
Bereshchenko O, Mancini E, Moore S, Bilbao D, Månsson R, Luc S, et al. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPα mutant AML. Cancer Cell. 2009;16:390–400.
Wagner K, Zhang P, Rosenbauer F, Drescher B, Kobayashi S, Radomska HS, et al. Absence of the transcription factor CCAAT enhancer binding protein α results in loss of myeloid identity in bcr/abl-induced malignancy. Proc Natl Acad Sci USA. 2006;103:6338–43.
Ohlsson E, Hasemann MS, Willer A, Lauridsen FK, Rapin N, Jendholm J, et al. Initiation of MLL-rearranged AML is dependent on C/EBPα. J Exp Med. 2013;211:5–13.
Reckzeh K, Bereshchenko O, Mead A, Rehn M, Kharazi S, Jacobsen SE, et al. Molecular and cellular effects of oncogene cooperation in a genetically accurate AML mouse model. Leukemia. 2012;26:1527–36.
Togami K, Kitaura J, Uchida T, Inoue D, Nishimura K, Kawabata KC, et al. A C-terminal mutant of CCAAT-enhancer-binding protein α (C/EBPα-Cm) downregulates Csf1r, a potent accelerator in the progression of acute myeloid leukemia with C/EBPα-Cm. Exp Hematol. 2014 (in press).
Watanabe-Okochi N, Yoshimi A, Sato T, Ikeda T, Kumano K, Taoka K, et al. The shortest isoform of C/EBPβ, liver inhibitory protein (LIP), collaborates with Evi1 to induce AML in a mouse BMT model. Blood. 2013;121:4142–55.
Friedman AD. Leukemogenesis by CBF oncoproteins. Leukemia. 1999;13:1932–42.
Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G, et al. AML1-ETO downregulates the granulocytic differentiation factor C/EBPα in t(8;21) myeloid leukemia. Nat Med. 2001;7:444–51.
Liu P, Tarlé SA, Hajra A, Claxton DF, Marlton P, Freedman M, et al. Fusion between transcription factor CBFβ/PEBP2β and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041–4.
Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114:5352–61.
Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9 % of cases. Leukemia. 2011;25:557–60.
Vainchencker W, Delhoummeau F, Constantinescu SN, Bernard OA. New mutations and pathogenesis of myeloproliferative neoplasms. Blood. 2011;118:1723–35.
Harada H, Harada Y, Tanaka H, Kimura A, Inaba T. Implications of somatic mutations in the AML1 gene in radiation-associated and therapy-related myelodysplastic syndrome/acute myeloid leukemia. Blood. 2003;101:673–80.
Zhao LJ, Wang YY, Li G, Ma LY, Xiong SM, Weng XQ, et al. Functional features of RUNX1 mutants in acute transformation of chronic myeloid leukemia and their contribution to inducing murine full-blown leukemia. Blood. 2012;119:2873–82.
Cuenco GM, Ren R. Cooperation of BCR-ABL and AML1/MDS1/EVI1 in blocking myeloid differentiation and rapid induction of an acute myelogenous leukemia. Oncogene. 2001;20:8236–48.
Ptasinska A, Assi SA, Mannari D, James SR, Williamson D, Dunne J, et al. Depletion of RUNX1/ETO in t(8;21) AML cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia. 2012;26:1829–41.
Network The Cancer Genome Atlas Research. Genomic and epigenomic landscapes of adult de novo acute myeloid leuekemia. New Engl J Med. 2013;368:2059–74.
Döhner K, Tobis K, Bischof T, Hein S, Schlenk RF, Fröhling S, et al. Mutation analysis of the transcription factor PU.1 in younger adults (16–60 years) with acute myeloid leukemia: a study of the AML Study Group Ulm (AMLSG ULM). Blood. 2013;102:3850.
Musialik E, Bujko M, Kober P, Grygorowicz MA, Libura M, Przestrzelska M, et al. Comparison of promoter DNA methylation and expression levels of genes encoding CCAAT/enhancer binding proteins in AML patients. Leuk Res. 2014;38:850–6.
Wouters BJ, Jordà MA, Keeshan K, Louwers I, Erpelinck-Verschueren CA, Tielemans D, et al. Distinct gene expression profiles of acute myeloid/T-lymphoid leukemia with silenced CEBPA and mutations in NOTCH1. Blood. 2007;110:3706–14.
Alberich-Jordà M, Wouters B, Balastik M, Shapiro-Koss C, Zhang H, Di Ruscio A, et al. C/EBPγ deregulation results in differentiation arrest in acute myeloid leukemia. J Clin Invest. 2012;122:4490–504.
Zhang H, Alberich-Jorda M, Amabile G, Yang H, Staber PB, Di Ruscio A, et al. Sox4 is a key oncogenic target in C/EBPα mutant acute myeloid leukemia. Cancer Cell. 2013;24:575–88.
Dedhia PH, Keeshan K, Uljon S, Xu L, Vega ME, Shestova O, et al. Differential ability of Tribbles family members to promote degradation of C/EBPα and induce acute myelogenous leukemia. Blood. 2010;116:1321–8.
Liang KL, Rishi L, Keeshan K. Tribbles in acute leukemia. Blood. 2013;121:4265–70.
Radomska HS, Bassères DS, Zheng R, Zhang P, Dayaram T, Yamamoto Y, et al. Block of C/EBPα function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J Exp Med. 2006;203:371–81.
Radomska HS, Alberich-Jordà M, Will B, Gonzalez D, Delwel R, Tenen DG. Targeting CDK1 promotes FLT3-activated acute myeloid leukemia differentiation through C/EBPα. J Clin Invest. 2012;122:2955–66.
Perrotti D, Calabretta B. Post-transcriptional mechanisms in BCR/ABL leukemogenesis: role of shuttling RNA-binding proteins. Oncogene. 2002;21:8577–83.
Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, et al. C/EBPβ is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–9.
Jones LC, Lin ML, Chen SS, Krug U, Hofmann WK, Lee S, et al. Expression of C/EBPβ from the C/ebpα gene locus is sufficient for normal hematopoiesis in vivo. Blood. 2002;99:2032–6.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Friedman, A.D. C/EBPα in normal and malignant myelopoiesis. Int J Hematol 101, 330–341 (2015). https://doi.org/10.1007/s12185-015-1764-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-015-1764-6