
Abstract
Macrophages differentiated from human induced pluripotent stem cells (hiPSCs) provide an alternative new tool overcoming some of the limitations of existing models for human macrophages, such as human macrophage-like cell lines and primary monocyte-derived macrophages. A combination of different cytokines and growth factors can differentiate hiPSCs toward myeloid lineage. Here we describe a simple multistep protocol for differentiating hiPSCs into functional macrophages. This includes derivation of three germ-line containing embryoid bodies (EBs) from iPSCs, generation of myeloid precursors from EBs, and finally maturation of myeloid precursors into functional macrophages. Technical procedure and specific culture conditions associated with each of these steps are discussed in detail.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Macrophages are a key component of the innate immune system, populate different tissues and organs during development, and are maintained throughout life by local proliferation and steady-state recruitment [1]. These resident macrophage populations play a central role in a variety of tissue-specific physiological and pathophysiological processes [2]. Macrophages also provide first-line defense against invading pathogens, orchestrate innate immune responses, and initiate adaptive immunity [3]. Thus, characterization of macrophage function is a major focus of many investigators, in the context of both fundamental biology as well as human diseases.
Immortalized or leukemia-derived human macrophage-like cell lines and primary monocyte-derived macrophages (MDM) differentiated from human blood are two commonly used model systems to study human macrophage functions. Although useful, these systems have inherent limitations. Existing macrophage-like cell lines often show chromosomal abnormalities and do not recapitulate all the phenotypic and functional characteristics of primary macrophages [4, 5]. By contrast, although MDMs are currently the gold standard, they show high levels of variability on the basis of genetic makeup and physiological state of individual donors. There are also significant practical and ethical constraints in terms of how frequently and what volumes of blood can be drawn from individual donors, as well as in genetic analysis of such material.
Development of human induced pluripotent stem cell (hiPSC) technology provides a novel tool for disease modeling and to study human cellular function in a controlled environment [6]. Adult somatic cells can be reprogrammed into an embryonic pluripotent state by forced expression of a defined set of transcription factors [7]. These cells retain their original genetic architecture, able to self-renew and differentiate into virtually any cell types in the presence of optimal conditions. Recently, our group and others have developed methods to differentiate hiPSCs into macrophage-like cells that share characteristics of primary human macrophages and overcome some of the limitations of the existing models in human macrophage biology [8,9,10,11].
Macrophages are highly resistant to genetic manipulation and induce a robust antiviral response to exogenous nucleic acid [12, 13]. By contrast iPSCs are immunologically unresponsive and more amenable to genetic manipulation [14]. Once genetically modified at iPSC stage, they can be subsequently differentiated into macrophages without inducing a response to foreign nucleic acid [15]. Macrophages differentiated from patient-derived iPSCs harboring rare genetic mutations phenocopy original macrophage defects observed in patients [16,17,18]. Similarly, large numbers of iPSCs from healthy donors have been differentiated into macrophages to study how common genetic variation influences innate immune responses to pathogens. Furthermore, iPSC-derived macrophages are especially useful to study human-adapted pathogens for which currently no animal models are available [8, 19]. In a more translational context, proof-of-principle evidence is now emerging that iPSC-derived macrophages can be potentially exploited to treat certain types of cancer by cellular therapy [20]. Different tissue-resident macrophages are difficult to access especially from healthy individuals. Human iPSC-derived macrophages follow a similar developmental pathway as described for tissue-resident populations in vivo, and can be further differentiated into specific tissue-resident populations [21,22,23]. Thus hiPSC provide an alternative source for tissue-specific macrophage populations which are difficult to obtain from primary tissues.
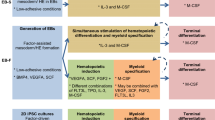
Here we provide a simple, stepwise protocol for differentiating hiPSCs that can be scaled up to produce large numbers of functional macrophages according to individual requirements. The protocol follows a continuous culture method that produces individual batches of macrophages every 5–7 days that can be maintained for several months, particularly convenient for repeated experiments. Some variations of this protocol have been reported by our group and others, but the current protocol is technically less challenging, reproducible, and economic and can be easily adapted for use by nonspecialist laboratories. The protocol involves several key steps: (1) expansion of iPSCs in feeder-free conditions using chemically defined medium, (2) adapting iPSCs in feeder-dependent culture, (3) formation of three germ layers (ectoderm, mesoderm, and endoderm) containing embryoid bodies (EBs), (4) generation of myeloid precursor cells from EBs in the presence of IL-3 and M-CSF , and (5) terminal differentiation of myeloid precursors into mature macrophages in the presence of higher concentrations of M-CSF (Fig. 1).

Directed differentiation of macrophages from human iPSCs: (a) A schematic diagram showing specific culture conditions required for each differentiation step from iPSCs to generation of macrophages, as well as phase-contrast photomicrographs of each differentiation step (b). (c) Phase-contrast micrographs showing distinct size and morphologies of EBs generated in 10 cm2 dish, 96 round-bottomed wells and AggreWell plates (reproduced and adapted from [8])
Macrophages generated by this protocol closely share gene expression profiles of MDMs [9], express well-established markers of human macrophages (Fig. 2), and can be polarized into classical or alternatively activated phenotypes after stimulation with IFN-γ and IL-4 , respectively (Fig. 3). Functionally, these macrophages respond to microbial stimulation and support pathogen infection [8, 9].

Comparison of mRNA expression of selected macrophage-specific markers between undifferentiated iPS cells and fully differentiated macrophages: RNA was extracted and sequenced from undifferentiated iPS cells; differentiated iPSDMs and log2-normalized expression value for indicated macrophage surface markers were plotted. Data from three independent replicate is presented (reproduced and adapted from [8])

hiPS-derived macrophages express pan-macrophage lineage markers in the resting state and induce specific activation markers after classical and alternative activation: (a) Histograms showing expression of established markers of resting Mφs CD14, CD16, CD44, CD64, CD200, CD206, CD54, CD68, CD11c, and CD163 and activation markers HLA-DR and CD86 in unstimulated iPS-derived Mφs. (b) Histogram showing expression of classical activation (M1) marker HLA-DR, DP, and DQ and alternative activation marker and CD206 after overnight stimulation with 20 ng/mL IFN-γ and 50 ng/mL IL-4 , respectively (reproduced and adapted from [8])
2 Materials
2.1 Human iPSC Culture in Feeder-Free System
-
1.
Essential 8 (E8) medium: This reagent comes in two components—490 mL base media at 4 °C and 10 mL supplement at −20 °C. To reconstitute the media thaw whole 10 mL supplement at room temperature, add in to 490 mL base media, and store at 4 °C. Alternatively, aliquot the supplement in small volumes and store at −80 °C; reconstitute small volumes of media as required using same as above (see Notes 1 and 2 ).
-
2.
Recombinant human vitronectin (rhVTN-N): Aliquot vitronectin in appropriate volumes in sterile tubes and store at −80 °C (stable up to 1 year).
-
3.
D-PBS without calcium and magnesium.
-
4.
Water for embryo transfer.
-
5.
Rock inhibitor Y-27632 dihydrochloride: Prepare a 10 mM stock of Rock inhibitor in ultrapure water and store at −20 °C in small aliquots. Final concentration of Rock inhibitor required in cell culture medium is 10 μM; this can be achieved by adding 1 μL of 10 mM stock per 1 mL of culture medium.
2.2 Human iPSC Culture on Inactivated Mouse Embryonic Fibroblast
-
1.
Irradiated mouse embryonic fibroblast: Store frozen stock vials of irradiated MEFs in liquid nitrogen or −80 °C.
-
2.
0.1% Gelatine solution: Add 500 mg porcine gelatine powder into 500 mL endotoxin-free embryo transfer water and mix gently. Place the bottle in a 56 °C water bath for 30–45 min with regular mixing to ensure that gelatine is dissolved. Once gelatine is fully dissolved transfer solution to a 500 mL sterile vacuum filter unit and filter sterilize. Store gelatine solution at 4 °C for up to 2 months.
-
3.
Human iPSC base medium: Advanced DMEM/F12 medium; 20% knockout serum replacement (KSR), 2 mM L-glutamine; 100 IU/mL penicillin-streptomycin (Pen/Strep); 0.1 mM β-mercaptoethanol (2 ME); filter sterilize and store at 4 °C (see Note 3 ).
-
4.
Dispase: Add 500 mg of dispase powder into 500 mL advanced DMEM/F12 medium and mix well to dissolve. Once powder is fully dissolved transfer solution into a 500 mL sterile vacuum filter unit and filter sterilize. Aliquot in appropriate volumes and store at −20 °C for up to 8 months.
-
5.
Collagenase : Add 500 mg of collagenase IV powder into 500 mL of supplemented hiPSC base medium and mix well to dissolve. Once powder is fully dissolved transfer solution into a 500 mL sterile vacuum filter unit and filter sterilize. Aliquot in appropriate volumes and store at −20 °C for up to 8 months.
-
6.
0.1% Bovine serum albumin (BSA) solution: Add 100 mg endotoxin-free tissue culture-grade BSA powder into 100 mL D-PBS and mix well; keep solution at 37 °C water bath until BSA powder is fully dissolved, filter sterilize, and store at 4 °C for up to 2 months.
-
7.
Recombinant human basic fibroblast growth factor (rhFGF, 25 μg/mL stock concentration): Add 1 mL of 0.1% BSA solution directly into vial containing 25 μg lyophilized FGF and mix gently. Make 8 μL aliquots in sterile tubes and store at −80 °C for up to 1 year (see Note 4 ).
2.3 Embryoid Body, Myeloid Precursors, and Macrophage Differentiation
-
1.
Recombinant human interleukin 3 (rhIL-3, 0.5 mg/mL stock concentration): Add 1 mL of 0.1% BSA solution directly into vial containing 0.5 mg lyophilized rhIL-3 to achieve 0.5 mg/mL stock concentration. Mix gently, aliquot in sterile tubes at desired volume, and store at −80 °C, stable up to 1 year (see Note 4 ).
-
2.
Recombinant human macrophage colony-stimulating factor (rhM-CSF, 1 mg/mL stock concentration): Add 500 μL 0.1% BSA solution directly into vial containing 0.5 mg lyophilized rhM-CSF to achieve 1 mg/mL stock concentration. Mix gently, aliquot in desired volumes, and store at −80 °C (stable up to 1 year) (see Note 5 ).
-
3.
Myeloid precursor base medium: X-VIVO 15 serum-free medium; 2 mM L-glutamine; 100 IU/mL penicillin-streptomycin (Pen/Strep); 0.1 mM β-mercaptoethanol (2 ME); filter sterilize and store at 4 °C (see Note 6 ).
-
4.
Macrophage differentiation base medium: RPMI 1640; 10% heat-inactivated fetal calf serum; 2 mM L-glutamine; 100 IU/mL penicillin-streptomycin; filter sterilize and store at 4 °C.
3 Methods
Perform all cell culture in a class two microbiological cabinet under aseptic conditions. We recommend after thawing the iPSCs from frozen stock, cells are cultured in feeder-free conditions using E8 medium for initial expansion. Once the cells have adapted to feeder-free culture and expanded in sufficient numbers they are transferred into feeder-dependent culture conditions before proceeding with subsequent differentiation steps.
3.1 General Considerations
-
1.
Human iPSCs are delicate and more difficult to maintain in culture compared to most other conventional human cell lines. They show a natural tendency to spontaneously differentiate into fibroblast like cells. Cytokines and growth factors present in the medium allow them to be kept in pluripotent state; hence, daily change of media is essential.
-
2.
iPSCs grow in individual colonies. During handling, it is critical that iPSCs remain as small clumps and not broken down into single cells. Hence, iPSC colonies should be always treated gently; do not pipette too harshly or too many times as this will result in single-cell colonies. Use wide-bore stripettes, and centrifuge slowly and with shorter duration where possible.
-
3.
It is important to ensure that iPSC colonies remain separate in culture and do not fuse to each other during expansion to prevent spontaneous differentiation . However, low levels of spontaneous differentiation are sometimes unavoidable especially on feeder-dependent culture. Culture can be cleaned up by carefully selecting only pluripotent colonies during passage steps—this will eventually clean up the culture after few passages.
3.2 hiPSC Culture in Feeder-Free System
-
1.
Prepare vitronectin-coated maintenance plate at least 2 h before plating the hiPSCs. Thaw 250 μL aliquot of vitronectin at room temperature and dilute it in 6 mL of D-PBS. Add 1 mL of diluted vitronectin into each well of 6-well tissue culture plates or add whole 6 mL into 100 mm tissue culture dish. Make sure that the vitronectin solution covers the whole surface of the plastic vessel and incubate at room temperature for at least 2 h or at 4 °C overnight tightly sealed with Parafilm. If necessary, sealed plates can be stored at 4 °C for up to 5 days without removing the vitronectin solution (see Note 7 ).
-
2.
Prepare a 15 mL centrifuge tube with 9 mL E8 media supplemented with 1 μM Rock inhibitor for each vial of iPSC that will be defrosted. Thaw the vial at 37 °C in water bath. Gently transfer the defrosted cells into 9 mL E8 media and slowly invert the tube to evenly mix the cells and to dilute out DMSO. DMSO is toxic for cells at room temperature; therefore the thawing process needs to be as quick as possible; keep frozen vial in water bath for minimum time required to defrost but do not warm up cells to 37 °C.
-
3.
Centrifuge the tube at 290 × g for 3 min at 20 °C. Resuspend the cell pellet into desired volume of E8 medium supplemented with 10 μM Rock inhibitor (final concentration). Plate 1 mL of cell suspension into each wells of a 6-well plate. Content of one frozen vial (equivalent to 1 well of ~70–80% confluent 6-well plate or 1/10th of a 100 mm dish) can be distributed into 2–3 wells of a 6-well plate with 1 mL medium. Alternatively, if a specific iPSC line shows good recovery and growth characteristics, cells can be seeded less densely—for example one vial can be directly seeded onto a 100 mm dish using 8 mL of total media. Generally it is better to seed the colonies densely after thawing to ensure better recovery (see Notes 8 and 9 ).
-
4.
Place the plate into a humidified 37 °C incubator with 5% CO2. Once inside the incubator gently rock the plate in different directions to make sure that colonies are evenly distributed throughout the surface of the dish. Keep the plate undisturbed for at least 24 h to allow colonies to attach to the vitronectin-coated surface.
-
5.
After 24 h majority of the colonies will attach to the surface. Remove all media from the dish to discard any unattached colonies and dead cells, and add 2–3 mL of fresh E8 media without Rock inhibitor for each well of a 6-well plate or 10–12 mL for each 100 mm dish (see Note 10 ).
-
6.
Change medium every day and replace with fresh E8 medium until cells are ~70–80% confluent and ready for passaging.
-
7.
For passaging, start by preparing vitronectin-coated plates as before.
-
8.
Once cells are sufficiently confluent, remove the spent media and gently wash the plate twice with an appropriate volume (~5 mL) of D-PBS.
-
9.
Remove the D-PBS and add required volume of 5 mM PBS-EDTA solution to cover the entire surface of the dish (1 mL for each well of a 6-well plate, and 8 mL for 100 mm dish).
-
10.
Look under the microscope every 2 min to confirm that colonies are detaching and have changed their morphologies but do not wait too long such that the colonies are fully detached or broken up into single cells.
-
11.
Carefully remove PBS-EDTA solution without disturbing iPSC colonies and add same volume of fresh E8 media. Gently pipette up and down using a 10 mL stripette to dislodge the colonies from the surface. If necessary repeat this step two more times to recover as many iPSC colonies as possible.
-
12.
Collect the media and detached iPSC colonies into a new 15 mL centrifuge tube and centrifuge at 290 × g for 3 min at 20 °C. Resuspend the colonies in 2 mL of fresh E8 media supplemented with 10 μM Rock inhibitor. If colonies are too big at this stage pipette up and down 2–3 times to break them into smaller clumps.
-
13.
Transfer colonies in a desired splitting ratio into a fresh vitronectin-coated dish with appropriate volume of E8 media supplemented with 10 μM Rock inhibitor. Generally we recommend a splitting ratio of 1:10; but this depends on growth rates of individual iPSC lines and how frequently cells are passaged. Thus, the splitting ratio should be adjusted between 1:5 and 1:10 according to the requirement of individual user.
-
14.
After 24 h remove medium and add fresh E8 medium (without Rock inhibitor); change medium every day until next passage.
-
15.
To freeze iPS cells, freshly prepare the freezing mixture—10% DMSO in KSR (9 mL KSR + 1 mL DMSO) at room temperature. After lifting off iPSC colonies as described in Subheading 3.2, resuspend the cell pellet into desired volume of freezing mixture and quickly transfer 1 mL cell suspension into each cryovial. We recommend one 100 mm dish (70–80% confluent) to divide into ten cryovials and each well of a 6-well plate into two cryovials for freezing. Immediately transfer the cryovials into a Mr. Frosty or CoolCell container and place them into −80 °C freezer overnight. After 24 h transfer the cryovials into liquid nitrogen for long-term storage.
3.3 Culturing hiPSCs in Feeder-Dependent System
-
1.
At least 24 h before transferring the iPSCs from feeder-free culture, prepare feeder plates using inactivated mouse embryonic fibroblasts (MEFs). First coat the tissue culture plates by adding 2 mL of 0.1% gelatine solution into each well of 6-well plates or 8 mL for 100 mm dishes; ensure that whole surface of the dish is covered and then incubate at room temperature for at least 2 h.
-
2.
Thaw one vial (2 × 106 cell) of irradiated mouse embryonic fibroblasts into 9 mL of human iPSC basal medium in a 15 mL centrifuge tube and spin at 290 × g at 20 °C. Resuspend the cell pellet in desired volume of human iPSC base medium.
-
3.
Remove the gelatine solution from the plate, seed the MEFs, and place them in 37 °C incubator overnight. One vial of MEF (2 × 106 cell) is sufficient for a whole 6-well plate or one 100 mm dish.
-
4.
Lift off the iPSC colonies growing under feeder-free conditions as described in Subheading 3.2. Resuspend these colonies into the required volume of human iPSC base medium supplemented with 4 ng/mL bFGF and 10 μM Rock inhibitor (final concentrations).
-
5.
Remove medium and any unattached MEFs from the feeder plate and transfer iPSC colonies onto the feeder layer. Usually a splitting ratio of 1:10 works well at this stage. Place the dish inside the incubator, redistribute colonies evenly on feeder layer, and keep undisturbed for at least 24 h.
-
6.
After 24 h a majority of colonies will have attached to the feeder layer. Remove medium and add fresh human iPS base medium supplemented with 4 ng/mL bFGF but no Rock inhibitor. Change medium every day until colonies are ready to passage (see Note 10 ).
-
7.
For passaging, thaw collagenase , dispase aliquots at room temperature, and mix them in 1:1 ratio in a tube.
-
8.
Remove medium from iPSCs growing on feeder layer, wash once with D-PBS, and add collagenase-dispase mix on the dish to cover the whole surface. We recommend 2 mL enzyme mix for each well of a 6-well feeder plate and 8 mL for 100 mm dish.
-
9.
Incubate at 37 °C for 30–60 min until pluripotent colonies start to lift off but feeder layer and any differentiated cells remain attached. The duration of incubation varies between iPSC lines and can take up to 90 min depending on colony size and level of differentiation . Start checking under a microscope for floating colonies after 20 min and every 5–10 min subsequently. It is important not to overdigest to prevent colonies from breaking up into small fragments.
-
10.
Gently mix the collagenase-dispase solution up and down few times using a 10 mL stripette to dislodge loosely adherent colonies from the feeder layer. Collect collagenase-dispase mixture along with all floating colonies into a new 50 mL and add at least same volume of human iPSC basal medium to neutralize enzyme activity.
-
11.
Centrifuge harvested colonies at 290 × g for 3 min at 20 °C and discard the supernatant. Wash at least two times by resuspending colonies in iPSC base medium followed by centrifugation. It is important to make sure that all traces of enzymes are removed before replating the cells onto a new feeder plate.
-
12.
After the last wash, if required, break the colonies into smaller sizes, gently resuspend in desired volume of iPSC base medium supplemented with bFGF and Rock inhibitor, transfer to new feeder plates in a desired splitting ratio, and continue as before.
3.4 Differentiation of iPSCs into Macrophages
-
1.
Once iPSC colonies are 70–80% confluent on feeder-dependent culture detach them from the feeder layer using collagenase-dispase method . Transfer all the floating iPSC colonies with enzyme mix into a fresh 50 mL tube and add at least twice the volume of iPSC base medium to neutralize enzyme function.
-
2.
Keep the tube upright in a tube rack undisturbed for 3–5 min to allow colonies to settle. A large pellet of iPSC colonies will become visible at the bottom of the tube and medium on the top will be clearer. Do not centrifuge at this stage as fragmentation of colonies into smaller pieces will reduce the efficiency of EB formation in subsequent steps.
-
3.
Aspirate medium from the top without disturbing iPSC colonies in the bottom of the tube. Wash 2–3 times by adding excess volumes of iPSC base media and by allowing the colonies to settle.
-
4.
Resuspend iPSC colonies in iPSC base medium without any cytokine supplement, and distribute them to desired numbers of 100 mm low-adherent bacteriological dishes. We recommend dividing one 100 mm dish of feeder-dependent iPSC culture into four 100 mm low-adherent bacteriological dishes for EB formation (see Note 11 ).
-
5.
Add extra medium as necessary to make the final volume of each 100 mm low-adherent bacteriological dish ~30 mL. Transfer the dishes to a humidified 37 °C incubator and leave undisturbed for 4 days to allow EB formation.
-
6.
On day 5, the EBs should have formed and be visible with the naked eye. Harvest them by collecting all the media into 50 mL tubes using a 10 mL stripette and leaving the tubes in an upright position for 3–5 min to let the EBs settle by gravity. Do not centrifuge at this stage. Carefully aspirate the medium along with single cell and cell debris and gently resuspend EBs into desired volume of myeloid precursor base medium supplemented with IL-3 and M-CSF . Distribute the EBs onto gelatine-coated 100 mm tissue culture-treated dishes with final volume of ~12 mL media in each dish.
-
7.
After 4–5 days, the majority of EBs will attach to the gelatine-coated surface and start to spread out as a stromal layer around the EB. It is possible that some EBs are still not attached to the surface at this stage; in such a case add additional 12–15 mL of cytokine-supplemented myeloid precursor base media and leave the EBs in culture for another 4–5 days (see Note 12 ). At this stage many small apoptotic looking floating cells will arise in the medium; these are released from underlying EBs, but are not myeloid precursors and should be regularly removed from the culture.
-
8.
Change the medium every 4–5 days for next 2–3 weeks by harvesting all media from the EB dish and pass through a 70 μm cell strainer. Discard the flow through as this will mostly contain contaminating small cells and debris. Any floating EBs will be retained by the cell strainer and should be transferred back into the culture dish; invert the cell strainer and directly pipette the medium onto the bottom surface of the strainer back into the same dish.
-
9.
Approximately 3–4 weeks after transferring the EBs to gelatinized plates, the smaller apoptotic looking cells will disappear from the culture to be replaced by larger blast-like cells with dendrite-like structure. These are the myeloid precursor cells that could be further differentiated into mature macrophages as described below. From step 8 onwards this is a continuous culture; myeloid precursors can be harvested every 4–5 days and as long as cytokine-supplemented fresh medium are added to EBs they will produce further precursors . This process can be continued for 6–8 months after which precursor number drops significantly (see Notes 13 – 15 ).
-
10.
Harvest the myeloid precursor cells by removing spent medium from EB plates. Filter through a 70 μm cell strainer and collect the flow through in 50 mL tubes as it will contain the precursor cells. Centrifuge at 290 × g for 3 min at 20 °C and resuspend the pellet in macrophage differentiation base medium supplemented with 100 ng/mL M-CSF .
-
11.
Count cells and plate ~150,000 precursor cells in each well of a 6-well plates or 500,000 cells in a 100 mm tissue culture dish (see Note 16 ). Culture cells at 37 °C incubator for another 6 days to differentiate into mature macrophages. From day 7 onwards fully mature macrophages will be ready for phenotypic assessment or downstream functional assays.
4 Notes
-
1.
All solutions and media should be prepared inside a class 2 microbiological safety cabinet using standard aseptic techniques; unless otherwise specified prepare all reagents at room temperature. Reconstitute all cytokines in 0.1% BSA solution in D-PBS, aliquot immediately in appropriate volume, and store at −80 °C until required. Store all base media at 4 °C and do not keep reconstituted media longer than 2 weeks. Always freshly add cytokines into base media immediately before use.
-
2.
E8 media are light sensitive . Keep individual components and reconstituted media protected from light.
-
3.
Immediately before culturing iPSCs on mouse feeder add required volume of bFGF (rhFGF2 basic 146aa) into iPSC base media to obtain 4 ng/mL final concentrations. In addition, during first-time transfers of hiPSCs onto feeder layers and during all subsequent passaging steps also add Rock inhibitor into hiPSC base medium to obtain 10 μM final concentrations.
-
4.
During reconstitution and aliquoting of cytokines, pipette carefully to avoid excessive frothing.
-
5.
Before transferring the EBs onto gelatinized plates add the required volume of rhIL-3 (25 ng/mL final concentration) and rhM-CSF (50 ng/mL final concentration) into myeloid precursor base medium. This can be easily achieved by further diluting cytokine aliquots in appropriate volumes of 0.1% BSA solution before adding to culture medium.
-
6.
Before culturing myeloid precursors for terminal differentiation into macrophages add required volume of rhM-CSF into macrophage differentiation base media to obtain100 ng/mL final concentration.
-
7.
Ensure that vitronectin-coated plates are tightly sealed with Parafilm to prevent evaporation of solution and ensure that the entire surface of the plastic dish is fully covered at all times.
-
8.
Human iPSCs are sensitive to temperature changes. Always use media and solutions that are pre-warmed to room temperature. Warm media bottles at room temperature only and never warm in 37 °C water bath.
-
9.
During initial seeding of iPSC colonies on vitronectin-coated plates and feeder layers, use minimum volumes of media for first 24 h. This enhances the contact of colonies with the surface and facilitates attachment.
-
10.
Rock inhibitor prevents apoptosis and spontaneous differentiation of iPSCs. Addition of Rock inhibitor during initial plating and subsequent passaging of iPSCs facilitates attachment and recovery. However, Rock inhibitor can interfere with cellular growth and colony expansion and must be removed from media after first 24 h.
-
11.
It is critical that during EB formation iPSC colonies are plated on low-adherent bacteriological dishes without coating, and no bFGF is added at this stage. EBs generated in this method may be heterogeneous in size and shape, but that does not interfere with their ability to produce myeloid precursors in subsequent steps. There are other methods to generate equal-size EBs using AggreWell and hanging-drop methods, but these are technically more involved and not discussed here.
-
12.
It is not essential that EBs should attach to the gelatine-coated surface in order to produce myeloid precursors, but attachment makes it easier for weekly media change and harvest of precursors. Occasionally when EB sizes are too small or too large they may not attach to gelatine-coated plates; do not discard such plates as they will generate myeloid precursors as usual.
-
13.
Efficiency of myeloid precursor generation from EB is highly dependent on required levels of nutrients and cytokine concentration at all times. Care should be taken that media never become too yellow at any point during culture. This could be achieved either by changing media more frequently or adding additional media as necessary.
-
14.
Serum components significantly reduce efficiency of EB to precursor differentiation ; only serum-free culture media should be used at this stage.
-
15.
Recombinant cytokines from commercial vendors often contain significant levels of endotoxin which reduces precursor differentiation and also activates them immunologically.
-
16.
During precursor-to-macrophage differentiation, myeloid precursors divide 2–3 times before differentiating into mature macrophages. Therefore, the final number of macrophages should be at least double compared to the initial number of precursors seeded.
References
Wynn TA, Chawla A, Pollard JW (2013) Macrophage biology in development, homeostasis and disease. Nature 496:445–455
Murray PJ, Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11:723–737
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216
Adati N, Huang MC, Suzuki T et al (2009) High-resolution analysis of aberrant regions in autosomal chromosomes in human leukemia THP-1 cell line. BMC Res Notes 2:153
Schildberger A, Rossmanith E, Eichhorn T et al (2013) Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflamm 2013:697972
Grskovic M, Javaherian A, Strulovici B, Daley GQ (2011) Induced pluripotent stem cells—opportunities for disease modelling and drug discovery. Nat Rev Drug Discov 10:915–929
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676
Hale C, Yeung A, Goulding D et al (2015) Induced pluripotent stem cell derived macrophages as a cellular system to study salmonella and other pathogens. PLoS One 10:e0124307
Alasoo K, Martinez FO, Hale C et al (2015) Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Sci Rep 5:12524
Karlsson KR, Cowley S, Martinez FO et al (2008) Homogeneous monocytes and macrophages from human embryonic stem cells following coculture-free differentiation in M-CSF and IL-3. Exp Hematol 36:1167–1175
van Wilgenburg B, Browne C, Vowles J, Cowley SA (2013) Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS One 8:e71098
Lyakh LA, Koski GK, Young HA et al (2002) Adenovirus type 5 vectors induce dendritic cell differentiation in human CD14(+) monocytes cultured under serum-free conditions. Blood 99:600–608
Muruve DA, Petrilli V, Zaiss AK et al (2008) The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452:103–107
Rossi R, Hale C, Goulding D et al (2012) Interaction of Salmonella typhimurium with dendritic cells derived from pluripotent embryonic stem cells. PLoS One 7:e52232
Yeung ATY, Hale C, Lee AH et al (2017) Exploiting induced pluripotent stem cell-derived macrophages to unravel host factors influencing Chlamydia trachomatis pathogenesis. Nat Commun 8:15013
Panicker LM, Miller D, Awad O et al (2014) Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells 32:2338–2349
Panicker LM, Miller D, Park TS et al (2012) Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc Natl Acad Sci U S A 109:18054–18059
Jiang Y, Cowley SA, Siler U et al (2012) Derivation and functional analysis of patient-specific induced pluripotent stem cells as an in vitro model of chronic granulomatous disease. Stem Cells 30:599–611
van Wilgenburg B, Moore MD, James WS, Cowley SA (2014) The productive entry pathway of HIV-1 in macrophages is dependent on endocytosis through lipid rafts containing CD4. PLoS One 9:e86071
Senju S, Koba C, Haruta M et al (2014) Application of iPS cell-derived macrophages to cancer therapy. Oncoimmunology 3:e27927
Haenseler W, Sansom SN, Buchrieser J et al (2017) A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Rep 8:1727–1742
Muffat J, Li Y, Yuan B et al (2016) Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med 22:1358–1367
Takata K, Kozaki T, Lee CZW et al (2017) Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for modeling tissue-resident macrophage differentiation and function. Immunity 47:183–198
Acknowledgments
CM was supported by a British Heart Foundation Cambridge Cardiovascular Research Excellence Pump-priming Grant; CH and SM are supported by Wellcome Trust core funding to the Wellcome Trust Sanger Institute. The authors thank Profs. Gordon Dougan, Antonio Vidal-Puig, and Siamon Gordon for encouragement and support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, a link is provided to the Creative Commons license and any changes made are indicated. The images or other third party material in this chapter are included in the work's Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work's Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2018 The Author(s)
About this protocol

Cite this protocol
Mukherjee, C., Hale, C., Mukhopadhyay, S. (2018). A Simple Multistep Protocol for Differentiating Human Induced Pluripotent Stem Cells into Functional Macrophages. In: Rousselet, G. (eds) Macrophages. Methods in Molecular Biology, vol 1784. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7837-3_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7837-3_2
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-7836-6
Online ISBN: 978-1-4939-7837-3
eBook Packages: Springer Protocols