
Abstract
Therapeutic options against the human immunodeficiency virus type 1 (HIV-1) continue to expand with the development of new drugs and new therapeutic strategies. Nevertheless, management of HIV-1 infected individuals has become increasingly complex. The emergence of drug-resistant variants, the growing recognition of the long-term toxicity of antiretroviral therapies and the persistence of viral reservoirs justify the continued efforts to develop new anti-HIV-1 strategies. Recent advances regarding the utility of RNA-mediated interference (RNAi) to specifically inhibit HIV-1 replication have opened new possibilities for the development of gene-based therapies against HIV-1 infection. Here, the recent advances in siRNA-based therapies are reviewed.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key Words:
17.1 1 Introduction: Alternatives to Conventional Antiretroviral Therapy
Since the first report on HIV-1-infected individuals in the New England Journal of Medicine in 1981 (1), nearly 40 million individuals have been infected. In 2006 alone four million people were infected, and over three million died of the acquired immunodeficiency syndrome (AIDS) (www.unaids.org). A unique feature of HIV-1 is the establishment of a pool of latently infected cells very early during primary infection, resulting in the indefinite establishment of HIV-1 infection in all infected individuals. This feature of HIV-1 infection places it in sharp contrast with almost all other viral infections, in which the initial rounds of viral replication do not establish a permanent reservoir of infection. Although a few HIV-1 infected individuals have remained healthy for nearly 18 years (2, 3) to date not a single spontaneous cure of HIV-1 infection has been confirmed, and infected individuals have either died or remained infected.
Therefore, HIV-1 poses a greater challenge to the development of classic vaccination or antiviral strategies geared to ultimately lead to the eradication of the virus. A safe and effective HIV-1 vaccine to stop the spread of HIV-1 has not yet been developed and the possibility of developing a successful vaccine in the near future remains to be seen (4).
The introduction of potent antiretroviral therapies has been an important achievement towards the control of HIV-1 infection and AIDS (5, see Table 17.1). Therapies to combat HIV-1 infection have resulted in a dramatic decrease in AIDS-associated morbidity and mortality in developed countries (6). Despite the development of successful therapeutic strategies employing treatment combinations, or highly active antiretroviral therapy (HAART), it has not been possible to eradicate HIV-1 in infected individuals, mainly due to the persistence of viral reservoirs (7, 8, 9, 10). The HIV-1 reservoir is not eradicated even after extended antiretroviral therapy that can reduce viremia to undetectable levels (<50 viral RNA copies per milliliter of plasma). Therefore, individuals infected with HIV-1 need to receive antiretroviral therapy for many years, if not for life. Current treatments target viral enzymes, such as the reverse transcriptase (RT) and protease, as well as the envelope glycoprotein gp41 (Table 17.1). The most recent antiretroviral approved for clinical use blocks the binding of HIV-1 to its co-receptor CCR5 in host cells (11). Despite the success of potent combination regimens, the development of HIV-1 drug resistance constitutes a major hurdle towards long-term efficacy of current antiretroviral therapies (12). Moreover, just several years after the implementation of HAART, the increase of HIV-1 drug resistance has required the initiation of a continuous effort in the development of new drugs, therapy strategies and viral targets. So far, 22 antiretroviral drugs have been approved and several others are in clinical or preclinical development (Table 17.1). Current limitations of HAART are not restricted to drug resistance. Toxicity and side effects, such as hyperlipidemia, hyperglycemia, hypersensitivity, pancreatitis, lipoatrophy, anemia and neutropenia, rash, diarrhoea, gastrointestinal distress, insulin resistance, immune reconstitution syndrome, renal dysfunction, hepatotoxicity and an increased risk of liver cirrhosis and myocardial infarction have all been described in response to treatment with antiretroviral drugs (13). Moreover, the likelihood of side effects increases due to the improved lifespan as a result of the success of HAART. Finally, coinfection with hepatitis C, hepatitis B, tuberculosis or malaria complicates the treatment regimens for HIV-1 infected individuals.
The limitations and challenges of current antiretroviral therapy justify the continued effort to develop new anti-HIV-1 strategies. Fire et al. first discovered that introducing long double-stranded RNA (dsRNA) into the nematode Caenorhabditis elegans led to the targeted degradation of homologous mRNA, revealing the existence of a fundamental mechanism now known as RNAi through which gene expression can be regulated (14). Later findings by Elbashir et al. showed that RNAi also occurs in mammalians cells (15). Importantly, this study made the extraordinary demonstration that cell transfection of synthetic 21 base pairs (bp) short interfering RNA (siRNA) duplexes can mediate RNAi in a sequence-specific manner; this finding enabled the specific regulation of gene expression in a variety of biological systems, including diseased cells.
In this review, I will focus on the progress in research employing RNAi to disrupt the disease process caused by HIV-1. Given that an exogenous RNA can be targeted without affecting cellular functions, one of the most promising applications of RNAi is in the treatment of infectious diseases. Nevertheless, there are some obstacles to the development of an anti-HIV-1 RNAi-based therapeutic agent. One major problem of all antiretroviral therapies is the emergence of resistant variants, and the enormous genomic heterogeneity of HIV-1 may hinder the efficacy of single defined siRNAs. Coexpression of multiple siRNAs could reduce the emergence of single-siRNA-resistant viruses, with an effect comparable to that achieved by HAART, which combines three- or four-anti-HIV-1 drugs in treatment. An alternative strategy to counter the possibility of siRNA-resistant viruses would be to target transcripts of cellular cofactors essential for HIV-1 infectivity or replication (e.g., specific cell surface receptors). The side effects of downregulating cellular targets over the long term, however, are currently unknown. Delivery remains a major hurdle for RNAi therapy, as siRNAs are unable to cross the mammalian cell membrane without aid. Gene-therapy vectors are capable of stably expressing siRNA precursors, although well-documented hazards have been observed after the introduction of foreign vector sequences into chromosomal DNA. Finally, off-target effects, such as the targeting of genes sharing partial homology to the siRNA and the stimulation of the innate immune system, need to be considered and avoided. Although the innate immune system is efficiently triggered only by dsRNAs more than 30 bp, high concentrations of smaller siRNAs may be able to activate this pathway. Nevertheless, there is no evidence that the activation of the innate immune system influences the degree or specificity of siRNAs.
17.2 2 Targeting HIV-1 Replication by RNAi
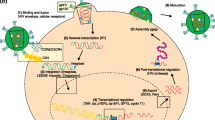
The HIV-1 life cycle begins by viral binding via the viral envelope to the cellular receptor CD4 in conjunction with a coreceptor, either CXCR4 in the case of T-cell-tropic virus or CCR5 for macrophage-tropic virus (Fig. 17.1). HIV-1 infects cells of the immune system, specifically CD4+ cells, which include T lymphocytes, monocytes and macrophages. As a result, in the absence of effective vaccination or therapy, a slow and continued depletion of CD4+ T cells ensues and there is a progression towards AIDS. Fusion of the viral and cellular membranes provides the HIV-1 genome, two RNA molecules of positive polarity, access to the interior of the cell. The genomic viral RNA is then converted into double-stranded DNA by the HIV-1 RT (Fig. 17.1). The viral DNA forms a pre-integration complex along with the viral integrase, which is then transported to the nucleus where the DNA is integrated into the host genome as a provirus. HIV-1 replication can be divided into early and late phases. During the early phase, spliced transcripts encoding two essential proteins, Tat and Rev, are synthesized. Tat further activates viral transcription, whereas Rev interacts with Rev-responsive elements (RRE) to facilitate nuclear export of unspliced and singly spliced mRNAs. These late mRNAs encode the remaining viral proteins, including Gag, Pol and Env, that assemble at the cell surface together with the viral genome to form virions that are released from the cell by budding. The viral protease continues to process the viral polyproteins into their mature form, condensing the viral genomic RNA core and yielding infectious particles (Fig. 17.1).

Schematic representation of the HIV-1 replication cycle. Virus attachment, reverse transcription, integration into the cellular genome, transcription, translation, virus assembly, virus budding and virus maturation are consecutively shown by arrows. siRNAs or shRNAs that target HIV-1 RNA might induce the cleavage of preintegrated genomic RNA or interfere with HIV-1 RNA transcripts postintegration and block progeny virus production
Soon after the demonstration that synthetic siRNAs were able to induce the RNAi mechanism in mammalian cells (15), several studies reported that HIV-1 gene expression and replication ex vivo could be inhibited by virus-specific synthetic siRNAs (16, 17, 18, 19, 20, 21, 22) or expressed siRNAs (16, 18) that were targeted to early or late phases of virus replication. siRNAs against several HIV-1 components (Gag, Env, Pol, the HIV-1 long terminal repeat, Vif, Nef, Tat and Rev) were confirmed to effectively target these genes. Moreover, CD4+ T cells (18, 23) and macrophages (24, 25), which are the natural targets for HIV-1 infection, were also found to be functional for RNAi. However, an important question arose from these pioneer studies: is the virion-associated incoming genomic viral RNA targeted by RNAi? Since HIV-1 is capable of being integrated in the host genome as well as infecting resting CD4+ T cells to create a pool of latently infected cells (4), the capacity to target incoming viral RNA by siRNAs has important therapeutic implications (Fig. 17.1) (26). If it is not possible to target incoming viral genomic RNA, it will be virtually impossible to prevent the formation of integrated provirus and, as a result, to sterilize cells from infection. Targeting the incoming viral RNA may have several advantages, as the RNAi machinery would only have to deal with two or a few (in the case of superinfection) genomic viral RNAs. However, after a provirus is established, several thousands of viral transcripts are generated de novo in the infected cell and the targeted degradation of these may be more difficult for the RNAi machinery. Data has been conflicting as to whether RNAi can target the incoming genomic viral RNA of infecting HIV-1 particles. Several studies have reported degradation of the incoming RNA genome in cells transfected with artificial siRNAs or stably expressing siRNAs (18, 19, 20, 27). However, other studies have reported an absence of RNAi-mediated degradation of the genomic viral RNA in siRNAs-transfected or siRNA-producing cells (22, 28, 29). Moreover, a study with the non-segmented, negative-strand RNA virus respiratory syncytial (RSV) showed that genomic and antigenomic viral RNAs encapsidated in the virus nucleocapsid protein were not susceptible to siRNA-mediated silencing, but viral mRNAs were highly susceptible (30).
The mechanisms underscoring the efficiency of specific siRNAs in targeting genes is not completely known. RNAi efficiency is known to be influenced by local RNA structure of the target sequence (31). Randomly selected siRNAs against a target sequence showed a large variation in their efficiency (32). It has been argued that some regions of genomic viral RNA might be less accessible to siRNAs when the RNA is contained in the virus reverse transcription complex; therefore, some siRNAs might be more effective than others in inhibiting this early step in virus replication (26). Recently, a more exhaustive study was performed to address this issue (33). In this study, the transduction efficiency of a lentiviral vector, as measured by the number of successful integration events, was determined in a cell line stably expressing an siRNA targeting the HIV-1 Nef sequence. The results indicated a similar transduction efficiency for vectors regardless of the presence or absence of the Nef target sequence in their genome. Moreover, no reduced transduction efficiencies were observed in the presence of multiple other stably expressed siRNAs targeting the vector genome, or when synthetic siRNA targeting Nef was transiently transfected prior to transduction. These results highlight the difficulties of designing therapeutic RNAi strategies aimed at preventing HIV-1 proviral integration. Silencing Tat, a factor that must be expressed before efficient provirus transcription can occur, may make it easier to target other viral transcripts by virtue of their lower abundance when Tat function is compromised (26).
Synthetic siRNAs mediate strong and specific but transient suppression of gene expression (18, 19, 20, 28). The transient reduction in gene expression by siRNAs severely restricts its applications in a therapeutic setting of a persistent infection, such as HIV-1 infection. However, this limitation was quickly overcome with the use of plasmids or viral vectors that expressed siRNAs from polymerase-III transcription units. The sense and antisense strands of an siRNA were expressed from two different promoters and RNAi was triggered upon annealing of the two strands (16). A more potent gene silencing was achieved when the sense and antisense strands were expressed as a single transcript with the ability to form a duplex hairpin structure (34). Indeed, HIV-1 replication could be efficiently inhibited with such short hairpin RNAs (shRNAs) in stably transduced cell lines (34, 35, 36, 37). An additional benefit in using shRNAs is that these can be processed by Dicer like the endogenous microRNAs (miRNAs). Tat siRNA delivered as a pre-miRNA precursor was 80% more effective in reducing HIV-1 p24 antigen production than Tat siRNA expressed as a conventional shRNA. Modeling shRNAs after endogenous miRNAs can increase the antiviral potency of RNAi (38).
As discussed in this section, an exogenously induced RNAi response in mammalian cells mediated by siRNAs or shRNAs can inhibit HIV-1 replication. These results raise the question of whether HIV-1 interacts with the cellular RNAi machinery, and understanding the potential physiological interaction between HIV-1 and the RNAi machinery could significantly contribute to the design of an effective RNAi-based therapy. RNAi constitutes a key component of the innate immune response to viral infection in both plants and invertebrate animals (39), and has been postulated to have a similar protective function in mammals (40). In plants and invertebrate animals, long dsRNA serves as the initial trigger for the RNAi mechanism; the initial dsRNA is then processed by Dicer into siRNAs. However, in mammalian cells, long dsRNA sequences (more than 30 bp in length) are potent inducers of the interferon (IFN) response (41). It remains unclear whether the introduction of long dsRNA into mammalian somatic cells is capable of resulting in the production of siRNAs (39). Nevertheless, RNAi can be induced in mammalian cells by the nuclear endogenous expression of RNAs that are intermediates in the miRNA biogenesis pathway (i.e., siRNAs and shRNAs). The human genome encodes several hundred different miRNA molecules that are believed to be key players in the posttranscriptional regulation of many aspects of cellular differentiation, and RNAi has been postulated to exist in mammals only as a mechanism of post-transcriptional regulation “programmed” by endogenously encoded miRNA, rather than being involved in intrinsic antiviral immunity (39).
All RNA viruses, except retroviruses, produce long dsRNA molecules in infected cells that represent essential intermediates in genomic RNA production. Many DNA viruses also generate long dsRNA, as their small packed genomes have convergent transcription promoters. Some studies have attempted to identify siRNAs in virus-infected human cells. Pfeffer et al. (42) failed to identify any viral siRNA in cells infected by DNA viruses, such as human cytomegalovirus, Kaposi sarcoma-associated herpes virus, Epstein-Barr virus and mouse herpes virus, as well as HIV-1 and the RNA viruses for yellow fever and hepatitis C. This report, however, did identify several virally encoded miRNA molecules in the DNA virus. In contrast, Bennasser et al. (43) reported that HIV-1 gives rise to an siRNA that can inhibit HIV-1 replication, and additionally proposed that the HIV-1 Tat protein relieves this inhibition by blocking the function of the cellular Dicer, which plays a key role in both siRNA and miRNA biogenesis. Moreover, the Ebola virus VP35 protein has been shown to be a suppressor of RNAi in mammalian cells and its suppressor activity is functionally equivalent to that of the HIV-1 Tat protein (44). VP35 can replace HIV-1 Tat and thereby support the replication of an HIV-1 variant deficient for Tat. These results support the hypothesis that RNAi is indeed part of the innate antiviral response in mammalian cells. However, in a recent report, the stable expression of physiological levels of Tat did not globally inhibit miRNA production or expression in HIV-1 infected human cells (45). Strong evidence for RNAi as part of the antiviral innate immune in plants comes from the demonstration that almost all plant viruses encode one or more suppressor of RNA silencing (SRS) proteins, which target several key steps in the RNAi response (40). Similarly, several viruses that target invertebrates (specifically nematodes and insects) also encode SRS proteins. Flock house virus, a member of the nodavirus family that infect both insects and vertebrate cells, encodes a viral SRS that inhibits Dicer function (46). Further evidence of the controversial relationship between HIV-1 and cellular RNAi machinery comes from the recent report by Triboulet et al. (47),which provides evidence for a physiological role of the miRNA-silencing machinery in controlling HIV-1 replication. This study showed that HIV-1 infection and replication were more efficient in peripheral blood mononuclear cells from HIV-1-infected donors and latently infected cells knocked-down for Dicer or Drosha, a miRNA processing factor. Moreover, HIV-1 actively suppressed the expression of the polycistronic miRNA cluster miR-17/92, and this suppression was found to be required for efficient viral replication. As suggested in this study, these findings may help address the current challenge of how to activate latent viral reservoirs in HIV-1 therapy. Virally encoded miRNAs have been discovered in herpes viruses (39), and human cytomegalovirus has been shown to evade the host immune system by targeting host genes with virally encoded miRNAs (48). Whether RNA viruses, including HIV-1, encode miRNAs still remains controversial (48). The therapeutic implications of this molecular feature are intriguing, as targeting these viral miRNAs might constitute an antiviral therapy while mimicking their role could provide a means of immunosuppressive therapy (48). Latent infection is one of the most important characteristics required for the in vivo survival of all HIV-1 strains and the major obstacle in eradicating virus infection with HAART (7–10). A recent study showed that cellular miRNAs potently inhibit HIV-1 production in resting primary CD4+ T cells (49); the 3′ ends of HIV-1 mRNAs were found to be targeted by a cluster of cellular miRNAs including miR-28, miR-125b, miR-150, miR-223 and miR-382, which are enriched in resting CD4+ T cells compared to activated CD4+ T cells (see Chap. 20). Because specific inhibitors of these miRNAs substantially counteracted their effects on the target mRNAs (49), a combined miRNA inhibitor panel could be used to activate latent HIV-1 for therapeutic purposes. In agreement with the former report, the cellular miRNAs miR-196, miR-296, miR-351, miR-431 and miR-448 have been shown to be capable of inhibiting hepatitis C virus replication and infection (50). These miRNAs are upregulated by IFNα/β, suggesting that cellular miRNAs may be components of the mammalian innate immune response (50).
17.3 3 Obstacles to Therapy
17.3.1 3.1 HIV-1 Escaping Inhibition by RNAi
This review highlights the evidence showing that RNAi provides a robust method for specifically inhibiting the expression of targeted HIV-1 genes, and its promise as a novel and broadly applicable approach to antiviral therapy. However, clinical application of RNAi faces several challenges, specifically the potential for viral escape. One of the main advantages of the RNAi mechanism is that it is highly sequence specific. Pioneer studies confirmed the specificity of RNAi by evaluating the activity of an siRNA with one or more mismatches relative to the target RNA sequence (18); these studies showed that a mismatch may be sufficient to reduce the silencing effect, suggesting that an unspecific antiviral response induced by siRNA was not involved in the silencing effect, but also showing that HIV-1 may easily escape inhibition by siRNAs. Indeed, HIV-1 has been observed to promptly escape suppression by effective siRNAs (36, 51, 52).
Unlike eukaryotic DNA polymerases, the HIV-1 RT lacks proofreading activity, and its error rate has been estimated at 10−4 to 10−5 mutations per nucleotide and cycle of replication (Table 17.2) (53). If one assumes that 109–1010 viral particles are produced each day in an infected person (54, 55), these must be the product of at least 107–108 replication cycles. Given the length of the HIV-1 genome (approximately 10,000 nucleotides) and its high recombination rate (56), it is likely that every single possible point mutation (and likely many double mutations) will occur at least once each day in an infected individual (57, 58). Although specific combinations of multiple mutations may be rare, it is clear that the degree of potential genetic change drives the diversification of HIV-1 in response to the selective pressure of host immune responses or antiretroviral therapy (Table 17.2).
HIV-1 strains have diversified extensively through mutation and recombination since their initial transmission to humans many decades ago in central Africa. Phylogenetic analysis of numerous isolates obtained from diverse geographic origins has allowed the division of HIV into types, groups, subtypes, sub-subtypes, circulating recombinant forms (CRFs) and unique recombinant forms (URFs). HIV-1 is divided into three groups, M, O and N (59). Most sequences within HIV-1 group M, which accounts for the majority of infections worldwide, fall into a limited number of discrete clades, allowing the classification into subtypes and sub-subtypes. Near 30 circulating genetic forms of the HIV-1 group M are presently recognized, including 11 subtypes and sub-subtypes, and nearly 20 CRF. The diverse HIV-1 subtypes, CRFs and the inter-subtype mosaic genomes further exacerbate the problem of designing effective siRNAs.
Boden et al. (51) characterized the potency and durability of virus-specific RNAi in cell lines that stably expressed shRNA targeting the HIV-1 transactivator protein gene Tat. They found that the antiviral activity of Tat shRNA was abolished due to the emergence of viral species harboring a point mutation in the shRNA target region. During the first three weeks, HIV-1 replication was reduced by 95% in cells expressing Tat shRNA compared to control cells. By day 25, however, viral titers increased, indicating loss of Tat shRNA-mediated antiviral activity, and sequencing of this virus stock revealed that the emerged viral species contained a nonsynonymous mutation at nucleotide position 9 of the targeted sequence. Das et al. (36) used retroviral transduction to stably introduce vectors expressing siRNAs directed against the HIV-1 Nef gene into human T cells; HIV-1 escape variants that were resistant to siRNA-Nef appeared after several weeks of culture, and these RNAi-resistant viruses contained point mutations, double point mutations or partial or complete deletion of the Nef gene target sequence. The complete inactivation of the accessory Nef gene has a relatively minor impact on the replication capacity of HIV-1 ex vivo. Interestingly, Westerhout et al. (52) used human T cells that stably express an siRNA-Nef target sequence to show that HIV-1 can also escape siRNA-mediated Nef inhibition by a point mutation outside the target sequence. This mutation changed local RNA folding, such that the target sequence becomes inaccessible to the RNAi machinery. Recently, Sabariegos et al. (60) showed that optimal HIV-1 gene silencing by siRNA requires precise complementarity with most of the target sequence and that only a few substitutions at the 5′ and 3′ ends are partially tolerated. Viral escape was simulated by systematically introducing single-nucleotide substitutions in all 19 HIV-1 residues targeted by an effective siRNA directed against the RT coding region. All mutant viruses that were tested replicated better in the presence of the siRNA-RT than in the presence of the wild type virus. The antiviral activity of the siRNA-RT was completely abolished by single substitutions in 10 (positions 4 to 11, 14, and 15) out of 16 positions tested (substitutions at 3 of the 19 positions rendered nonviable viruses). With the exception of one substitution, substitutions at either the 5′ or 3′ end were better tolerated by the RNA interference machinery and only partially affected siRNA-RT inhibition.
Since the emergence of resistant virus variants poses a serious problem for using RNAi in a therapeutic setting, different strategies have been developed to counteract viral escape. One obvious strategy that will successfully both inhibit a diversity of HIV-1 isolates and protect against the emergence of viral escape would be to identify viral conserved sequences for targeting. Targeting highly conserved sequences may hamper the development of escape mutants, as these variants may be highly compromised for viral fitness. Indeed, Lee et al. (37) targeted highly conserved HIV-1 Vif sequences and successfully suppressed a variety of primary viral isolates from five different viral clades. This study also showed that tolerance to target sequence mismatches may depend on the sequence of the siRNA tested (37). Nevertheless, our knowledge of HIV-1 variability and experience from over one decade of antiretroviral therapy have taught us that is unrealistic to try to target HIV-1 with a unique siRNA because the rapid development of resistance will occur. The high error rate of HIV-1 RT and the rapid viral turnover means that for each of the 19 nucleotides targeted by an si- or shRNA, several potential escape variants will be already present before the start of therapy (57). Moreover, HIV-1 may escape RNAi through silent mutations, which, in most cases, will not have any cost on the viral fitness. To counteract this strategic weakness, co-expression of multiple siRNAs or shRNAs that target conserved RNA sequences could reduce the emergence of single siRNA-resistant virus with a comparable effect to that achieved by the multiple anti-HIV drug combination approach employed by HAART. Delivery of multiple siRNAs could induce highly active antiretroviral gene silencing (HAAGS) (61).
The combination of multiple shRNAs against conserved HIV-1 genomic regions effectively inhibits HIV-1 replication. Expression of three different shRNAs from a single lentiviral vector resulted in similar levels of inhibition per shRNA compared to single shRNA vectors (62). In this study, the three shRNAs targeted Gag and Pol HIV-1 coding regions; moreover, when cells transduced with a double shRNA viral vector were infected, virus escape was delayed (62). There are several methods currently in use that are capable of expressing multiple effective siRNAs. One possibility is by inserting multiple shRNA-expression cassettes into a viral vector. However, repeats of the same regulatory sequences (e.g., the U6 or H1 polymerase III promoter) may cause genetic instability and reduced titer of the vector system (63). Moreover, some studies have shown that it is possible to saturate the RNAi pathway by high levels of expression of shRNAs (64, 65) resulting in cellular toxicity, particularly with the U6 promoter [64]. Polymerase III promoters other than U6 and H1 has been used (66). Another approach has been the use of long-hairpin RNAs (lhRNAs), from which multiple siRNAs can be produced. lhRNAs greater than 50 bp in length can be expressed in cells and create multiple siRNAs via Dicer-mediated processing without inducing the IFN pathway (67). Several reports described efficient RNAi induction by lhRNAs against HIV-1 (68, 69). Nishitsuji et al. (69) showed that a 50 bp lhRNA against a conserved HIV-1 integrase region suppressed HIV-1 replication in a variant resistant to a shorter shRNA for the same target. Similarly, lentiviral vectors expressing 50, 53, or 80 bp lhRNAs targeting contiguous sequences within the Tat and Rev genes inhibited viral replication against both non-mutant and mutant variants of HIV-1 (70).
An alternative to target multiple conserved viral genomic regions is to use a second generation of siRNAs that recognize the mutated target sites (60, 69, 71) by using siRNAs or shRNAs that target the most likely escape variants. Sabariegos et al. tested a second generation siRNA that compensates for a fully resistant mutation at position 9 of the target sequence (60). Nishitsuji et al. isolated single point mutation resistant viruses that emerged in lentiviral vector transduced cells expressing shRNAs against the HIV-1 U3 region, integrase and Tat genes (69) and restored viral inhibition by using a second generation shRNA that matched escape mutants (69). The use of cell culture infections to determine which target site mutations result in replication competent escape variants will allow the design of siRNAs capable of inhibiting these escape variants. To reduce the number of second generation siRNAs to be designed, this strategy should also target a conserved region of the HIV-1 coding sequence and the new siRNAs should also be capable of inducing RNAi. Recently, Escherichia coli endoribonuclease III (RNase III) or mammalian Dicer was used to cleave dsRNA into endoribonuclease-prepared siRNA (esiRNA) (72), which generate a variety of siRNAs that efficiently and specifically target multiple sites in the cognate RNA (73, 74). In this study, esiRNAs targeting the HIV-1 RT coding region reduced viral replication by 90% in a dose dependent and sequence specific manner. Importantly, esiRNAs obtained from the prototypic RT sequence of the HXB2 strain and from highly mutated RT sequences showed similar degrees of viral inhibition, suggesting that the heterogeneous population of esiRNAs could overcome individual mismatches in the RT sequence. These results demonstrated the ability of esiRNAs to function as potent HIV-1 inhibitors. Moreover, sequence targets do not need to be highly conserved to reach a high level of viral replication inhibition. Nevertheless, this work was performed in cell culture and its translation to an in vivo setting may be difficult.
Another recently explored strategy has been to combine an shRNA with an anti-HIV-1 ribozyme and an RNA decoy. A triple combination lentiviral construct comprised of a U6-driven TAR RNA decoy with a U16 snoRNA for nucleolar localization, a U6 promoted shRNA targeted to both Tat and Rev, and a VA1 promoted chimeric anti-CCR5 trans-cleaving hammerhead ribozyme efficiently transduced human progenitor CD34+ cells and improved suppression of HIV-1 over 42 days compared to a single anti-Tat/Rev shRNA or double combinations of shRNA/ribozyme or decoy (75). This triple combination is about to enter human clinical trials for AIDS/lymphoma patients using autologous hematopoietic progenitor cells as the targets for vector insertion. A second trial in which the same construct will be inserted in autologous T lymphocytes will most likely initiate in late 2007 or early 2008 (66).
In order to prevent provirus establishment and to augment the genetic barrier for viral escape, cellular receptors or co-factors involved in the initial phase of infection may be targeted. The combination of siRNAs or shRNAs directed against both HIV-1 and cellular transcripts may result in a more potent and robust inhibition of viral replication. Nevertheless, targeting a cellular cofactor required by the virus should be performed with caution as this approach has the potential to harm host cells. Several cellular factors involved in the HIV-1 life-cycle have been explored as possible RNAi targets: the CD4 receptor and coreceptors CCR5 and CXCR4 (17, 76, 77), integration factors like BAF-1, Emerin and LEDGF/75 (78, 79, 80), transcriptional factors such as NF-κB, PAK-1 and cyclin (22, 81, 82), and Furin which is involved in Env maturation (81). Since HIV-1 actively suppressed the expression of the polycistronic miRNA cluster miR-17/92 and this suppression was found to be required for efficient viral replication (47), nuclear expression of some miRNAs from this cluster may be another alternative to inhibit HIV-1 replication (66). In vivo suppression of genes such as CD4 could be limited due to roles in normal immune function, making HIV-1 coreceptors more attractive alternatives to target host proteins. CCR5 may be a potential coreceptor target, as a homozygous mutation in CCR5 effectively conferred protection from HIV-1 without any serious deleterious effects in human immune function, and heterozygous individuals with 50% decrease in CCR5 surface expression have lower plasma viral load and a substantially prolonged course of disease (83). A potent and noncytotoxic shRNA directed to CCR5 stably down-regulates CCR5 when introduced via CD34+ hematopoietic stem cell transplant in non-human primates (84) and these cells were less susceptible to simian immunodeficiency virus (SIV) infection ex vivo. Recently, several CCR5 antagonists examined in clinical trials were able to reduce plasma viral loads by more than one order of magnitude during treatment (11). Although, CCR5 antagonists hold great promise, the long-term toxicity associated with the impairment of CCR5 function remains to be addressed. Targeting cellular factors will require extensive toxicity studies.
17.3.2 3.2 In Vivo Delivery
Delivery remains a major hurdle for RNAi therapy, as siRNAs are unable to cross the mammalian cell membrane without aid. Most of the ex vivo transfection methods used for delivering siRNAs can not be used in vivo. There are two strategies for delivering siRNAs in vivo. As discussed in the previous section, one is to stably express siRNA precursors, such as shRNAs, from viral vectors using gene therapy technology; the other is to deliver synthetic siRNAs naked or by complexing or covalently linking the siRNA with lipids, aptamers, peptides or proteins (85). Intranasal administration of naked siRNAs either in saline or with excipients such as 5% dextrose or lung surfactants reduced the viral load of respiratory syncytial virus (RSV) and parainfluenza virus in pediatric or immuno-compromised individuals by more than three orders of magnitude (86, see Chap. 15). Similarly, siRNA intranasally administered in a non-human primate model of severe acute respiratory syndrome (SARS) corona virus infection significant inhibited viral replication in the lung (87). These studies clearly demonstrated the utility of RNAi to treat viral respiratory infections. Liposomes, vesicles with an aqueous compartment enclosed in a phospholipid bilayer that can fuse with cell membranes and enhance drug delivery into cells, have been extensively used to deliver siRNA in vitro and in vivo. In vitro transfection of siRNA using lipid-based delivery agents is a routine laboratory procedure. Lipid-based formulations effectively delivered siRNA to the liver in animal models of hepatitis B and Ebola virus infection (88, 89). Locally injected liposomes have also delivered siRNA effectively to target cells in the eye and nervous system, and to tumors (85). Topical delivery of siRNAs using liposomes may be especially effective. Intravaginal application of siRNAs protected mice from lethal herpes simple virus type 2 (HSV-2) sexually transmitted infection (90). The cervical/vaginal mucosa is the main port of HIV-1 entry in women, thus an effective topical microbicide against HIV-1 may be useful to prevent sexual transmission. Since the cellular coreceptor CCR5 is required for infection by the majority of primary HIV-1 isolates, a lipid-formulated siRNA against the CCR5 transcripts will be an excellent candidate for anti-HIV-1 microbicide.
siRNAs can be complexed with cationic peptides and polymers to form stable nanoparticles via ionic interactions with their negatively charged phosphate backbones (91). Similarly, one study showed that a protamine antibody fusion protein delivered non-covalently bound siRNA to HIV-1-infected primary CD4+ T cells in vitro with high efficiency (92). In this approach, the Fab fragment of an HIV-1 envelope antibody mediates receptor-specific binding to cells expressing the HIV-1 envelope protein, illustrating the potential for antibodies to also direct siRNA selectively into cells in vivo.
Given the HIV-1 life cycle, in which the persistence of a latent reservoir of resting infected cells makes eradication of the virus from infected individuals extremely difficult, the most promising strategy for in vivo delivery of siRNA is the use of gene therapy approaches. Because HIV-1 predominately infects T lymphocytes and macrophages, blood CD34+ hematopoietic stem cell transplant could be the best strategy, in which progeny cells would stably express siRNAs that down-regulate viral or cellular genes that are targets for HIV-1 infection. As noted before, HIV-1 can infect resting non-dividing CD4+ T cells, thus lentiviral vectors should be employed over murine retroviral vectors, as HIV-1-based lentiviral vectors have been shown to be particularly suited for the transduction of non-dividing cells, such as hematopoietic progenitor cells (93). These HIV-1-based vectors seem to be preferable candidates for gene therapy development in the treatment of HIV-1-infected individuals (94). However, shRNAs targeting highly conserved HIV-1 sequences may be an obstacle in vector production, as it has been reported that expression of shRNAs that target also the delivering vector can result in a reduction of the transduction titer (23), suggesting that these shRNAs could cross-react with critical sequences of the vector. Nevertheless, new lentiviral vectors have been developed to overcome this issue, and the vector backbone may be modified by point mutations to resist RNAi-mediated degradation during vector production (95). However, introducing RNAi-resistant mutations within the lentiviral vector backbone adds the potential risk of transfer of resistance to the wild type HIV-1 genome. One solution that has been suggested would be to incorporate stop codons within these vector sequences that should not affect the RNA/DNA function, yet would confer full resistance to RNAi (96). Recombination will result in an RNAi-resistant yet replication-defective HIV-1 variant due to the stop codons. Alternatively, it has also been shown that the incorporation of target sequences for endogenous miRNAs within the lentiviral genome can severely reduce potential recombination or mobilization (97). The HIV-1 genome is highly prone to recombination, and recombinants arise very frequently (56). Lentiviruses, unlike murine retroviruses, are more prone to integrate distally from promoters within intron sequences, potentially limiting their overall oncogenicity (98). One of the main concerns with current gene therapy approaches is insertional mutagenesis from preferential integration of retroviral vectors into actively transcribed genes, including proto-oncogenes. This has led to hematological malignancies in young individuals with severe immunodeficiency (SCID) disease treated with retroviral-based gene therapy (99). This problem could be addressed by designing vectors that integrate into specific, well-defined regions of the genome. The partially random insertion of transgenes into chromosomal DNA of hematopoietic cells may induce clonal competition, which could potentially trigger leukemia or sarcoma (100, 101).
Lentiviral vectors derived from HIV-1, HIV-2/SIV, or feline immunodeficiency virus (FIV) have been shown to be capable of stably transducing many cell types, including hematopoietic stem cells (102, 103). Interestingly, HIV-1 vectors have been shown to cross-package by FIV and are capable of stably transducing and protecting human primary blood mononuclear cells from HIV-1 infection (104). FIV packaged HIV-1 vectors reduce the likelihood of immune recognition, or seroconversion, due to exposure to HIV-1 structural proteins. A continuous effort is focused on producing effective, safe and high-titer lentiviral vectors to deliver RNAi.
17.3.3 3.3 Immunostimulation and Off-Target Toxicity by RNAi
Several important issues related to in vivo safety, toxicity or side effects warrant close attention before RNAi may be considered a valid alternative for treating HIV-1 infection or other diseases. In mammalian cells, dsRNA is recognized by dsRNA sensors such as Toll-like receptors (TLRs), the dsRNA-dependent protein kinase R (PKR) and retinoic-acid-inducible gene-I (RIG-I), which are components of the innate immune system. This recognition leads to the activation of the IFN-regulatory transcription factors and NF-kB, which in turn results in the expression of the IFNs (41) that subsequently activate the transcription of hundreds of IFN-stimulated genes (ISGs) through the JAK-STAT pathway. Many ISGs encode proteins with antiviral activities, including PKR. The IFN response plays a crucial role in antiviral immunity in vertebrates, particularly against RNA viruses that generate dsRNA molecules during their life cycle. The IFN response could significantly influence the in vivo application of siRNA owing to the off-target effects and toxicities associated with immune stimulation. siRNA molecules less than 30 bp in length are generally considered incapable of inducing IFN pathways (15). However, synthetic siRNAs formulated in non-viral delivery vehicles can be potent inducers of IFNs and inflammatory cytokines both in vivo in mouse and ex vivo in human blood cells (105). The immunostimulatory activity of formulated siRNAs and the associated toxicities may be dependent on the nucleotide sequence (106). Although some of the off-target effects can be reduced using lower concentration of siRNAs, the IFN response was observed even at low concentrations (107). These studies emphasize the relevance of examining the immunostimulatory effects of any potential therapeutic siRNA in human immune cells prior to clinical applications. Replacement of the 2′-hydroxyl uridines with either 2′-fluoro, or 2′-deoxy or 2′-O methyl uridines can abrogate immune recognition of siRNAs by TLRs without compromising siRNA silencing potency (108, 109, 110). In addition, modified nucleotides may protect siRNAs from nuclease degradation and ameliorate their pharmacokinetic parameters in vivo (111).
Similar to synthetic siRNAs, endogenously expressed shRNAs also activated the IFN pathway (112). Of particular concern is the development of a multiple shRNA or lhRNAs approach against HIV-1 because these molecules are longer than 30 bp. Endogenously (nuclear) expressed shRNAs were recently shown to evade detection by TLRs, RIG-1 and PKR when integrated in CD34+ progenitor hematopoietic stem cells (113). Endogenously expressed lhRNA encoding an effective Nef-specific shRNA was capable of inhibiting HIV-1 production without inducing the type I IFN genes (68). Endogenously produced dsRNA is suggested to be less active than exogenous dsRNA in inducing the IFN response (114). Extended shRNAs (e-shRNAs) of 40–44 bp that encode two effective siRNAs against conserved HIV-1 sequences, Pol and Nef, did not induce the IFN response in e-shRNA transfected 293T cells (63). A simple strategy for avoiding activation of the IFN response by dsRNA has been described (67), where modified hairpin-RNAs (mhRNAs) of more than 100 bp with multiple specific point-mutations within the sense strand and transcribed from the U6 or tRNA(Val) promoters can produce RNAi without inducing the IFN pathway genes (67, 115). The expression of lhRNAs also appears to be well tolerated and does not induce IFN gene activation in vivo when delivered to mice via by hydrodynamic tail-vein injection (116).
Another potential side effect of siRNAs and shRNAs is lethality associated with overdosing. A previous study in which mice were treated with high doses of a viral vector expressing shRNAs that resulted in high copy numbers per cell led to fatality due to oversaturation of the mi/siRNA pathways (65). This potential risk should be assessed prior to an eventual clinical RNAi application. Cells should be transduced with few or a single vector copy to avoid high expression levels that may induce unwanted side effects. Overdosing could also induce off-target effects in which siRNAs or shRNAs silence partially complementary transcripts through a miRNA-like mechanism. Cytotoxic effects of shRNAs in human T lymphocytes as a result of overexpression have also been reported (64). The risk of adverse effects of siRNAs would increase in situations where shRNA expression is to be maintained in a living organism for a long period, such as for intracellular immunization against HIV-1. In addition, transduced T cell lines and differentiated primary human T cells are relatively different from the hematopoietic CD34+ stem cells that will be transduced in an ultimate gene therapy therapeutic setting. Stem cells will develop into different lineages, and expression of shRNAs could influence their development. As suggested before (96), saturation of the miRNA pathway may disturb hematopoiesis, and indeed, miRNAs are involved in the regulation of genes that control hematopoiesis in the mouse (117). Whether in vivo gene silencing by siRNA or shRNAs can disrupt the endogenous miRNA pathway remains to be addressed. Recently, target genes were shown to be effectively silenced in the mouse and hamster liver by systemic administration of synthetic siRNA without any demonstrable effect on miRNA levels or activity (118). siRNAs targeting two hepatocyte-specific genes (apolipoprotein B and factor VII) were administered to mice and achieved efficient (80%) silencing of mRNA transcripts without significant changes in the levels of three hepatocyte-expressed miRNAs (miR-122, miR-16 and let-7a) (118). Moreover, multiple administrations of an siRNA targeting the hepatocyte-expressed gene Scap in hamsters achieved long-term mRNA silencing without significant changes in miR-122 levels (118). This study may advance the use of siRNAs as a safe therapeutic alternative.
Another potential side effect for siRNAs and shRNAs is the silencing of cellular mRNAs that share partial homology to the siRNA or shRNA sequences through an miRNA-like mechanism. This off-target effect requires complementarity between the siRNA seed region and the 3′untranslated region of a target mRNA (119, 121, 122). Any effective siRNA or shRNA may in theory have numerous potential off-target mRNAs, thus off-target silencing may not be easily eliminated by siRNA sequence selection. Moreover, the combining of multiple shRNAs would increase the number of off-target mRNAs potentially affected. Off-target transcript silencing may limit the specificity of siRNAs for therapeutic applications. One study showed that 2′-uridine modifications of siRNAs can reduce siRNAs off-target effects (123), and similarly, chemical modification also reduced off-target phenotypes in growth inhibition studies. Key to the modification was a 2′-O-methyl ribosyl substitution at position 2 in the guide strand, which reduced the silencing of most off-target transcripts exhibiting complementarity to the seed region of the siRNA guide strand (124).
Ex vivo selection of siRNAs to verify the absence of unwanted off-targets should help to define strategies to either enhance or avoid the non-specific effects of siRNAs in order to develop safe therapeutics. Studies with an appropriate animal model should also help preclinical assessment of safety and efficacy. HIV-1 research is largely restricted to in vitro, ex vivo or clinical studies, all limited in their ability to rapidly assess new strategies to treat virus infection. The humanized Rag2(−/−) gammac(−/−) SCID mouse model sustains long-term multi-lineage hematopoiesis and is capable of mounting immune responses, achieved by the intraperitoneal injection of CD34+ precursor cells into a newborn Rag2(−/−) gammac(−/−) mouse (125, 126). In this model, injecting p53 shRNA-transduced CD34+ cells resulted in stable expression and down-modulation of p53 in the mature T-cell offspring. Infection of HIV-1 in this mouse model resulted in high viremia and CD4+ T cell depletion (127). Therefore, this in vivo animal model and/or others developed in the near future should be valuable to evaluate RNAi-based strategies aiming to prevent or treat HIV-1 infection.
17.4 4 Conclusions
The discovery of RNAi has provided us with powerful new tools for biological research and drug discovery, and RNAi is currently advancing from basic research to clinical trials. It has also expanded the direction of a new field of therapeutic strategies with the potential to treat a wide range of different diseases, including HIV-1. Several decades ago, adaptive immunity allowed the possibility of preventing the spread of many pathogenic human viruses. Nevertheless, the discovery of an effective preventive or therapeutic vaccine against HIV-1 remains elusive. RNAi therapeutics is rapidly arriving into clinical studies for many diseases, including several which are currently untreatable or difficult to treat with conventional drugs. Although promising studies and results have emerged over the recent years, the challenge remains as to whether RNAi will be suitable for treating or preventing HIV-1 infection.
References
Gottlieb, M.S., Schroff, R., Schanker, H.M. et al. (1981) Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N Engl J Med 305, 1425–1431.
Kirchhoff, F., Greenough, T.C., Brettler, D.B., Sullivan, J.L., and Desrosiers, R.C. (1995) Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med 332, 228–232.
Learmont, J.C., Geczy, A.F., Mills, J. et al. (1999) Immunologic and virologic status after 14 to 18 years of infection with an attenuated strain of HIV-1. A report from the Sydney Blood Bank Cohort. N Engl J Med 340, 1715–1722.
Johnston, M.I. and Fauci, A.S. (2007) An HIV vaccine--evolving concepts. N Engl J Med 356, 2073–2081.
Ho, D.D. (1995) Time to hit HIV, early and hard. N Engl J Med 333, 450–451.
Walensky, R.P., Paltiel, A.D., Losina, E. et al. (2006) The survival benefits of AIDS treatment in the United States. J Infect Dis 194, 11–19.
Chun, T.W., Stuyver, L., Mizell, S.B. et al. (1997) Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 94, 13193–13197.
Finzi, D., Hermankova, M., Pierson, T. et al. (1997) Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300.
Wong, J.K., Hezareh, M., Gunthard, H.F. et al. (1997) Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278, 1291–1295.
Ibanez, A., Puig, T., Elias, J., Clotet, B., Ruiz, L., and Martinez, M.A. (1999) Quantification of integrated and total HIV-1 DNA after long-term highly active antiretroviral therapy in HIV-1-infected patients. AIDS 13, 1045–1049.
Este, J.A. and Telenti, A. (2007) HIV entry inhibitors. Lancet 370, 81–88.
Johnson, V.A., Brun-Vezinet, F., Clotet, B. et al. (2007) Update of the Drug Resistance Mutations in HIV-1: 2007. Top HIV Med 15, 119–125.
Deeks, S.G. (2006) Antiretroviral treatment of HIV infected adults. BMJ 332, 1489.
Fire, A., Xu, S., Montgomery, M.K., Kostas, S.A., Driver, S.E., and Mello, C.C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811.
Elbashir, S.M., Harborth, J., Lendeckel, W., Yalcin, A., Weber, K., and Tuschl, T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498.
Lee, N.S., Dohjima, T., Bauer, G. et al. (2002) Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol 20, 500–505.
Novina, C.D., Murray, M.F., Dykxhoorn, D.M. et al. (2002) siRNA-directed inhibition of HIV-1 infection. Nat Med 8, 681–686.
Jacque, J.M., Triques, K., and Stevenson, M. (2002) Modulation of HIV-1 replication by RNA interference. Nature 418, 435–438.
Coburn, G.A. and Cullen, B.R. (2002) Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J Virol 76, 9225–9231.
Capodici, J., Kariko, K., and Weissman, D. (2002) Inhibition of HIV-1 infection by small interfering RNA-mediated RNA interference. J Immunol 169, 5196–5201.
Park, W.S., Miyano-Kurosaki, N., Hayafune, M. et al. (2002) Prevention of HIV-1 infection in human peripheral blood mononuclear cells by specific RNA interference. Nucleic Acids Res 30, 4830–4835.
Surabhi, R.M. and Gaynor, R.B. (2002) RNA interference directed against viral and cellular targets inhibits human immunodeficiency virus Type 1 replication. J Virol 76, 12963–12973.
Banerjea, A., Li, M.J., Bauer, G. et al. (2003) Inhibition of HIV-1 by lentiviral vector-transduced siRNAs in T lymphocytes differentiated in SCID-hu mice and CD34+ progenitor cell-derived macrophages. Mol Ther 8, 62–71.
Song, E., Lee, S.K., Dykxhoorn, D.M. et al. (2003) Sustained small interfering RNA-mediated human immunodeficiency virus type 1 inhibition in primary macrophages. J Virol 77, 7174–7181.
Lee, M.T., Coburn, G.A., McClure, M.O., and Cullen, B.R. (2003) Inhibition of human immunodeficiency virus type 1 replication in primary macrophages by using Tat- or CCR5-specific small interfering RNAs expressed from a lentivirus vector. J Virol 77, 11964–11972.
Stevenson, M. (2003) Dissecting HIV-1 through RNA interference. Nat Rev Immunol 3, 851–858.
Joshi, P.J., North, T.W., and Prasad, V.R. (2005) Aptamers directed to HIV-1 reverse transcriptase display greater efficacy over small hairpin RNAs targeted to viral RNA in blocking HIV-1 replication. Mol Ther 11, 677–686.
Hu, W.Y., Myers, C.P., Kilzer, J.M., Pfaff, S.L., and Bushman, F.D. (2002) Inhibition of retroviral pathogenesis by RNA interference. Curr Biol 12, 1301–1311.
Nishitsuji, H., Ikeda, T., Miyoshi, H., Ohashi, T., Kannagi, M., and Masuda, T. (2004) Expression of small hairpin RNA by lentivirus-based vector confers efficient and stable gene-suppression of HIV-1 on human cells including primary non-dividing cells. Microbes Infect 6, 76–85.
Bitko, V. and Barik, S. (2001) Phenotypic silencing of cytoplasmic genes using sequence-specific double-stranded short interfering RNA and its application in the reverse genetics of wild type negative-strand RNA viruses. BMC Microbiol 1, 34–40
Westerhout, E.M. and Berkhout, B. (2007) A systematic analysis of the effect of target RNA structure on RNA interference. Nucleic Acids Res 35, 4322–4330.
Holen, T., Amarzguioui, M., Wiiger, M.T., Babaie, E., and Prydz, H. (2002) Positional effects of short interfering RNAs targeting the human coagulation trigger Tissue Factor. Nucleic Acids Res 30, 1757–1766.
Westerhout, E.M., ter Brake, O., and Berkhout, B. (2006) The virion-associated incoming HIV-1 RNA genome is not targeted by RNA interference. Retrovirology 3, 57.
Brummelkamp, T.R., Bernards, R., and Agami, R. (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550–553.
Boden, D., Pusch, O., Lee, F., Tucker, L., and Ramratnam, B. (2004) Efficient gene transfer of HIV-1-specific short hairpin RNA into human lymphocytic cells using recombinant adeno-associated virus vectors. Mol Ther 9, 396–402.
Das, A.T., Brummelkamp, T.R., Westerhout, E.M. et al. (2004) Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol 78, 2601–2605.
Lee, S.K., Dykxhoorn, D.M., Kumar, P. et al. (2005) Lentiviral delivery of short hairpin RNAs protects CD4 T cells from multiple clades and primary isolates of HIV. Blood 106, 818–826.
Silva, J.M., Li, M.Z., Chang, K. et al. (2005) Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet 37, 281–1288.
Cullen, B.R. (2006) Is RNA interference involved in intrinsic antiviral immunity in mammals. Nat Immunol 7, 563–567.
Voinnet, O. (2005) Induction and suppression of RNA silencing: insights from viral infections. Nat Rev Genet. 6, 206–220.
Garcia-Sastre, A. and Biron, C.A. (2006) Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312, 879–882.
Pfeffer, S., Sewer, A., Lagos-Quintana, M. et al. (2005) Identification of microRNAs of the herpesvirus family. Nat Methods 2, 269–276.
Bennasser, Y., Le, S.Y., Benkirane, M., and Jeang, K.T. (2005) Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 22, 607–619.
Haasnoot, J., de Vries, W., Geutjes, E.J., Prins, M., de Haan, P., and Berkhout, B. (2007) The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog 3, e86.
Lin , J. and Cullen, B.R. (2007) Analysis of the Interaction of Primate Retroviruses with the Human RNA Interference Machinery. J Virol 81:12218–26
Li, H., Li, W.X., and Ding, S.W. (2002) Induction and suppression of RNA silencing by an animal virus. Science 296, 1319–1321.
Triboulet, R., Mari, B., Lin, Y.L. et al. (2007) Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 315, 1579–1582.
Stern-Ginossar, N., Elefant, N., Zimmermann, A. et al. (2007) Host immune system gene targeting by a viral miRNA. Science 317, 376–381.
Huang, J., Wang, F., Argyris, E. et al. (2007) Cellular microRNAs contribute to HIV-1 latency in resting primary CD4(+) T lymphocytes. Nat Med 13, 1241–1247.
Pedersen, I.M., Cheng, G., Wieland, S., et al. (2007) Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449, 919–922.
Boden, D., Pusch, O., Lee, F., Tucker, L., and Ramratnam, B. (2003) Human immunodeficiency virus type 1 escape from RNA interference. J Virol 77, 11531–11535.
Westerhout, E.M., Ooms, M., Vink, M., Das, A.T., and Berkhout, B. (2005) HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res 33, 796–804.
Mansky, L.M. and Temin, H.M. (1995) Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol 69, 5087–5094.
Ho, D.D., Neumann, A.U., Perelson, A.S., Chen, W., Leonard, J.M., and Markowitz, M. (1995) Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123–126.
Wei, X., Ghosh, S.K., Taylor, M.E. et al. (1995) Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373, 117–122.
Jung, A., Maier, R., Vartanian, J.P. et al. (2002) Multiply infected spleen cells in HIV patients. Nature 418, 144.
Coffin, J.M. (1995) HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267, 483–489.
Domingo, E., Martin, V., Perales, C., Grande-Perez, A., Garcia-Arriaza, J., and Arias, A. (2006) Viruses as quasispecies: biological implications. Curr Top Microbiol Immunol 299, 51–82.
Leitner, T., Foley, B., Hahn, B. et al. (2007) HIV Sequence Compendium 2006/2007. Published by Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, LA-UR number 07-4826. 2007.
Sabariegos, R., Gimenez-Barcons, M., Tapia, N., Clotet, B., and Martinez, M.A. (2006) Sequence homology required by human immunodeficiency virus type 1 to escape from short interfering RNAs. J Virol 80, 571–577.
Martinez, M.A., Clotet, B., and Este, J.A. (2002) RNA interference of HIV replication. Trends Immunol 23, 559–561.
ter Brake, O., Konstantinova, P., Ceylan, M., and Berkhout, B. (2006) Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Mol Ther 14, 883–892.
Liu, Y.P., Haasnoot, J., and Berkhout, B. (2007) Design of extended short hairpin RNAs for HIV-1 inhibition. Nucleic Acids Res 35, 5683–5693.
An, D.S., Qin, F.X., Auyeung, V.C. et al. (2006) Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol Ther 14, 494–504.
Grimm, D., Streetz, K.L., Jopling, C.L. et al. (2006) Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441, 537–541.
Scherer, L.J., Frank, R., and Rossi, J.J. (2007) Optimization and characterization of tRNA-shRNA expression constructs. Nucleic Acids Res 35, 2620–2628.
Akashi, H., Miyagishi, M., Yokota, T. et al. (2005) Escape from the interferon response associated with RNA interference using vectors that encode long modified hairpin-RNA. Mol Biosyst 1, 382–390.
Konstantinova, P., de Vries, W., Haasnoot, J., ter Brake, O., and de Haan, P., Berkhout, B. (2006) Inhibition of human immunodeficiency virus type 1 by RNA interference using long-hairpin RNA. Gene Ther 13, 1403–1413.
Nishitsuji, H., Kohara, M., Kannagi, M., and Masuda, T. (2006) Effective suppression of human immunodeficiency virus type 1 through a combination of short- or long-hairpin RNAs targeting essential sequences for retroviral integration. J Virol 80, 7658–7666.
Sano , M., Li, H., Nakanishi, M., and Rossi, J.J. (2007) Expression of long anti-HIV-1 hairpin RNAs for the generation of multiple siRNAs: Advantages and limitations. Mol Ther 2007 499:745–7
ter Brake, O. and Berkhout, B. (2005) A novel approach for inhibition of HIV-1 by RNA interference: counteracting viral escape with a second generation of siRNAs. J. RNAi Gene Silencing 1, 56–65.
Gimenez-Barcons, M., Clotet, B., and Martinez, M.A. (2007) Endoribonuclease-Prepared Short Interfering RNAs Induce Effective and Specific Inhibition of Human Immunodeficiency Virus Type 1 Replication. J Virol 81, 10680–10686.
Kawasaki, H., Suyama, E., Iyo, M., and Taira, K. (2003) siRNAs generated by recombinant human Dicer induce specific and significant but target site-independent gene silencing in human cells. Nucleic Acids Res 31, 981–987.
Myers, J.W., Jones, J.T., Meyer, T., and Ferrell, J.E. Jr. (2003) Recombinant Dicer efficiently converts large dsRNAs into siRNAs suitable for gene silencing. Nat Biotechnol 21, 324–328.
Li, M. and Rossi, J.J. (2005) Lentiviral vector delivery of siRNA and shRNA encoding genes into cultured and primary hematopoietic cells. Methods Mol Biol 309, 261–272.
Martinez, M.A., Gutierrez, A., Armand-Ugon, M. et al. (2002) Suppression of chemokine receptor expression by RNA interference allows for inhibition of HIV-1 replication. AIDS 16, 2385–2390.
Qin, X.F., An, D.S., Shen, I.S., and Baltimore, D. (2003) Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci U S A 100, 183–188.
Llano, M., Saenz, D.T., Meehan, A. et al. (2006) An essential role for LEDGF/p75 in HIV integration. Science 314, 461–464.
Maertens, G., Cherepanov, P., Pluymers, W. et al. (2003) LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J Biol Chem 278, 33528–33539.
Jacque, J.M. and Stevenson, M. (2006) The inner-nuclear-envelope protein emerin regulates HIV-1 infectivity. Nature 441, 641–645.
Nguyen, D.G., Wolff, K.C., Yin, H., Caldwell, J.S., and Kuhen, K.L. (2006) “UnPAKing” human immunodeficiency virus (HIV) replication: using small interfering RNA screening to identify novel cofactors and elucidate the role of group I PAKs in HIV infection. J Virol 80, 130–137.
Chiu, Y.L., Cao, H., Jacque, J.M., Stevenson, M., and Rana, T.M. (2004) Inhibition of human immunodeficiency virus type 1 replication by RNA interference directed against human transcription elongation factor P-TEFb (CDK9/CyclinT1). J Virol 78, 2517–2529.
Berger, E.A., Murphy, P.M., and Farber, J.M. (1999) Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 17, 657–700.
An, D.S., Donahue, R.E., Kamata, M. et al. (2007) Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci U S A 104, 13110–13115.
de Fougerolles, A., Vornlocher, H.P., Maraganore, J., and Lieberman, J. (2007) Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov 6, 443–453.
Bitko, V., Musiyenko, A., Shulyayeva, O., and Barik, S. (2005) Inhibition of respiratory viruses by nasally administered siRNA. Nat Med 11, 50–55.
Li, B.J., Tang, Q., Cheng, D. et al. (2005) Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat Med 11, 944–951.
Morrissey, D.V., Lockridge, J.A., Shaw, L. et al. (2005) Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotechnol 23, 1002–1007.
Geisbert, T.W., Hensley, L.E., Kagan, E. et al. (2006) Postexposure protection of guinea pigs against a lethal Ebola virus challenge is conferred by RNA interference. J Infect Dis 193, 1650–1657.
Palliser, D., Chowdhury, D., Wang, Q.Y. et al. (2006) An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 439, 89–94.
Juliano, R.L. (2005) Peptide-oligonucleotide conjugates for the delivery of antisense and siRNA. Curr Opin Mol Ther 7, 132–136.
Song, E., Zhu, P., Lee, S.K. et al. (2005) Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol 23, 709–717.
Naldini, L., Blomer, U., Gallay, P. et al. (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267.
Morris, K.V. and Rossi, J.J. (2006) Lentivirus-mediated RNA interference therapy for human immunodeficiency virus type 1 infection. Hum Gene Ther 17, 479–486.
Ter Brake, O. and Berkhout, B. (2007) Lentiviral vectors that carry anti-HIV shRNAs: problems and solutions. J Gene Med 9, 743–750.
Ter Brake, O. (2007) Development of an RNAi based gene therapy against HIV-1. Amsterdam: Doctoral Thesis, University of Amsterdam.
Brown, B.D., Venneri, M.A., Zingale, A., Sergi Sergi, L., and Naldini, L. (2006) Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med 12, 585–591.
Wu, X., Li, Y., Crise, B., and Burgess, S.M. (2003) Transcription start regions in the human genome are favored targets for MLV integration. Science 300, 1749–1751.
Hacein-Bey-Abina, S., von Kalle, C., Schmidt, M. et al. (2003) A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 348, 255–256.
Baum, C. (2007) Insertional mutagenesis in gene therapy and stem cell biology. Curr Opin Hematol 14, 337–342.
Kustikova, O.S., Geiger, H., Li, Z. et al. (2007) Retroviral vector insertion sites associated with dominant hematopoietic clones mark “stemness” pathways. Blood 109, 1897–1907.
Gervaix, A., Schwarz, L., Law, P. et al. (1997) Gene therapy targeting peripheral blood CD34+ hematopoietic stem cells of HIV-infected individuals. Hum Gene Ther 8, 2229–2238.
Yam, P.Y., Li, S., Wu, J., Hu, J., Zaia, J.A., and Yee, J.K. (2002) Design of HIV vectors for efficient gene delivery into human hematopoietic cells. Mol Ther 5, 479–484.
Morris, K.V., Gilbert, J., Wong-Staal, F., Gasmi, M., and Looney, D.J. (2004) Transduction of cell lines and primary cells by FIV-packaged HIV vectors. Mol Ther 10, 181–190.
Sioud, M. (2007) RNA interference and innate immunity. Adv Drug Deliv Rev 59, 153–163.
Judge, A.D., Sood, V., Shaw, J.R., Fang, D., McClintock, K., and MacLachlan, I. (2005) Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol 23, 457–462.
Sioud, M. and Sorensen, D.R. (2003) Cationic liposome-mediated delivery of siRNAs in adult mice. Biochem Biophys Res Commun 312, 1220–1225.
Chiu, Y.L. and Rana, T.M. (2003) siRNA function in RNAi: A chemical modification analysis. RNA 9, 1034–1048.
Layzer, J.M., McCaffrey, A.P., Tanner, A.K., Huang, Z., Kay, M.A. and Sullenger, B.A. (2004) In vivo activity of nuclease-resistant siRNAs. RNA 10, 766–771.
Sioud, M. (2006) Single-stranded small interfering RNA are more immunostimulatory than their double-stranded counterparts: a central role for 2′-hydroxyl uridines in immune responses. Eur J Immunol 36, 1222–1230.
Sioud, M. (2005) On the delivery of small interfering RNAs into mammalian cells. Expert Opin Drug Deliv 2, 639–651.
Kariko, K., Bhuyan, P., Capodici, J., and Weissman, D. (2004) Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J Immunol 172, 6545–6549.
Robbins, M.A., Li, M., Leung, I. et al. (2006) Stable expression of shRNAs in human CD34+ progenitor cells can avoid induction of interferon responses to siRNAs in vitro. Nat Biotechnol 24, 566–571.
Konstantinova, P., ter Brake, O., Haasnoot, J., de Haan, P., and Berkhout, B. (2007) Trans-inhibition of HIV-1 by a long hairpin RNA expressed within the viral genome. Retrovirology 4, 15.
Watanabe, T., Sudoh, M., Miyagishi, M. et al. (2006) Intracellular-diced dsRNA has enhanced efficacy for silencing HCV RNA and overcomes variation in the viral genotype. Gene Ther 13, 883–892.
Weinberg, M.S., Ely, A., Passman, M., Mufamadi, S.M., and Arbuthnot, P. (2007) Effective anti-hepatitis B virus hammerhead ribozymes derived from multimeric precursors. Oligonucleotides 17, 104–112.
Chen, C.Z., Li, L., Lodish, H.F., and Bartel, D.P. (2004) MicroRNAs modulate hematopoietic lineage differentiation. Science 303, 83–86.
John, M., Constien, R., Akinc, A. et al. (2007) Effective RNAi-mediated gene silencing without interruption of the endogenous microRNA pathway. Nature 449, 745–747.
Jackson, A.L., Bartz, S.R., Schelter, J. et al. (2003) Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 21, 635–637.
Jackson, A.L., Burchard, J., Schelter, J. et al. (2006) Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12, 1179–1187.
Birmingham, A., Anderson, E.M., Reynolds, A. et al. (2006) 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods 3, 199–204.
Lin, X., Ruan, X., Anderson, M.G. et al. (2005) siRNA-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res 33, 4527–4535.
Cekaite, L., Furset, G., Hovig, E., and Sioud, M. (2007) Gene expression analysis in blood cells in response to unmodified and 2′-modified siRNAs reveals TLR-dependent and independent effects. J Mol Biol 365, 90–108.
Jackson, A.L., Burchard, J., Leake, D. et al. (2006) Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 12, 1197–1205.
Gimeno, R., Weijer, K., Voordouw, A. et al. (2004) Monitoring the effect of gene silencing by RNA interference in human CD34+ cells injected into newborn RAG2−/− gammac−/− mice: functional inactivation of p53 in developing T cells. Blood 104, 3886–3893.
Legrand, N., Weijer, K., and Spits, H. (2006) Experimental models to study development and function of the human immune system in vivo. J Immunol 176, 2053–2058.
Baenziger, S., Tussiwand, R., and Schlaepfer, E. (2006) Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2−/−gamma c−/− mice. Proc Natl Acad Sci U S A 103, 15951–15956.
Acknowledgements
MAM is supported by the Spanish Minsiterio de Educación y Ciencia (MEC) project BFU2006-01066/BMC, Fondo de Investigación Sanitaria (FIS) project PI070098, and Fundación para la investigación y la prevención del SIDA en España (FIPSE 36549/06).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Humana Press, a part of Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Martínez, M. (2009). Progress in the Therapeutic Applications of siRNAs Against HIV-1. In: Sioud, M. (eds) siRNA and miRNA Gene Silencing. Methods in Molecular Biology, vol 487. Humana Press. https://doi.org/10.1007/978-1-60327-547-7_17
Download citation
DOI: https://doi.org/10.1007/978-1-60327-547-7_17
Published:
Publisher Name: Humana Press
Print ISBN: 978-1-60327-546-0
Online ISBN: 978-1-60327-547-7
eBook Packages: Springer Protocols