
Abstract
Peptide and protein aberrant lipidation patterns are often involved in many diseases including cancer and neurological disorders. Peptide lipidation is also a promising strategy to improve pharmacokinetic and pharmacodynamic profiles of peptide-based drugs. Self-adjuvanting peptide-based vaccines commonly utilise the powerful TLR2 agonist PamnCys lipid to stimulate adjuvant activity. The chemical synthesis of lipidated peptides can be challenging hence efficient, flexible and straightforward synthetic routes to access homogeneous lipid-tagged peptides are in high demand. A new technique coined Cysteine Lipidation on a Peptide or Amino acid (CLipPA) uses a ‘thiol-ene’ reaction between a cysteine and a vinyl ester and offers great promise due to its simplicity, functional group compatibility and selectivity. Herein a brief review of various synthetic strategies to access lipidated peptides, focusing on synthetic methods to incorporate a PamnCys motif into peptides, is provided.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
9.1 Introduction
The market for peptide based therapeutics has been constantly growing since the late 1990s with 140 peptide drugs currently estimated to be undergoing clinical trials and 500 therapeutic peptides in pre-clinical development (Fosgerau and Hoffmann 2015; Kaspar and Reichert 2013; Otvos and Wade 2014). Biologically active peptides are excellent drug candidates due to high receptor selectivity, binding affinity, potency and relatively low toxicity (Fosgerau and Hoffmann 2015; Trabocchi and Guarna 2014). However, the therapeutic potential of peptides can be limited due to their poor chemical and physical stability, short plasma half-life, and low oral bioavailability (Fosgerau and Hoffmann 2015; Trabocchi and Guarna 2014). Peptide drug delivery to the site of action is often challenging and improved technologies to overcome this obstacle are highly desirable (Lewis and Richard 2015). Structural and functional modifications of native peptides using chemical techniques have been used to generate compounds with higher affinity, improved enzymatic stability and/or efficacy compared to the parent peptide (Trabocchi and Guarna 2014). Peptide backbone modifications, cyclization, unnatural amino acid insertion, PEGylation, glycosylation, phosphorylation and lipidation are common techniques to improve the physicochemical and pharmacological profiles of bioactive peptides. (Zhang and Bulaj 2012)
Peptide lipidation is an effective strategy to modify the pharmacokinetic and pharmacodynamic properties of lead peptide therapeutics and has proven to be successful with several marketed peptides including liraglutide (Victoza®) (Jackson et al. 2010; Knudsen et al. 2000) and insulin detemir (Levemir®) (Zhang and Bulaj 2012; Home and Kurtzhals 2006; Le Floch 2010). Incorporation of lipid units onto a peptide backbone can dramatically increase enzymatic stability (Simerska et al. 2011), receptor selectivity and potency (Ward et al. 2013), bioavailability (Hamman et al. 2005; Park et al. 2011; Renukuntla et al. 2013; Karsdal et al. 2015) and drug delivery potential (membrane permeability) (Zhang and Bulaj 2012; Simerska et al. 2011).
This review describes the impact of lipidation on peptide-based drug development and summarises the most recent strategies to incorporate a lipid moiety onto a peptide using chemical techniques. A brief discussion on naturally occuring lipidated proteins and peptides and the potential for lipidation to create bioactive therapeutics is covered. The highlight of this perspective relates to synthetic approaches to incorporate PamnCys-based Toll-like receptor 2 (TLR2) lipidated ligands into peptides with the potential to generate self-adjuvanting vaccine constructs.
9.1.1 Protein Lipidation in Nature
Protein lipidation is one of the most important post- and co-translational modifications controlling protein affinity to cellular membranes and influencing protein regulatory and signalling functions (Mejuch and Waldmann 2016; Resh 2013). Altered lipidation patterns are associated with various diseases including cancer, neurological diseases, diabetes, infections (bacterial, fungal and viral) (Resh 2012).
Protein acylation phenomena encompasses a broad range of saturated and unsaturated fatty acids of different length creating proteins with a unique set of functions. Protein-bound lipid types and lipid-protein linkages vary in nature. Covalent attachment of unique fatty acid chains is controlled by the action of specific transferases affording a broad range of lipidated proteins including N-myristoylated, S- or N-palmitoylated, and cholesterol- and isoprenol-enriched moieties (Fig. 9.1). Glycosylphosphatidylinositol (GPI), and phosphatidylethanolamine (PE) conjugation to proteins has also been described (Resh 2013).

Protein modifications with various lipids found in nature
Lipid addition occurs at N- and C- terminal sites of proteins or within the protein sequence directed by specific amino acids such as cysteine, serine, threonine, and lysine (Hannoush 2015). Lipidation can be irreversible when formed via an amide bond using an N-terminal glycine or cysteine moiety (N-myristoylation and N-palmitoylation , respectively) or reversible when a thioester bond is formed between the fatty acid the thiol of the cysteine residue (S-palmitoylation), Fig. 9.1 (Resh 2013; Chamberlain and Shipston 2015).
Proteins can exist in a mono-lipidated state or with multiple-lipid group addition. Membrane proteins such as MARCKS, GPCRs, and K-Ras4B are monolipidated proteins enriched with myristoyl, palmitoyl and farnesyl motifs, respectively. The Hedgehog (Hh) family of proteins which are associated with developmental processes (Lee et al. 2016) are modified with palmitate and cholesterol; similarly, the Src family kinases are myristoylated and palmitoylated and plasma membrane H-Ras and N-Ras proteins are farnesylated and palmitoylated (Resh 2013).
Irreversible protein modification with myristic acid, a 14-carbon fatty acid, is the most prevalent in nature and accounts for 0.5–0.8% of all lipidated eukaryotic proteins. It can occur both co- and post-translationally at the N-terminal glycine and is catalysed by N-myristoyltransferase (NMT), Fig. 9.1a (Resh 2013; Wright et al. 2010; Resh 2016). N-Myristoylation at the Nε of lysine was also observed for interleukin 1α (Stevenson et al. 1993) and tumour necrosis factor alpha (TNF) (Stevenson et al. 1992); However, enzymes involved in these acylation processes are yet to be identified (Resh 2016). N-Myristoylated proteins such as c-Src, BID, PAK2, or gelsolin play important roles in various biological processes including cellular transformation and effecting protein localization (Hannoush 2015; Wright et al. 2010). N-Myristoylation is involved in pathogen survival and altered myristoylation patterns are linked to carcinogenesis (Wright et al. 2010).
S-Palmitoylation is the most common form of protein S-acylation affording reversibly-tagged proteins with a 16-carbon palmitic acid unit (Chamberlain and Shipston 2015; Resh 2016). S-Palmitoylation can occur at the cysteine moiety located in the proximity of either the N -or C-terminus of proteins, Fig. 9.1b. Attachment of stearic acid (C18:0) and monounsaturated omega-9 oleic acid (C18:1) via the thiol group of a cysteine residue has also been described (Chamberlain and Shipston 2015).
Due to labile nature of the thioester bond used to link a fatty acid with a protein backbone, a dynamic equilibrium between protein S-acylation and deacylation with distinct turnover rates occurs that influences intracellular localization, membrane association, and the regulatory functions of a diverse family of proteins. S-Acylation of cellular proteins is mediated via S-acyl transferases from the zDHHC protein family. However, only scant information is available on the S-acyl thioesterases involved in protein deacetylation and the dynamic S-acylation process (Chamberlain and Shipston 2015). It is proposed that enzymes from the serine hydrolase family including acyl protein thioesterases (APTs) (Davda and Martin 2014), and protein palmitoyl thioesterases (PPTs) (Lin and Conibear 2015) may be involved (Chamberlain and Shipston 2015).
S-Acylation facilitates stable membrane binding of peripheral proteins and mediates protein targeting to specific endoplasmic reticulum (ER) subdomains. Protein S-acylation controls trafficking and localization of cellular proteins, and improves protein stability in addition to regulating cellular signalling receptors (Chamberlain and Shipston 2015).
The Hedgehog protein family are critical proteins with roles in embryonic development and tumorigenesis (Resh 2016; Pepinsky et al. 1998). These mature signalling proteins are dually lipidated comprising a palmitate unit which is incorporated through an amide bond at N-terminal cysteine (N-palmitoylation) via the action of hedgehog acyltransferase (Hhat), a member of a membrane-bound O-acyltransferases (MBOAT) protein superfamily (Konitsiotis et al. 2015; Matevossian and Resh 2015), and a cholesterol moiety covalently attached to the C-terminal glycine via its 3β-hydroxyl group, Fig. 9.1c (Resh 2013, 2016). N-Palmitoylation is essential for signalling activity of Hh proteins during development while the cholesterol unit aids the signalling functions (Resh 2013, 2016). Aberrant Hh signalling pathways result in birth defects in humans including microencephaly, cyclopia, absent nose or cleft palate. The development of breast, prostate and lung cancer has also been associated with Hh signaling anomalies (Gupta et al. 2010).
Another member of MBOAT superfamily is porcupine (Porcn) transferase which mediates attachment of a monounsaturated cis-Δ9-palmitoleate unit via a side chain of serine residue to a secreted Wnt glycoprotein family (Resh 2016; Hofmann 2000; Nile and Hannoush 2016; Shindou et al. 2009). This post-translational lipid attachment plays a crucial role in regulating signalling during embryonic development and tissue homeostasis, Fig. 9.1d (Resh 2016; Nile and Hannoush 2016). It has been recently reported that Wnts palmitoylation is reversible; notum hydrolase, which participates in deacylation, affords an inactive form of Wnts with inhibited signalling ability (Resh 2016; Nile and Hannoush 2016; Zhang et al. 2015; Kakugawa et al. 2015). Targeting Wnt signalling pathways using synthetic modulators including small molecules and peptides is therefore a promising tool to inhibit Wnt-driven diseases such as cancer (Nile and Hannoush 2016; Anastas and Moon 2013).
Ghrelin O-acyltransferase (GOAT), another MBOAT enzyme, mediates the covalent attachment of octanoic acid onto Ser-3 of the 28-amino acid peptide hormone ghrelin (Fig. 9.1e) (Resh 2016; Yang et al. 2008; Gutierrez et al. 2008; Kojima et al. 1999; Müller et al. 2015). Ghrelin octanoylation is essential for the secretion of insulin and growth hormone, and hormone activity including appetite stimulation, adiposity and cardiovascular functions (Resh 2016; Gutierrez et al. 2008; Müller et al. 2015; Sato et al. 2015). Therefore, ghrelin is an attractive target in novel therapies to treat obesity and diabetes (Müller et al. 2015; Sato et al. 2015).
Protein prenylation refers to a post-translational attachment of isoprenoid lipids. Incorporation of farnesyl (C15) and geranylgeranyl (C20) groups is effected by formation of a thioether bond using a cysteine moiety in the C-terminal proximity of the protein via protein farnesyltransferase (FT) and geranylgeranyltransferase 1 (GGT 1), Fig. 9.1f, g, respectively (Wang and Casey 2016). The fully processed lipidated protein contains a prenylated cysteine residue with a methylated carboxylic acid moiety, at the protein C-terminus. Members from HRAS, KRAS, NRAS, prelamin A, lamin B, and RAS-related GTPases are examples of protein families incorporating isoprenoid lipids within their structures (Wang and Casey 2016). Prenylation controls the oncogenic activity of many proteins including farnesylated RAS proteins that are involved in 30% of human cancers (Wang and Casey 2016).
Another common eukaryotic post-translational lipid modification is the attachment of a complex glycosylphosphatidylinositol anchor to the C-terminus of proteins (Paulick and Bertozzi 2008; Ferguson et al. 2009). GPI comprises a phosphoethanolamine linker, a highly conserved glycan core (mannose(α1-2)mannose(α1-6)mannose(α1-4)glucosamine(α1-6)myo-inositol) and phospholipid tail which links the GPI anchor to the cell membrane (Paulick and Bertozzi 2008; Ferguson et al. 2009). The sugar-rich domain can be further modified with the addition of various groups including other glycans, sialic acid and phosphoethanolamine moieties affording functionally diverse glycoforms of GPI anchors (Paulick and Bertozzi 2008; Ferguson et al. 2009). The lipid portion of the GPI moiety differs depending on the protein which it is attached to and the organism it originates from. The GPI anchor of human erythrocyte acetylcholinesterase for example, comprises three fatty acids in various states of saturation and lengths ranging from 16 to 22 carbons (Fig. 9.2) (Paulick and Bertozzi 2008; Ferguson et al. 2009; Deeg et al. 1992; Roberts et al. 1988b, a). The exact structure-activity relationship of GPI-anchored proteins is poorly understood due to the complex nature of the GPI anchor structure. GPI-anchored proteins are multifunctional ; these proteins have been identified in receptors, hydrolytic enzymes, adhesion and regulatory molecules etc (Paulick and Bertozzi 2008; Ferguson et al. 2009).

Atg8 and LC3 proteins found in yeast and mammals respectively, contain a phospholipid moiety, namely phosphatidylethanolamine (PE) that is post-translationally anchored to a C-terminal glycine residue via numerous steps of ubiquitination-like reactions catalysed by autophagy-related (Atg) proteins (Resh 2013). It has been reported that increased levels of PE enhance autophagy, a cytoprotective mechanism responsible for degradation of toxic proteins and potentially harmful and damaged organelles (Feng et al. 2014; Rockenfeller et al. 2015). Modulating autophagy can be used for the treatment of human disorders including cancer, diabetes, and Alzheimer’s and Parkinsons’ disease therefore new autophagy controllers are strongly desirable (Feng et al. 2014; Rockenfeller et al. 2015).
In summary, regulating the action of lipidated proteins may lead to potential therapies to treat infectious disease and human pathologies. Targeting NMT, Hedgehog acyltransferase, FT and GGT 1 inhibitors may play a role in anticancer therapies (Wang and Casey 2016; Berndt et al. 2011). Effective techniques to modulate prenylation patterns can be used in hepatitis D and C viruses (HDV and HCV) treatment (Koh et al. 2015; Cory et al. 2015; Ye et al. 2003), premature ageing disorders such as Hutchinson-Gilford progeria syndrome (HGPS) (Gordon et al. 2014; Young et al. 2013) in addition to neurodegenerative pathologies like multiple sclerosis and Alzheimer’s disease (Wang and Casey 2016; Gao et al. 2016).
9.1.2 Nature-Derived Lipopeptides with Therapeutic Potential
Lipopeptides isolated from microorganisms such as fungi and bacteria show great therapeutic promise in the development of novel antimicrobial (Cochrane and Vederas 2016), antifungal, antitumor, and anti-inflammatory agents. In case of the plipastatins they can also act as potential therapies for neurological diseases (Dey et al. 2015).
Bacillus and Paenibacillus spp. produce lipopeptides of various structures including cyclic cationic and non-cationic lipopeptides where ring formation mostly occurs via the ester or amide bond and engages the C-terminal carboxylic acid residue (Cochrane and Vederas 2016). The presence of both, d- and l-amino acids together with non-natural amino acids in these lipopeptide sequences is common and improves peptide stability against enzymatic degradation. Branched saturated or unsaturated fatty acids with diverse structures with the main chain varying mostly between C11 to 14 carbons are mostly incorporated into the Nα-terminal side of the peptides and often feature a β-hydroxyl moiety in their structure (Cochrane and Vederas 2016; Jacques 2011).
Polymyxins, octapeptins, pelgipeptins, and paenibacterins exhibit non-proteinogenic 2,4-diaminobutyric acid (Dab) residues that amplify the cationic character of these peptides (Cochrane and Vederas 2016). Examples of non-cationic cyclic lipopeptides include the iturin-, surfactin-, fengycin-, fusaricidin-, marihysin-, and kurstakin-families (Fig. 9.3) (Cochrane and Vederas 2016).

Linear cationic lipopeptides derived from Bacillus and Paenibacillus spp. such as cerexins and tridecaptins display promising antibacterial activity against Gram-positive and Gram-negative microbes (Fig. 9.3). A more detailed description of exact structures and biological activities for Bacillus and Pseudomonas spp. derived lipopeptides has recently been published (Cochrane and Vederas 2016; Jacques 2011; Mnif and Ghribi 2015).
Lipopeptides isolated from Pseudomonas spp., which mainly include the viscosins, amphisins and tolaasins in addition to syringomycins, are mostly known for their antiviral and antimicrobial properties (Mnif and Ghribi 2015; Raaijmakers et al. 2006). These structurally diverse cyclic peptides differ in the chain length and comprise 9-25 residues in the form of natural and non-natural amino acids including allo-threonine (allo-Thr), allo-isoleucine (allo-Ile), 3-hydroxyaspartic acid, Dab and homoserine (Hse). 4-Chlorothreonine is the amino acid responsible for the antifungal activity of syringomycin (Fig. 9.4) (Grgurina et al. 1994). The fatty acid moiety attached to the N-terminus of the peptide chain varies in length and composition and, similar to Bacillus-derived peptides, often features the β-hydroxyl unit. The lactone ring is generally formed between the carboxylic acid of the C-terminal amino acid and the hydroxyl group of either Ser, Thr or allo-Thr present within the peptide chain (Mnif and Ghribi 2015; Raaijmakers et al. 2006).

Chemical structures of non-natural amino acids found in lipopeptides isolated from Pseudomonas spp.
Other microbial sources of biologically active lipidated peptides with promising therapeutic potential found in nature include strains of Acremonium, Streptomyces, and Actinoplanes (Mnif and Ghribi 2015).
Lipopeptides exhibit a broad spectrum of activities against many pathogens and some naturally-derived compounds, as in the case of daptomycin, polymyxin B or colistin, have already received the Food and Drug Administration (FDA) approval. Daptomycin (Cubicin) isolated from Streptomyces roseosporus is a 13-amino acid, cyclic lipopeptide, containing decanoic acid at the Nα-amino group of the N-terminal l-tryptophan. Daptomycin exhibits potent activity against Gram-positive pathogens (Fig. 9.5) (Debono et al. 1987; Vilhena and Bettencourt 2012).

Chemical structure of daptomycin (cubicin)
Polymyxins are mixed peptide antibiotics produced by Bacillus polymyxa and are considered to be the last-line of defence agents against Gram-negative organisms; their use is limited due to concerns with nephrotoxicity (Stansly and Schlosser 1947; Benedict and Langlykke 1947). The general structure of polymyxins comprises a cyclic heptapeptide core attached to a tripeptide unit containing a lipid portion at the Nα-terminal site of the linear fragment (Velkov et al. 2016). Polymyxins are mixtures of structurally similar peptides. Members of the polymyxin B family mostly differ in the fatty acid component of the antibiotics. Examples include (S)-6-methylheptanoic acid for polymyxin B1 and B2 respectively (Velkov et al. 2016; Orwa et al. 2001). Colistin A and colistin B are highlighted examples of the polymyxin E family; these antibiotics differ in the substitution of d-phenylalanine to d-leucine at position six of polymyxin B (Fig. 9.6) (Velkov et al. 2016; Brink et al. 2014).

Chemical structure of polymyxin B1, B2 and colistin A and B
The concept of protein lipidation is clearly not uncommon in nature hence application of this strategy to the therapeutic arena offers enormous potential for the generation of effective peptide-based drug candidates. Therefore, development and synthetic optimisation of naturally derived lipopeptides may afford fine-tuned therapeutics, which are less toxic, more potent and capable of treating multidrug-resistant infections. Interestingly, it has been reported that the attachment of aliphatic chains of various length (C12-C16) can modulate antimicrobial and antifungal activity of otherwise inert short peptides (Makovitzki et al. 2006). Therefore, peptide lipidation can be used as an effective strategy to generate peptide drug leads with clinical potential.
9.1.3 Peptide Lipidation to Generate Peptide-Based Therapeutics
Peptide lipidation can modulate the physicochemical and pharmacological properties of bioactive peptides generating therapeutically useful targets. Increased lipophilicity of peptides due to the presence of fatty acids affects the secondary structure and receptor and membrane binding characteristics of peptides; accordingly lipidation alters absorption, distribution, metabolism, and excretion (ADME) properties and therefore is an attractive tool to convert peptides into drug candidates (Zhang and Bulaj 2012). The most notable examples of clinically relevant lipidated peptides include long-acting insulin detemir (Levemir®) (Home and Kurtzhals 2006; Le Floch 2010) and liraglutide (Victoza®) (Jackson et al. 2010; Knudsen et al. 2000), a glucagon-like peptide-1 (GLP-1) receptor agonist, which are both used to treat diabetes (Fig. 9.7).

Primary sequence of GLP1(7-37), liraglutide and insulin detemir
The prolonged activity of insulin detemir is due to the presence of C14 myristic acid incorporated into lysine-29 of the B chain of a modified insulin peptide sequence where the threonine-30 residue was removed (Fig. 9.7) (Le Floch 2010; Kurtzhals 2007).
Liraglutide is a long-acting analogue of GLP-1(7-37) where Lys-34 was replaced with Arg and Lys-26 was acylated with a C16 fatty acid attached to γ-glutamic acid as a spacer. The palmitic acid moiety plays a crucial role in delaying liraglutide absorption and extending the half-life of the drug which has been estimated to be 13 hours after subcutaneous injection compared to approximately 2 minutes for the native GLP-1 (Rigato and Fadini 2014; Elbrond et al. 2002). In addition, renal clearance of the drug is reduced due to the shielding effect of the fatty acid moiety; liraglutide binds to plasma albumin via the fatty acid group preventing drug degradation by dipeptidyl peptidase-4 (DPP-4) (Malm-Erjefalt et al. 2010; Watson et al. 2010). Lipidation of potent, but unstable GLP-1(7-37), much improved the pharmacokinetic profile of the peptide making it suitable for once-daily administration (Elbrond et al. 2002; Ryan and Hardy 2011). Liraglutide (Saxenda®) has been recently approved by the FDA and the European Medicines Agency (EMA) for adjunctive treatment of obesity (December 2014 and March 2015, respectively) (Iepsen et al. 2015; Bray 2015; Tomlinson et al. 2016).
It has been reported that the type and composition of the fatty acid attached to a bioactive peptide as well as the nature of the spacer between the peptide chain and the fatty acid moiety influences its activity and plasma half-life (Knudsen et al. 2000; Madsen et al. 2007; Lau et al. 2015).
Structure-activity studies of liraglutide analogues revealed the importance of the length, composition, polarity and bulkiness of the fatty acid moiety as well as the type of spacer between the active molecule and the lipid tail on half-life calculations (in vivo in pigs) and potency using the cloned human GLP-1 model (Knudsen et al. 2000; Madsen et al. 2007). Linear fatty acids ranging from C10 to C18 (1) incorporated into the liraglutide sequence using various linkers including α-, d-γ-glutamic acid, 4-aminobutanoic acid (GABA), β-alanine and triethyleneglycol were evaluated (Fig. 9.8a) (Madsen et al. 2007). Interestingly, prolonged activity increased with the fatty acid chain length starting from 0.8 hours for C10, increasing to 5.1 h (C11), 7.6 (C12), 9 h (C14), 16 h (C16) and 21 h (C18); receptor potency was only affected when the acid chain length was longer than 16 carbons (Madsen et al. 2007). The study underlined the importance of the spacer between the active peptide and the fatty acid and revealed the complete loss of receptor potency when palmitic acid was directly bound to Lys-26 (Madsen et al. 2007). Liraglutide analogues containing α- or d-γ-glutamic acid (2 and 3), GABA (4) or β-Ala (5), as linkers in place of the native γ-Glu demonstrated similar activities and half-life values to those of liraglutide; unlike the triethylene glycol linker (6) which caused a 25-fold decrease in activity (Fig. 9.8b) (Knudsen et al. 2000; Madsen et al. 2007). Increasing the polarity of the fatty acid component by introducing one or more ether groups (7-9) or inserting hydroxyl group at the omega terminus (10) decreased the protraction of the analogues possibly due to reduced interactions with the fatty acid sites present on albumin, Fig. 9.8c (Madsen et al. 2007). Modification of the C16 palmitic acid in the liraglutide sequence with 2-hexyldecanoyl acid (11) which is equivalent to 16 carbon atoms led to slightly improved protraction (18 hours versus 16 h) and a significant decrease in potency of the analogue. Incorporation of more bulky phenyl- and cyclohexyl rings (12 and 13, respectively) in place of palmitate, or palmitate replacement with a pentylbenzenesulfonyl group (14) was not beneficial in regards to improved potency and half-life values compared to the original molecule (Fig. 9.8d) (Madsen et al. 2007).

Selected modifications of an acyl component (a, c, d) and spacer (b) investigated during the structure-activity study on liraglutide (Madsen et al. 2007)
Further derivatization of the liraglutide structure resulted in the development of semaglutide (Lau et al. 2015; Nauck et al. 2016). Semaglutide is the once-weekly GLP-1(7-37) analogue currently in phase 3 clinical development for the treatment of type 2 diabetes (Lau et al. 2015; Nauck et al. 2016). Extending the half-life of semaglutide to 165 hours was realised through systematic study of the fatty acid chain type and the spacer attached to liraglutide (Lau et al. 2015). The superior effect of a C18 octadecanedioic acid moiety attached to Lys-26 and a long spacer unit composed of γ-Glu attached to two 8-amino-3,6-dioxaoctanoic acid moieties provided the optimal lead candidate (Fig. 9.9). Non-natural modification of Ala-8 with 2-aminoisobutyric acid (Aib) allowed for additional shielding of the molecule from degradative DPP-4 action (Lau et al. 2015).

Primary sequence of semaglutide
The therapeutic potential of peptides as drugs is often hampered by undesirable ADME profiles; peptides are subjected to rapid proteolytic cleavage in the digestive system and are unable to cross the epithelial layer (Karsdal et al. 2015; Di 2015). Oral administration of peptide-based therapeutics is therefore limited. Many strategies to enhance oral delivery of peptides have been described in the literature. Generally, they include attachment of permeation enhancers (such as glycosides, lipids and PEG) and/or targeting proteolytic enzyme inhibitors. Exploration of multifunctional polymers as a polymeric matrix to provide controlled drug release and drug encapsulation in polymeric nanoparticulate systems has also been reported. Using ligand-specific binding and uptake techniques which employ vitamin B12, biotin, folate, and lectins to name a few, as drug carriers was also demonstrated. A more detailed discussion on these topics is covered elsewhere (Park et al. 2011; Karsdal et al. 2011, 2015). A brief discussion of lipidation phenomena affecting oral bioavailability with selected examples of biologically active peptides is described herein.
Chemical modification of the 32-amino acid salmon calcitonin (sCT) with an N-palmitoylated cysteine moiety attached to Cys-1 and Cys-7 of sCT via disulphide bonds greatly improved the bioavailability of the orally administrated native peptide (Wang et al. 2003). Significant levels of sCT could still be detected in rat plasma up to 12 hours after oral administration of lipidated-sCT compared to undetectable levels after 1 hour when the same dose of native sCT was used (Wang et al. 2003). In this report, a method termed ‘reversible aqueous lipidization’ (REAL) was used that allows for selective conjugation of a protein to a fatty acid via reversible disulphide linkage in aqueous solution using the water soluble N-palmitoyl cysteinyl 2-pyridyl disulphide reagent 15 (Scheme 9.1a) and the protein thiol (Ekrami et al. 1995). The REAL technique was applied to the lipidation of other therapeutic peptide drugs including Bowman-Birk protease inhibitor (BBI) (Ekrami et al. 1995), desmopressin (Wang et al. 1999; Wang et al. 2002) and octeotride (Yuan et al. 2005).

Peptide lipidation to improve oral bioavailability was also applied to the endogenous opioid peptide leu-enkephalin (ENK) using a modified REAL technique wherein 3,4-bis(decylthiomethyl)-2,5-furandione 16 was used to introduce a lipophilic moiety onto the Nα-amino group of the N-terminus (Scheme 9.1b) (Wang et al. 2006).
It has been reported that incorporation of lauric acid to the N-terminal pyroglutamyl group of thyrotropin-releasing hormone (TRH) significantly improved peptide penetration across the upper small intestine (Muranishi et al. 1991; Tanaka et al. 1996).
There is ongoing interest in developing an insulin formulation that could bypass the requirement for daily subcutaneous insulin injection for the management of diabetes (Wong et al. 2016; Ramesan and Sharma 2014). Promising reports on improved stability of mono- and di-palmitoylated insulin analogues in mucosal tissue homogenates compared to native insulin (Hashimoto et al. 1989; Hashizume et al. 1992) prompted further research into the effects of lipidation on the pharmacokinetic profile of insulin (Asada et al. 1994, 1995). The effect of acylation on the stability and absorption of insulin from the small and large intestines was examined using mono- and di-acylated bovine insulin analogues (Asada et al. 1994, 1995). Mono-acylated analogues were constructed via incorporation of caproic (C6), lauric (C12) and palmitic acid (C16) at the Nα-amino group of Phe-1 of the insulin B chain; di-acylated analogues were prepared by modification of both the Nα-amino group of Phe-1 and the Nε-amino group of Lys-29 of insulin B chain, with two copies of C6, C12, or C16 fatty acids (Fig. 9.10a). Mono-acylated analogues were found to be more stable in small intestinal fluid at 37 °C (Asada et al. 1994) and increased absorption of caproic acid-modified analogues from the large intestine was observed compared to the native compound (Asada et al. 1995).

Acylation of insulin and insulin analogues incorporating arginine residues at various sites within the insulin sequence, with various saturated and unsaturated fatty acids attached to the B chain showed improved solubility at moderately acidic pH inducing long-acting basal control of glucose levels (Flora 2002).
Hexyl-insulin monoconjugate 2 (HIM2) is an insulin analogue that can be administrated as an oral semisolid formulation in hard gelatin capsules (Clement et al. 2002; Still 2002; Kipnes et al. 2003; Clement et al. 2004). HIM2 was created by chemical modification of recombinant insulin by covalent attachment of an amphiphilic oligomer consisting of a lipophilic alkyl unit (C6) and a hydrophilic PEG moiety covalently bound to the Nε amino group of the Lys-29 (B chain) (Fig. 9.10b) (Clement et al. 2002, 2004; Still 2002; Kipnes et al. 2003).
Despite various scientific efforts, formulation of orally available insulin and other peptide-based drugs remains a challenging task (Lewis and Richard 2015; Hamman et al. 2005; Renukuntla et al. 2013; Karsdal et al. 2011, 2015).
Peptide lipidation has also been used to mimic the post-translational processes of sterol or lipid attachment facilitating protein association with cell membranes and subsequent initiation of protein activation or deactivation processes (Mejuch and Waldmann 2016; Resh 2013; Avadisian and Gunning 2013). This nature-derived strategy is often designed to generate lipid-anchored drugs including lipidated peptide inhibitors with improved in vivo half-life and cell-penetrating potential. The lipid moiety attached to a peptide allows drug anchoring within the cell membrane and enabling action on soluble cytosolic proteins and membrane-bound/associated proteins (Avadisian and Gunning 2013; Rajendran et al. 2012). Cholesterol and fatty acids of various chain lengths such as C8-caprylic, C12-lauric, and C16-palmitic are often utilized as lipid motifs that are covalently attached to a peptide inhibitor via ester, ether, amide or carbamate bonds (Avadisian and Gunning 2013; Zhao et al. 2012; Wexler-Cohen and Shai 2009; Remsberg et al. 2007; Rajendran et al. 2008a, b; Porotto et al. 2010; Johannessen et al. 2011; Avadisian et al. 2011).
This ‘lipid anchoring technique’ allowing for subcellular drug delivery by drug conjugation to a lipid via a linker, was recently used to effectively inhibit the action of endosomal β-secretase (Rajendran et al. 2008a, b). β-Secretase inhibitors may be useful for the treatment of Alzheimer’s disease by blocking the enzyme involved in amyloid formation. The lipid-anchored inhibitors consist of three main parts which include the pharmacophore (‘message’), the lipid anchor (‘address’), and the linker which conjugates both parts together and allows for optimal flexibility of the pharmacophore within the lipid bilayer to bind with the target (Rajendran et al. 2012). Simons et al. (Rajendran et al. 2008a, b) showed that that conjugation of a sterol to the β-secretase inhibitor (Glu-Val-Asn-statine-Val-Ala-Glu-Phe) via a polyglycol linker resulted in greater efficacy; β-secretase cleavage of β-amyloid precursor protein (APP) was decreased resulting in reduced β-amyloid peptide formation (Fig. 9.11). Importantly, the cholesterol-enriched drug was readily internalized into endosomes and cholesterol-sphingolipid domains (rafts) within cellular membranes where β-secretase activity is observed (Rajendran et al. 2008a, b; Hicks et al. 2012; Cordy et al. 2006). Comparison of stearyl-, palmityl-, myristyl-, and oleyl-linked inhibitors revealed cholesterol- and palmitoyl-linked analogues to be superior in terms of raft partitioning ability (Rajendran et al. 2008a).

The lipidation site within the peptide chain is critical as it can determine the pharmacokinetic and pharmacodynamic properties of drug candidates by affecting the solubility and the self-aggregating potential of lipopeptides. Ward et al. (Ward et al. 2013) investigated lipidated glucagon-based peptides to identify acylated co-agonists for the glucagon and glucagon-like peptide 1 receptors (GCGR and GLP-1R, respectively). A number of palmitoylated and C-amidated glucagon analogues were prepared where Ser-2 was substituted with an Aib moiety to prevent enzymatic degradation by dipeptidyl peptidase-4. The Nε-amino group of Lys-12 or an introduced lysine residue that was used to replace the mid-region moieties of glucagon, namely Tyr-10 or Tyr-13, Leu-14 or Ser-16, Arg-17 or Gln-20, was explored to attach a palmitic acid via a γGlu-γGlu dipeptide spacer (Ward et al. 2013). The solubility and aggregate-forming potential of glucagon analogues in phosphate-buffered saline (PBS) (pH 7.4) was variable. Decreased solubility and increased aggregation was observed for the acylated analogue at position 14 which correlated with its reduced in vivo activity compared to the other analogues (Ward et al. 2013). Interestingly, the study also revealed an increased proportion of helical content for all C16 fatty acid-tagged analogues in addition to improved potency at glucagon and GLP-1 receptors for most of the palmitoylated analogues. This is the first indication of enhancing in vitro receptor potency through helix stabilization by lipidation (Ward et al. 2013). This finding further reinforced the importance of lipidation in the development of therapeutic peptides (Ward et al. 2013). It was observed that saturated fatty acids with longer chains (>C8) have greater conformation-stabilising potential compared with unsaturated or hydroxyl counterparts due to enhanced hydrophobic interactions with the peptide chains (Zhang and Bulaj 2012). Lipidation was also shown to be an effective tool to induce peptide oligomerization and self-assembly resulting in the formation of micelles, tubules, vesicles, mono- and bilayer structures that can be used in both the drug delivery and tissue engineering fields (Zhang and Bulaj 2012; Hutchinson et al. 2017; Hamley 2015).
Peptide lipidation is an effective strategy to increase the drugable potential of bioactive peptides and has been applied to many other biomolecules not mentioned in this report including angiotensin II (Maletínskâ et al. 1996; Maletinska et al. 1997), BBI (Honeycutt et al. 1996), desmopressin (Wang et al. 1999; Wang et al. 2002), galanin, (Saar et al. 2013; Robertson et al. 2010; Zhang et al. 2009), ghrelin (Bednarek et al. 2000), neuropeptide Y (NPY) (Green et al. 2011; Green et al. 2010), octreotide (Yuan et al. 2005), luteinizing hormone releasing hormone (LHRH) (Toth et al. 1994), tetragastrin (Fujita et al. 1998; Setoh et al. 1995; Yodoya et al. 1994), and more. Further details relating to the above mentioned lipidated analogues can be found in the recent review by Zhang and Bulaj (Zhang and Bulaj 2012).
9.1.4 PamnCys Ligand as Adjuvant for Peptide-Based Vaccines
There has been significant interest directed towards the development and synthesis of peptide vaccines as alternatives to conventional vaccines, where potentially toxic, whole live attenuated or killed microorganisms are used to elicit immune responses (Simerska et al. 2011; Moyle and Toth 2008; Li et al. 2014; Brown and Jackson 2005). One of the limitations of peptide-based vaccines is the lack of immunogenicity thus requiring the inclusion of an effective and safe adjuvant (Simerska et al. 2011; Moyle and Toth 2008; Khong and Overwijk 2016).
A less explored class of immune adjuvants are compounds stimulating innate-like T cells, semi-activated T cells with an invariant T cell receptor (TCR) represented by the invariant natural killer T cells (NKT) that recognize glycolipid antigens binding to the lipid antigen-presenting molecule CD1d (Fujii et al. 2003; Hermans et al. 2003). The most well-known CD1d ligand is α-galactosylceramide (α-GalCer, KRN 7000) (Godfrey and Kronenberg 2004) and studies on the use of α-GalCer conjugated to peptide antigens generating potent self-adjuvanting vaccine constructs have been reported (Anderson et al. 2014, 2015 Cavallari et al. 2014).
Toll-like receptors (TLRs) are transmembrane glycoproteins which play an important role in initiating an innate immunity response and developing the adaptive immune response (Gay and Gangloff 2007; Basto and Leitao 2014). Ten members of the human TLR family namely TLR1-TLR10 have been identified. TLR agonists vary and include viral genetic material, microbial nucleic acids and microbial membrane components (Mifsud et al. 2014). Stimulation of TLRs may therefore lead to potent therapies against infectious diseases and many TLR ligands have been evaluated as potential treatments of viral and bacterial infections (Basto and Leitao 2014; Mifsud et al. 2014; Zaman and Toth 2013; Khong and Overwijk 2016).
Lipopeptides derived from bacterial cell wall components including lipoproteins, peptidoglycans, lipoteichoic acid and lipopolysaccharides can activate Toll-like receptor 2 (TLR2) (Basto and Leitao 2014; Zaman and Toth 2013). Conjugation of lipids and liposaccharides to peptide antigens is therefore used to elicit an immune response and plays an important role in self-adjuvanting vaccine development (Simerska et al. 2011; Moyle and Toth 2008; Zaman and Toth 2013).
Common lipidated moieties employed in vaccine design to induce immunogenicity include synthetic analogues of lipoprotein components of Escherichia coli (Braun 1975) and Mycoplasma (Muhlradt et al. 1998; Muhlradt et al. 1997), namely S-[2,3-bis(palmitoyloxy)propyl]-N-palmitoyl-l-cysteine (Pam3Cys) (17) and S-[2,3-bis(palmitoyloxy)propyl]-l-cysteine (Pam2Cys) (18) (Zeng et al. 2002), respectively (Fig. 9.12) (Khong and Overwijk 2016).

Chemical structure of Pam3Cys (17), Pam2Cys (18), PamCys (19), N-acetylated PamCys (20) and UPam
Pam3Cys and Pam2Cys have been used as adjuvants in several peptide-based vaccine studies directed towards treating various infectious diseases including, HIV, HBV, hepatitis C (Chua et al. 2008; Chua et al. 2012; Eriksson and Jackson 2007), Lyme disease and influenza (Moyle and Toth 2008; Khong and Overwijk 2016; Zaman and Toth 2013; Chua et al. 2015; Tan et al. 2012) in addition to melanoma (Zom et al. 2014). Better water solubility and similar or improved immunogenicity shown by Pam2Cys compared to Pam3Cys (Zaman and Toth 2013; Jackson et al. 2004), makes this motif an even more interesting synthetic target for incorporation into peptide-based vaccines. Structure-activity studies carried out for Pam2Cys demonstrated enhanced activity by the natural (R) configuration at the asymmetric glyceryl carbon, in comparison to the (S) isomer, namely S-[2(R),3-bis(palmitoyloxy)propyl]-l-cysteine [(R)-Pam2Cys], and S-[2(S),3-bis(palmitoyloxy)propyl]-l-cysteine [(S)-Pam2Cys], respectively (Moyle and Toth 2008; Zaman and Toth 2013; Wu et al. 2010; Takeuchi et al. 2000). Conversely, incorporation of the (R/S) diastereoisomer of Pam3Cys within the MUC1 antitumor vaccine construct elicited immune responses similar to that of the same MUC1 glycopeptide comprising only the (R)-enantiomer (Shi et al. 2016).
It has been reported that the Pam2Cys fatty acid chain length plays a crucial role in determining TLR2 activation; the minimum carbon chain length required for immunogenic activity is C8 and the strength of immune response increases with carbon addition up to C16 (C18=C16>C12>C8) (Moyle and Toth 2008; Zaman and Toth 2013; Buwitt-Beckmann et al. 2005b; Chua et al. 2007). A more soluble derivative of Pam2Cys, namely Pam2CysSK4 showed the most promising activity amongst a range of adjuvants tested in the evaluation of a Chlamydia trachomatis vaccine (Cheng et al. 2011; Spohn et al. 2004). It has been reported that the presence of a serine moiety within the Pam2CysSK4 motif plays a role in enhanced agonist activity for TLR2 (Wu et al. 2010; Kang et al. 2009).
Further SAR studies on Pam2CysSK4 led to identification of a structurally simpler and water soluble monopalmitoylted analogue 19 and its Nα-amino acetylated variant 20 possessing strong TLR2-agonistic activities, comparable to that of Pam2CysSer, in human (but not murine) blood (Fig. 9.12) (Agnihotri et al. 2011; Salunke et al. 2012). The correct spacing between the ester-linked palmitate and the thioether was found to be crucial for activity of analogue 19 and replacement of the ethyl chain with a propyl chain resulted in loss of activity (Wu et al. 2010; Agnihotri et al. 2011; Salunke et al. 2012).
Replacement of the native amide bond within the Pam3Cys motif with an urea led to discovery of a novel TLR2 ligand termed UPam; substitution of the native N-palmitoyl chain of Pam3Cys with an N-tetradecylcarbamyl moiety afforded a ligand with improved immunostimulatory activity compared to the parent lipopeptide (Fig. 9.12) (Zom et al. 2014, 2016; Willems et al. 2014).
The use of a cationic lipidated peptide such as R4Pam2Cys to elicit T-cell immunity via TLR2 stimulation was recently described; the strategy relies on electrostatic attraction of the R4Pam2Cys moiety with soluble protein antigens obviating the need for covalent bond generation between the TLR2 ligand and the antigen (Chua et al. 2014).
The use of palmitic acid, lipoamino acids and other lipid-based immunopotentiators, as an alternative to PamnCys, covalently bound to synthetic (glyco)peptides to improve the self-adjuvanting effect of vaccine constructs has been reported and is reviewed elsewhere (Moyle and Toth 2008; Khong and Overwijk 2016; Basto and Leitao 2014; Zaman and Toth 2013; McDonald et al. 2015; Steinhagen et al. 2011).
9.1.5 Chemical Approaches for Incorporation of PamnCys Ligands
Finding efficient methods to conjugate antigens to lipopeptide adjuvants remains challenging (McDonald et al. 2015). A simple and low-cost synthetic approach for peptide-lipid conjugation to effectively activate TLR2 to afford synthetic material in significant quantities for biological evaluation, is highly desired. A synthetic strategy must be devised using techniques from the chemistry toolbox that are compatible with the presence of lipid, carbohydrate and peptide moieties often required for self-adjuvanting vaccines . Herein, the most recent advances in synthetic techniques used to incorporate TLR2 ligands based on the PamnCys moiety into (glyco)peptides are summarized.
A solution phase synthesis of a simple dipeptide by direct condensation of Nα-9-fluorenylmethoxycarbonyl (Fmoc)-protected S-(2,3-bis(hydroxyl)propyl)-l-cysteine with serine where the side chain hydroxyl is protected with a tert-butyl (tBu) ether was reported by Jung et al. (Metzger et al. 1991). Subsequent palmitoylation of S-glycerylcysteinyl hydroxyls using palmitic acid, N,N′-diisopropylcarbodiimide (DIC) and 4-(dimethylamino)pyridine (DMAP), followed by tBu protecting group removal from the serine side chain effectively provided Fmoc-Pam2CysSer (Metzger et al. 1991).
Danishefsky et al. (Kudryashov et al. 2001) employed a solution phase approach to successfully incorporate the Pam3Cys ligand into a trivalent Lewis Y antigen resulting in antibody production in animal models. However, more common approaches to incorporate the PamnCys motif into peptides when designing a synthetic vaccine mostly rely on Fmoc solid phase peptide synthesis (SPPS). In this case, the peptide-based vaccine construct is synthesized first followed by lipid attachment. This approach however may prove problematic when synthesizing long or difficult peptide sequences (Zeng et al. 2011).
Alternatively, a convergent or modular approach can be used requiring initial preparation of vaccine motifs that are later conjugated, mostly via a linker, affording a self-adjuvanting vaccine construct (Zeng et al. 1996, 2001, 2002, 2011; Harris et al. 2007; Buwitt-Beckmann et al. 2005a; Metzger et al. 1995). The choice of chemical linkage used for adjuvant-antigen conjugation is very important and may influence the bioactivity of the construct (Zeng et al. 2011).
9.1.5.1 Convergent and Modular Approaches to Self-Adjuvanting Vaccine Constructs
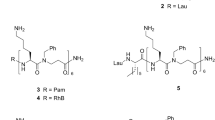
A fully synthetic convergent approach for the preparation of the minimal vaccine construct consisting of the S-[2(R),3-bis(palmitoyloxy)propyl]-N-palmitoyl-l-cysteine [(R)-Pam3Cys)], a helper T cell epitope and TN antigen (GalNAc) leading to high titres of IgG antibodies in mice was reported by Boons et al. (Buskas et al. 2005). In this example, the resin-bound and side chain protected peptide T cell epitope derived from an outer-membrane protein of Neisseria meningitides (Wiertz et al. 1992) was first synthesized using Fmoc SPPS using the extremely acid sensitive 4-(4-hydroxymethyl-3-methoxyphenoxy)butyryl-p-methylbenzhydrylamine (HMPB-MBHA) resin affording H2N-Y(tBu)AFK(Boc)Y(tBu)AR(Pbf)H(Trt)AN(Trt)VGR(Pbf)N(Trt)AFE(OtBu)LFLG-resin (21) (Scheme 9.2). To minimize racemization at cysteine, Pam3Cys was introduced into the epitope sequence using the Fmoc-S-[2(R),3-bis(palmitoyloxy)propyl]-l-cysteine (Fmoc-(R)-Pam2Cys-OH) 22 under the activation of (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP), 1-hydroxybenzotriazole (HOBt), N,N-iisopropylethylamine (iPr2NEt) in a mixture of N,N-dimethylformamide (DMF) and CH2Cl2. Subsequent acylation of the Fmoc-deprotected Nα-amino group of Cys with palmitic acid and using PyBOP and HOBt, followed by resin cleavage [2% trifluoroacetic acid (TFA) in CH2Cl2] gave the side-chain protected Pam3Cys-tagged lipidated peptide 23. Finally, condensation of 23 with a spacer containing tumour-associated TN antigen 24 activated by DIC, 1-hydroxy-7-azabenzotriazole (HOAt) and iPr2NEt and ultimate side chain protecting group removal using 95% TFA gave the target vaccine construct 25 (Scheme 9.2) (Buskas et al. 2005)

Convergent approach to a self-adjuvanting lipidated vaccine construct incorporating Pam3Cys TLR2 ligand by Boons et al. (Buskas et al. 2005). Reagents and conditions: (i) 22, PyBOP, HOBt, iPr2NEt, DMF/CH2Cl2 (1:5, v/v); (ii) 20% piperidine in DMF; (iii) CH3(CH2)14COOH, PyBOP, HOBt, DMF/CH2Cl2 (1:5); (iv) 2% TFA in CH2Cl2; (v) 24, DIC, HOAt, iPr2NEt, DMF/CH2Cl2 (2:1, v/v), 79%; (vi) TFA/H2O/1,2-ethanedithiol (EDT) (95:2.5:2.5, v/v/v)
Jackson et al. (Zeng et al. 2011) proposed a modular approach (Zeng et al. 2001) for the preparation of self-adjuvanting vaccine constructs, where standard Fmoc SPPS was used. On-resin incorporation of the Fmoc-Pam2Cys-OH (Zeng et al. 2002; Metzger et al. 1991; Jones 1975; Hida et al. 1995) via a diserine spacer to the Nε of an N-terminal lysine afforded lipidated CD4+ T(TH) cell epitope (Zeng et al. 1996, 2002, 2011). The lipid-tagged TH epitopes were then further N-terminally modified to facilitate a chemoselective ligation with complementary functional groups present at the target epitope modules affording oxime-, thioether-, and disulphide bond-linked lipidated vaccine constructs, ready for antibody response studies using animal models (Zeng et al. 2011).
Thus, Fmoc SPPS of TH epitopes containing N-terminal lysine with Nα-and Nε-amino groups orthogonally protected using 1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl (Dde) and Fmoc protecting groups respectively, were prepared affording TH constructs of general structure 26 (Scheme 9.3). Removal of the Fmoc protecting group using piperidine then allowed for peptide elongation via the exposed Nε-amino group to effect incorporation of the diserine spacer. Subsequently, the Fmoc-Pam2Cys-OH building block was attached using N-[(1H-benzotriazol-1-yl)(dimethylamino)methylene]-N-methylmethanaminium tetrafluoroborate N-oxide (TBTU), HOBt and iPr2NEt in CH2Cl2. The Fmoc protecting group of Pam2Cys moiety was then exchanged for the N-(tert-butoxycarbonyl) (Boc) (di-tert-butyl dicarbonate, Boc2O) allowing for orthogonal removal of the Dde from the N-terminal amino group of Lys using 2% hydrazine hydrate in DMF, providing lipidated TH construct 27. Boc-Cys(Trt)-OH or (Boc-aminooxy)acetic acid were then coupled to the lipidated epitope 27 with subsequent peptide cleavage from the resin using TFA to give Pam2Cys-tagged TH epitopes with sulphydryl- (28), or aminooxyacetyl-functionality (29) at the N-terminus, as handles for subsequent elongation with target epitopes (Scheme 9.3).

Synthesis of lipidated-TH epitopes incorporating Pam2Cys TLR2 ligand by Jackson et al. (Zeng et al. 2011). Reagents and conditions: (i) 20% piperidine in DMF; (ii) Fmoc-Ser(tBu)-OH, HBTU, HOBt, iPr2Net, DMF, then (i), repeated twice; (iii) Fmoc-Pam2Cys-OH, TBTU, HOBt, iPr2NEt, CH2Cl2, 16 h, then (i); (iv) Boc2O, DMF; (v) 2% hydrazine hydrate in DMF, 10 min; (vi) Boc-Cys(Trt)-OH, HBTU, HOBt, iPr2Net, DMF; (vii) (Boc-aminooxy)acetic acid, DMF; (viii) TFA/phenol/H2O/triisopropylsilane (iPr3SiH) (88:5:5:2)
The target epitopes were separately synthesized using Fmoc SPPS and their N-termini acylated with bromoacetic acid or cysteine while still bound to resin. TFA-mediated peptide cleavage from the resin subsequently afforded bromoacetyl-, and thiol-tagged epitopes 30 and 31, respectively. Alternatively, an additional serine residue was inserted at the N-terminus of the peptide sequence allowing for off-resin and sodium periodate-mediated serine oxidation affording an epitope with an N-terminal aldehyde handle 32. Chemoselective ligation between complementary-tagged TH and target epitopes in buffer solutions, namely 28 and 30 (aq buffer, pH 8), 28 and 31 (2,2′-dipyridyl disulphide), 29 and 32 (aq buffer, pH 3) gave thioether-, disulphide, and oxime-bond linked self-adjuvanting peptide-based vaccine constructs 33-35, ready for further bioanalysis (Zeng et al. 2011) (Scheme 9.4).

Schematic representation of modular approach to a self-adjuvanting lipidated vaccine constructs 33-35 incorporating Pam2Cys TLR2 ligand and different epitopes linked via thioether- (33), disulphide- (34), and oxime bond (35) by Jackson et al. (Zeng et al. 2011)
This modular approach (Zeng et al. 2001) ensured that attachment of the Pam2Cys motif at the Nε-amino group of Lys “in between” both epitopes orientating the vaccine constructs in a branched configuration. The Pam2Cys motif can also be incorporated at the Nα-amino group at the N-terminus of a vaccine construct; however, decreased immunogenic activity resulted following linear assembly, partially due to reduced solubility, compared to the branched vaccine counterparts (Zeng et al. 2002).
A new thioether ligation strategy to create self-adjuvanting peptide vaccine constructs using the Pam3CysSK4 moiety has been recently reported (Cai et al. 2013). This approach takes advantage of the complementary modified Pam3CysSK4 motif with a bromo-handle and thiol-containing antigen that are subsequently linked together via a thioether bond. Key to this approach was the initial preparation of an active intermediate Pam3CysSK4-K(COCH2Br)-OH 36 that was accessed by microwave-enhanced (MW) Fmoc SPPS. Herein, a Wang-resin was initially preloaded with lysine orthogonally protected with Fmoc at Nα and with a 1-(4,4-dimethyl-2,6-dioxo-cyclohexylidene)-3-methyl-butyl (ivDde) at Nε. Subsequent peptide chain elongation via the Nα-amino group followed by lipidation using Pam3Cys-pentafluorophenyl (Pfp) ester [HOBt in N-methyl-2-pyrrolidone (NMP) for 45 min at 50 °C] afforded resin-bound and side-chain protected Pam3CysS(OtBu)[K(Boc)]4-K(ivDde). The ivDde protecting group was then removed using hydrazine, and the Nε-amino group acylated with pentafluorophenyl bromoacetate. Subsequent TFA-mediated peptide cleavage gave Pam3CysSK4-K(COCH2Br)-OH 36. The key intermediate 36 was then converted into an active iodo-acetyl derivative using potassium iodide (KI) in urea/sodium acetate (NaOAc) mixture affording 37 (Scheme 9.5a). The iodo-acetyl moiety 37 was then ligated with several peptide epitopes that incorporated a thiol-terminated PEG spacer at their N-terminus. For example construct 38 was treated with 37 and trimethylamine (Et3N) in DMF at 40 °C affording construct 39 (Scheme 9.5b). The authors successfully applied this strategy for conjugation of a Pam3CysSK4 motif via a thioether linkage to B- and T-cell epitopes affording various self-adjuvanting vaccine constructs (Cai et al. 2013). The three-component construct 39 comprising P4 tetanus toxoid T cell epitope (Demotz et al. 1989; Monji and Pious 1997), linked via a PEG spacer with MUC1 glycopeptide comprising TN antigen, and a conjugated Pam3CysSK4 via a thioether linkage proved most efficacious (Cai et al. 2013).

Exemplified synthesis of three component synthetic vaccine incorporating Pam3Cys TLR2 ligand using thioether ligation strategy by Kunz et al. (Cai et al. 2013). Reagents and conditions: (i) MW Fmoc SPPS; (ii) (R)-Pam3Cys-OPfp, HOBt, NMP, 45 min, 50 °C; (iii) 2% hydrazine hydrate in DMF, 5 min, rt (repeated 3 x); (iv) BrCH2COOPfp, HOBt, 4 h, rt; (v) TFA/iPr3SiH/H2O (15:0.9:0.9, v/v/v); (vi) KI, 8 M urea/0.1M NaOAc, 30 min; (vii) NEt3, DMF, 40 °C
9.1.5.2 Native Chemical Ligation Approach to Self-Adjuvanting Vaccine Constructs
Native Chemical Ligation (NCL) (Dawson et al. 1994) enables synthetic access to long peptides and large biomolecules and has been used by our research group in numerous studies (Yang et al. 2013; Harris and Brimble 2015; Medini et al. 2015; Harris et al. 2015; Harris and Brimble 2013; Medini et al. 2016; Lee et al. 2011; Brimble et al. 2015; Son et al. 2014; Harris and Brimble 2010). NCL conjugates two synthetic partners containing complementary reactive sites, namely an N-terminal cysteine and a C-terminal thioester moiety via a thiol-catalysed chemoselective reaction affording a thioester-linked product; subsequent S→N transfer ensures the formation of a native peptide bond (Dawson et al. 1994). Brimble et al. (Harris et al. 2007) explored synthetic pathways to access Pam2Cys-linked thioester moiety that could be later incorporated into a long peptide via NCL. The initial effort to synthesise a more soluble derivative of Pam2Cys, namely Pam2CysSK4G thioester using tert-butyloxycarbonyl (Boc) SPPS resulted in unexpected cleavage of the palmitoyl esters during the final hydrofluoric acid (HF)-mediated peptide removal from the resin (Zeng et al. 2011). Successful synthesis of Pam2CysSK4G thioester was however completed using an alternative Fmoc SPPS strategy employing a sulfonamide ‘safety catch linker’ (Backes and Ellman 1999; Ingenito et al. 1999) and Fmoc-S-[2(S),3-bis(palmitoyloxy)propyl]-l-cysteine (Fmoc-(S)-Pam2Cys-OH) (40) as the building block (Scheme 9.6) (Harris et al. 2007). Loading of 4-sulfamylbutyryl aminomethyl polystyrene resin with Fmoc-Gly-OH was initially performed [DIC, N-methylimidazole (N-Melm) in DMF/CH2Cl2 mixture] followed by standard Fmoc SPPS affording side chain protected peptidyl-resin 41. Subsequent coupling of lipidated building block 40 (Metzger et al. 1991; Hida et al. 1995) was effected (PyBOP/HOBt) and the Fmoc protecting group was exchanged to Boc (Boc2O in DMF/CH2Cl2 mixture) to provide 42. Resin-bound 42 was then activated with iodoacetonitrile in NMP, with subsequent cleavage from resin using benzyl thiol (BnSH). Finally side chain protecting groups removal using TFA afforded the desired Pam2CysSK4G thioester 43 (Harris et al. 2007).

Synthesis of a C-terminal thioester derivative of the Pam2CysSKKKK using Fmoc SPPS by Brimble et al. (Harris et al. 2007). Reagents and conditions: (i) Fmoc-Gly-OH, DIC, N-Melm, CH2Cl2, DMF; (ii) Fmoc SPPS; (iii) 40, PyBOP, HOBt, CH2Cl2; (iv) 20% piperidine in DMF; (v) Boc2O, CH2Cl2, DMF; (vi) ICH2CN, NMP; (vii) BnSH, DMF; (viii) TFA/phenol/iPr3SiH/H2O (88:5:2:5, v/v/v/v)
Boons et al. (Ingale et al. 2006) were the first to demonstrate a successful synthesis of a three-component glycolipidated peptide vaccine by sequential NCL of the suitably prepared ligation fragments; Fmoc SPPS was employed to synthesise the T-cell epitope C(Acm)YAFKYARHANVGRNAFELFLG-thioester (44), the tumour-associated glycopeptide fragment derived from MUC-1 CTSAPDT(GalNAc)RPAP (45), and the TLR2 ligand Pam3CysSK4G-thioester (46). Due to limited success when ligation of 44 with 45 was undertaken using standard NCL conditions (phosphate buffer containing 6 M guanidinium hydrochloride, thiophenol, 37 °C), new methodology involving incorporation of 44 with 45 into liposomes to aid solubility was used. A film of dodecylphosphocholine (DPC), thioester 44 and thiol 45 were hydrated via incubating in a phosphate buffer (pH 7.5) for 4 h at 37 °C in the presence of tris(2-carboxyethyl)phosphine (TCEP) and ethylenediaminetetraacetic acid (EDTA) to suppress disulphide bond formation. The mixture was then sonicated and the resulting peptide/lipid suspension formed uniform 1 μm vesicles. Sodium 2-mercaptoethane sulfonate (MESNA) was subsequently added and ligation completed after 2 h at 37 °C affording 47 in high 78% yield after reversed-phase high-performance column chromatography (RP HPLC) purification (Ingale et al. 2006). Ligation of Pam3CysSK4G-thioester 46 with thiol 48, accessed by removal of the acetamidomethyl (Acm) protecting group from 47 [Hg(OAc)2], using liposome-mediated NCL afforded a three-component vaccine construct 49 in 83% yield after purification by chromatography (Scheme 9.7). The scope of this technique was later demonstrated by the synthesis of other self-adjuvanting vaccine constructs that differ in the composition of the (glyco)peptide and lipid component; some of the constructs proved highly immunogenic when tested in mice models (Ingale et al. 2006; Ingale et al. 2007; Lakshminarayanan et al. 2012; Abdel-Aal et al. 2014; Ingale et al. 2009).

Liposome-mediated NCL to the synthesis of three-component vaccine construct incorporating Pam3Cys TLR2 ligand by Boons et al. (Ingale et al. 2006). Reagents and conditions: (i) 200 mM sodium phosphate buffer (pH 7.5), DPC, TCEP, EDTA, sonication, extrusion, and then MESNA; (ii) Hg(OAc)2, 10% aq HOAc, 50 mM dl-dithiothreitol (DTT), 89%
The liposome-mediated NCL approach allowed for the generation of a native amide linkage between each of the required vaccine modules. However, the use of dodecylphosphocholine liposomes in ligation buffers can be limiting owing to the need for RP HPLC purification after each ligation step to isolate the product (McDonald et al. 2015; Ingale et al. 2006).
9.1.5.3 Fragment Condensation Approach to Self-Adjuvanting Vaccine Constructs
Kunz et al. (Kaiser et al. 2010) and Payne et al. (Wilkinson et al. 2010) described a fragment condensation approach to incorporate a Pam3Cys TLR2 ligand into mono- and per-glycosylated MUC1 glycopeptides respectively, using a PEG-based spacer to access fully synthetic vaccine constructs.
The Kunz approach involved initial synthesis of the lipidated, side-chain protected and the C-terminal carboxylic acid Pam3CysS(tBu)K(Boc)K(Boc)K(Boc)K(Boc) (50) unit using Fmoc SPPS. The MUC1 glycopeptides N-terminally modified with PEG linker, namely H2N(CH2CH2O)3CH2CH2CONH-PAH-GVT(sugar)-SAP-DTR-PAP-GST-AP-OH, comprising either TN- (51), T- (52) or 2,6-sialyl-T-antigen (53) at the singly glycosylated Thr-6 were then accessed via Fmoc SPPS. The fragment condensation was subsequently effected in solution and using N-[(dimethylamino)-1H-1,2,3-triazolo[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU)/HOAt and 4-methylmorpholine (NMM) in DMF which was followed by TFA-mediated protecting group removal and purification affording three novel vaccine constructs, 54, 55 and 56 in 25%, 21% and 20% yield, respectively (Scheme 9.8). Importantly, bio-assessment of TLR2 ligand-MUC1 assembly comprising T-antigen 55 showed the ability to elicit humoral immune response in mice (Kaiser et al. 2010).

Fragment condensation for the synthesis of the vaccine construct incorporating Pam3Cys TLR2 ligand by Kunz et al. (Kaiser et al. 2010). Reagents and conditions: (i) HATU, HOAt, NMM, DMF; (ii) TFA/iPr3SiH/H2O (10:1:1, v/v/v), 1.5 h
The Payne group employed the lipopeptide component with a PEG-like spacer at C-terminus, namely Pam3CysS(tBu)-CONH(CH2CH2O)2CH2COOH (57), and per-glycosylated full copies of the MUC1 VNTR domain epitope (GVT(sugar)-S(sugar)-APDT(sugar)-RPAPGS(sugar)T(sugar)APPAH), incorporating no copies (58) or multiple-copies of either TN- (59) or T-antigen (60), for convergent conjugation. All peptide fragments 57-60 were synthesized using Fmoc SPPS. The free carboxylic acid of the lipid partner 57 was pre-activated using pentafluorophenyl ester with ensuing fragment condensation with the requisite MUC1 epitopes 58, 59 or 60 using HOBt and iPr2NEt in DMF affording desired MUC1-Pam3Cys chimeras with no sugars 60, or containing five copies of either TN-or T-antigen, 62 and 63, respectively (Scheme 9.9) (Wilkinson et al. 2010). This fragment condensation approach was also used in other studies by the Payne group to synthesise multiple-component vaccine constructs incorporating Pam3Cys (Wilkinson et al. 2012; McDonald et al. 2014; Wilkinson et al. 2011). The fragment condensation strategy is a good alternative to the liposome-mediated NCL approach reported by Boons et al. (Ingale et al. 2006, 2007; Lakshminarayanan et al. 2012) with no requirements for solubilizing agents.

Fragment condensation approach for the synthesis of vaccine constructs incorporating Pam3Cys by Payne et al. (Wilkinson et al. 2010). Reagents and conditions: (i) pentafluorophenol, DIC, CH2Cl2;(ii) 58 or 59 or 60, HOBt, iPr2NEt, DMF; (iii) TFA/iPr3SiH (1:1, v/v)
9.1.5.4 Linear Approach to Self-Adjuvanting Vaccine Construct
The Boons group has recently reported a linear synthesis to access a three-component cancer vaccine composed of a B-cell epitope glycosylated with a sialyl-TN moiety, a TH epitope derived from polio virus (Leclerc et al. 1991) and a Pam3CysSK4 ligand. The key strategies employed by the Boons group included the use of microwave-enahanced Fmoc SPPS and on resin incorporation of the Fmoc-(S)-Pam2Cys-OH (40) building block onto the free Nα-amino group of the pre-synthesised glycopeptide construct containing deprotected hydroxyl groups of the sugar moiety (Thompson et al. 2015). Fmoc protecting group removal from the Fmoc-(S)-Pam2Cys-tagged vaccine construct (piperidine) could be then followed by Nα-amino group palmitoylation using palmitic acid, HATU, HOAt and iPr2NEt in DMF. Finally, TFA treatment afforded fully synthetic vaccine construct 64 incorporating the Pam3Cys TLR2 ligand (Fig. 9.13). Biological evaluation demonstrated induction of potent humoral and cellular immune responses in transgenic mice (Thompson et al. 2015).

A fully synthetic vaccine construct prepared using MW-enhanced linear Fmoc SPPS and incorporating Pam3Cys TLR2 ligand by Boons et al. (Thompson et al. 2015)
A three-component vaccine construct similar to that described above, but incorporating the unnatural TN moiety, namely α-O-GalNAc-α-methylserine in place of threonine, within the MUC1 epitope was recently accessed using the MW-enhanced Fmoc SPPS strategy previously reported by Boons et al. (Thompson et al. 2015; Martinez-Saez et al. 2016). This novel vaccine construct however, showed only comparable efficacy to that reported for the assembly containing native threonine.
As shown above, a linear approach for the synthesis of complex multi-component lipidated peptides containing only natural peptide bonds demonstrates the efficiency of the microwave-assisted Fmoc SPPS technique. However, longer and/or more hydrophobic lipopeptide constructs may still be difficult to access when using a linear SPPS and alternative synthetic routes for lipid incorporation are in demand.
9.1.5.5 TLR2 Ligand Conjugation Using Copper(I)-Catalysed Huisgen 1,3-Dipolar Cycloaddition
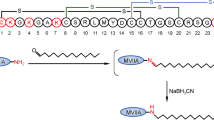
The need for large quantities of Fmoc-Pam2Cys building block required for SPPS conjugation poses a considerable obstacle due to the difficulty and cost involved in its synthesis. An alternative conjugation approach to incorporate the Pam2Cys moiety into a peptide could mitigate this conundrum. The copper(I)-catalysed Huisgen 1,3-dipolar cycloaddition of alkynes and azides to afford a 1,2,3-triazole conjugate (CuAAC ‘click chemistry’) offered promise for the conjugation of Pam2Cys with a peptide due to its tolerance of various functional groups and its complete regioselectivity to form 1,4-disubstituted products (Tornoe et al. 2002; Rostovtsev et al. 2002). The Brimble group therefore designed a Pam2Cys click building block containing an azide handle in place of the Nα-amino group of the cysteine residue which could be then clicked to a peptide functionalized with a propargyl moiety (Yeung et al. 2012). However, initial attempts to directly introduce an azide onto a free Nα-amino group of Pam2Cys using a diazotransfer reaction (Goddard-Borger and Stick 2007) proved unsuccessful, potentially due to obstruction of the reactive sites by the long palmitate groups (Yeung et al. 2012). A revised strategy was developed starting from an S-glyceryl cysteine intermediate 65 (Metzger et al. 1991; Pattabiraman et al. 2008) which was subjected to Nα-amino group deprotection (piperidine in CH2Cl2) to reveal the amino group for the ensuing diazotranfer reaction using imidazole-1-sulfonylazide.HCl, K2CO3 and CuSO4 .5H2O in MeOH (Goddard-Borger and Stick 2007) affording an azide-diol in 50% yield over 2 steps (Scheme 9.10a). Subsequent palmitoylation of the azide-diol [palmitic acid, DIC, and catalytic 4-(dimethylamino)pyridine (DMAP)] provided tBu-protected Pam2Cys azide 66 in 74% yield. Subsequent TFA treatment to remove the carboxyl protecting group gave the desired lipidated and azide-tagged ‘click’ ligation partner 67 in 84% yield (Yeung et al. 2012).

Synthesis of Pam2Cys azide 67 and Cu(I) ‘click’ conjugation of 67 with alkyne-modified peptides to get lipidated 70 and 71 by Brimble et al. (Yeung et al. 2012). Reagents and conditions: (i) piperidine, CH2Cl2, then imidazole-1-sulfonyl azide·HCl, K2CO3, CuSO4, MeOH, 50% over 2 steps;(ii) CH3(CH2)14COOH, DIC, DMAP, tetrahydrofuran (THF), 74%;(iii) TFA, 84%; (iv) CuI·P(OEt)3, iPr2NEt, DMF, 30 min
The synthesis of the alkyne-containing peptides for subsequent Cu(I) conjugation with 67 was undertaken using Fmoc SPPS (Yeung et al. 2012). Pentynoyl acid was coupled to the N-terminus affording 68 and propargylglycine (Pra) was used as an alkyne handle within the modified MUC1 peptide sequence, namely HGV-Pra-SAPDTRPAPGSTAPPA 69. The ‘click’ reaction of both alkyne-enriched peptides 68 and 69 using azide 67 was completed within 30 min as evidenced by RP HPLC using CuI·P(OEt)3 and iPr2NEt in DMF affording 1,2,3-triazole-linked Pam2Cys peptides 70 and 71, respectively. The amenability of the Pam2Cys azide to direct conjugation onto suitably modified peptides using the ‘click’ technique was successfully demonstrated (Scheme 9.10b) (Yeung et al. 2012). However, construct 70 was immunologically inactive possibly due to difference in the distance between the serine and the Pam2Cys (unpublished data). It has been reported that the exact length and geometry around the Cys-Ser unit is critical for activity of the Pam2CysSK4 motif (Wu et al. 2010; Kang et al. 2009).
Kunz et al. were the first to report CuAAC-assisted ligation of Pam3CysSK4 to a MUC1 glycopeptide to synthesise mono-, di- and tetra-valent MUC1 tandem repeat glycopeptide constructs to prepare of fully synthetic antitumour vaccines (Cai et al. 2011). The Kunz approach for the synthesis of monovalent MUC1 derivatives used Fmoc SPPS of MUC1 glycopeptide in which the N-terminal Nα-amino group was acylated with a PEG linker suitably modified with an azide handle affording construct 72. The ‘click’ synthetic partner 73 incorporated an alkyne group via a PEG spacer linking with the Pam3CysSK4 ligand by the Nε-amino group of the additional C-terminal lysine residue. The copper(I)-mediated reaction of the suitably prepared ‘click’ partners was then performed using copper acetate and Na ascorbate in H2O at 40 °C affording the monovalent vaccine construct 74 with >70% yield (Scheme 9.11) (Cai et al. 2011).

Synthesis of mono-valent ‘click’ construct 74 from azide-modified MUC1 antigen 72 and alkyne-modified Pam3Cys 73 and graphical representation of di- and tetra-valent vaccine constructs 75 and 76 by Kuntz et al. (Cai et al. 2011, 2014). Reagents and conditions: (i) copper acetate, Na ascorbate, H2O, 40 °C, >70%
The Nε-amino group of the C-terminal lysine linked to the Pam3CysSK4 moiety was later used as a point of attachment of additional lysine groups forming a multibranched lysine core which terminated with two or four copies of PEG-alkyne handles. Subsequent Cu(I) ‘click’ using the Pam3CysSK4 ligand incorporating two- or four alkyne groups and azide construct 72 afforded the desired di- (75) and tetra-valent (76) assemblies, respectively (Scheme 9.11) (Cai et al. 2011). Importantly, the tetravalent construct of general structure 76 synthesized using this strategy that incorporated the STN glycoside within the MUC1 sequence proved effective in inducing strong immune responses in mice including stimulation of killer cells (Cai et al. 2014).
The Sucheck group has reported the 1,2,3-triazole-mediated conjugation of a Pam3Cys ligand equipped with a C-terminal alkyne, with a 20-amino acid azide-tagged tandem repeat of MUC1 incorporating the TN unit (Sarkar et al. 2013). The alkyne-containing ‘click’ partner was available from Fmoc-Pam2Cys(OtBu) 77 by tert-butyl protection removal (TFA) followed by coupling with propargyl amine in the presence of PyBOP, HOBt and iPr2NEt in CH2Cl2. Fmoc protecting group removal and acylation of the revealed Nα-amino group with palmitic acid using PyBOP, HOBt, and iPr2NEt gave the alkyne functionalized Pam3Cys 78, Scheme 9.12a. The glycopeptide-azide was prepared via Fmoc SPPS on Fmoc-Ala-WANG resin using DIC/HOBt as coupling reagent and piperidine in DMF for Fmoc removal, affording resin-bound 79. The azido group was installed on-resin by coupling 6-azidohexanoic acid to the N-terminal proline residue of MUC1 followed by TFA-mediated peptide cleavage from the resin and acetyl deprotection of the TN hydroxyls (sodium methoxide in MeOH) to provide azide-containing MUC1 epitope 80 (Scheme 9.12b). The ‘click’ conjugation of both constructs, alkyne-functionalized Pam3Cys 78 and the azide-MUC1 component 80 was undertaken with CuSO4 .5H2O, Na ascorbate with the aid of a Cu(I) stabilizing agent tris[(1-benzyl-1H-1,2,3-triazol-4-yn)methyl]amine (TBTA) in water/MeOH/THF mixture affording Pam3Cys-MUC1 conjugate 81 quantitatively (Scheme 9.12c) (Sarkar et al. 2013).

Synthesis of Pam3Cys-MUC1-TN conjugate using ‘click’ chemistry by Sucheck et al. (Sarkar et al. 2013). Reagents and conditions: (i) TFA, 1 h, rt, then propargyl amine, PyBOP, HOBt, iPr2NEt, 4 Å molecular sieves, CH2Cl2, 4 h, rt, 66% over two steps; (ii) CH3CN/CH2Cl2/Et2NH (2:1:2), 2 h, rt, then CH3(CH2)14COOH, PyBOP, HOBt, iPr2NEt, 4 Å molecular sieves, CH2Cl2, 4 h, rt, 80% over two steps; (iii) 6-azidohexanoic acid, DIC, HOBt, NMP; (iv) TFA/thioanisole/EDT/H2O phenol (88:3:5:2:2, v/v/v/v/v); (v) NaOMe, MeOH, 2 h, rt, 100%; (vi) CuSO4·5H2O, Na ascorbate, TBTA, H2O/MeOH/THF (1:1:2), 40 h, rt, 100%
The Brimble group recently employed a Pam2CysSK4 motif for the synthesis of a series of lipopeptide-based TLR2 agonists using ‘click’ chemistry (Wright et al. 2013c). Incorporation of an acetylene handle at the C-terminal end of the Pam2CysSK4 construct would allowing for the chemoselective ‘click’ conjugation with an azide-tagged epitope. Unlike the previous study by the Brimble group (Yeung et al. 2012) this approach maintained the critical atomic distance between the Pam2Cys and adjacent serine moiety[159]. Additionally, both the self-adjuvanting lipopeptide construct and the epitope were directly conjugated via a 1,2,3-triazole unit in contrast to approach by Kunz et al. where a PEG linker spaced these units apart (Cai et al. 2011).
It has been reported that the immunogenicity of the antigen incorporated to a vaccine construct may be suppressed by the presence of a linker (Buskas et al. 2004). We were also interested if the location of the triazole between the antigen and the Pam2CysSK4 moiety affects the TLR2-mediated stimulation of innate immunity; antigen conjugation with the lipid at either the N- or C-terminus of the peptide antigen was therefore investigated. It has been reported that acetylation of the Nα-amino group of the monoacyl PamCys moiety improved TLR2 activity (Salunke et al. 2012) hence the effects of this modification were also evaluated in this study (Wright et al. 2013c).
A lipidated and C-propargylated ‘click’ partner 82, in addition to the N-acetylated analogue 83 were first synthesized (Scheme 9.13a). Synthesis began by the N-terminal coupling of the Fmoc-(S)-Pam2Cys-OH building block 40 prepared from l-cysteine (Zeng et al. 2002; Metzger et al. 1991; Jones 1975; Hida et al. 1995), to the resin-bound C-terminal propargylated H2N-S(tBu)K(Boc)K(Boc)K(Boc)K(Boc)-Pra-resin peptide synthesized using standard Fmoc SPPS (Wright et al. 2013c). The peptide was lipidated using 40 and conditions adapted from Albericio et al. [benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP), 2,4,6-collidine, CH2Cl2/DCM (1:1)] (Han et al. 1996). Subsequent Fmoc-deprotection, followed by TFA-mediated resin cleavage and RP HPLC purification afforded the desired construct 82. Acylation of the Nα-amino group of cysteine to give 83 was performed using a mixture of acetic anhydride and iPr2NEt in DMF, prior to peptide cleavage and purification (Wright et al. 2013c). A truncated fragment of ppUL83 protein, namely NLVPMVATV, derived from the cytomegalovirus (CMV) known to stimulate CD8+ cytotoxic T-cells (Kopycinski et al. 2010) was chosen as a model epitope for the ‘click’ reaction. Synthesis of two NLVPMVATV analogues incorporating an azide handle at either the N- or C-terminus was also required. For the preparation of an azide-tagged antigen at the N-terminal site of the peptide 84, Fmoc SPPS was employed starting from 4-(hydroxymethyl)phenoxypropanoic acid (HMPP) resin and coupling of 2-azidoacetic acid to the N-terminal Asn at the last step of the SPPS. Subsequent acid-mediated peptide cleavage from the resin followed by RP HPLC purification afforded the desired ‘click’ partner 84 (Scheme 9.13b).

Synthesis of Pam2Cys lipopeptide-based TLR2 agonists using ‘click’ chemistry by Brimble et al. (Wright et al. 2013c). Reagents and conditions: (i) 40, BOP, 2,4,6-collidine, CH2Cl2/DMF (1:1); (ii) 20% piperidine in DMF; (iii) Ac2O, iPr2NEt, DMF (only for 83); (iv) TFA/iPr3SiH/DODT/H2O (94:1:2.5:2.5, v/v/v/v), 4 h, rt; (v) CuSO4, Na ascorbate, DMSO, 10 min, 70 °C
For the synthesis of the NLVPMVATV analogue with the C-terminally-tagged azide moiety 2-azidoacetic acid was incorporated via the Nε-amino group of an inserted lysine moiety at the C-terminus of the peptide. An orthogonally protected lysine residue [Dde-Lys(Fmoc)] was coupled to the Rink-amide resin, followed by the selective Nε-Fmoc protecting group removal (20% piperidine in DMF) and coupling of the 2-azidoacetic acid moiety. Subsequent hydrazine hydrate-mediated Dde group deprotection allowed for the iterative peptide chain elongation using Fmoc SPPS through readily unmasked Nα-amino group of the lysine residue, affording construct 85 (Scheme 9.13b). Chemoselective conjugation of propargylated-, or propargylated and N-acetylated- Pam2CysSK4 motives 82 and 83, respectively with azidopeptides 84 and 85 under activation with CuSO4 and Na ascorbate in DMSO, gave 1,2,3-triazole-linked constructs 86-89 in good yields (30-40%) and high purities (>95% by RP HPLC) (Scheme 9.13c) (Wright et al. 2013c).
Biological evaluation of 82 and N-acetylated analogue 83 using fresh human blood and measuring the level of CD80 surface expression compared to commercially sourced Pam3CSK4 interestingly revealed no major difference in CD80 expression between both propargylated Pam2Cys analogues with free- (82) and N-acetylated-Nα-amine (83) in contrast to published reports (Salunke et al. 2012). Importantly, there were no preferences regarding the N- or C-terminus for the antigen conjugation with lipidated adjuvant via 1,2,3-triazole and similar CD80 expression levels were observed for both ‘clicked’ analogues 86 and 87 and activity of ‘clicked’ lipopetides was comparable with the activity of commercially available Pam3CysSK4 (Wright et al. 2013c). This efficient procedure can therefore be generally applied for rapid generation of lipopeptides providing access to vaccine constructs (Wright et al. 2013c).
9.1.5.6 Cysteine Lipidation on a Peptide or Amino acid (CLipPA)
The ‘thiol-ene ’ reaction, a radical-promoted alkylation of a thiol with an alkene has been gaining in popularity in polymer and material science (Lowe 2010; Lowe 2014) as well as providing an effective strategy for bioconjugation and for site-selective modification of protein and organic molecules (Dondoni and Marra 2012; Hoyle and Bowman 2010; Liu and Li 2012; Krall et al. 2016; Madder and Gunnoo 2016). The Brimble group have recently applied for the first time, a single step ‘thiol-ene ’ coupling to synthesise monoacyl lipopeptides that showed self-adjuvanting antigenic activity with potency comparable to that of the synthetically challenging Pam3Cys moiety (Wright et al. 2013a, b; Brimble et al. 2014). We envisaged lipid attachment via the ‘post-translational’ route where the desired peptide constructs incorporating a cysteine at the N-terminus are first synthesized followed by S-lipidation with inexpensive and commercially available vinyl palmitate using the ‘thiol-ene ’ reaction. The viability of the transformation was first tested by preparation of the S-palmitoylated, Nα-Fmoc protected cysteine, starting from commercially available Fmoc-Cys(Trt)-OH which thiol protecting group was removed (TFA) affording Fmoc-Cys-OH (90). This was followed by hydrothiolation of vinyl palmitate 91 using UV light at 365 nm and 2,2-dimethoxy-2-phenylacetophenone (DMPA) as photoinitiator in CH2Cl2 for 60 min. The S-palmitoylated, Nα-Fmoc protected cysteine 92 was obtained in satisfactory yield (44%) (Scheme 9.14a) (Wright et al. 2013a, b). Subsequent direct lipidation of short, unprotected peptides CysSK4 and Nα-acetylated CysSK4 using 91, DMPA and photoinitiation (365 nm), was examined. The study revealed the need for extraneous thiols to obviate problems of vinyl palmitate telomerization and mixed disulphide formation. The choice of solvent also proved critical for a successful reaction. The optimized ‘thiol-ene ’ conditions (DMPA, DTT as thiol additive, NMP, hν 365 nm) were then used to directly lipidate Nα-acetylated CysSK4 93 using vinyl palmitate (91) with high conversion (>90%) to the S-palmitoylated peptide 94 (Scheme 9.14b).

(a) Model ‘thiol-ene ’ reaction of Fmoc-Cys-OH (90) with vinyl palmitate (91). (b) Direct S-palmitoylation of 93 and antigenic peptide 95 using vinyl palmitate 91 and ‘thiol-ene’ reaction by Brimble et al. (Wright et al. 2013a; Wright et al. 2013b). Reagents and conditions: (i) 91 (2 equiv) DMPA (0.2 equiv), CH2Cl2, 1 h, hν 365 nm, 44%; (ii) 91 (5 equiv), DMPA (0.4 equiv), DTT (3 equiv), hν 365 nm
The utility of direct lipidation was explored using more structurally complex antigenic peptide substrate derived from the cytomegalovirus ppUL85 protein (Kopycinski et al. 2010) comprising an N-terminally CysSK4, motif Ac-CSKKKK-NLVPMVATV (95). Pleasingly, good conversion of 95 to S-palmitoylated peptide antigen 96 using the photoinitiated ‘thiol-ene ’ reaction, 91 and optimized conditions (DMPA, DTT, DMSO) was observed as judged by RP HPLC profile (Scheme 9.14b) (Wright et al. 2013a; Wright et al. 2013b). We therefore coined the term ‘Cysteine Lipidation on a Peptide or Amino acid (CLipPA)’ to describe this efficient transformation allowing for one step lipidation of Nα-protected cysteine derivatives using vinyl palmitate .
We subsequently focused on a detailed study to optimise conditions for highly selective and effective mono-S-palmitoylation of peptides using CLipPA technology (Yang et al. 2016). Our first goal was to provide optimal conditions for the synthesis of a lipidated Nα-protected cysteine building block that could be used directly in SPPS. The Nα-protecting group, radical initiator and activation method were revised. Treatment of Nα-protected Fmoc, Boc or Nα-acetylated cysteine with an excess of vinyl palmitate in the presence of DMPA or 2,2-azo-bis(2-methylpropioniytile (AIBN) as radical initiator in either CH2Cl2 or 1,2-dichloroethane as solvent and under thermal heating (reflux at 90 °C), microwave irradiation (100 W, 70 °C) or UV light (365 nm) was studied. The S-palmitoylated products obtained were readily purified by silica gel chromatography without the need for RP HPLC.
An optimal conversion of Nα-protected with Fmoc- or Boc cysteine 90 and 96 was observed under UV light activation, using excess DMPA (1 equiv) for 1 h in CH2Cl2 affording 92 and 97 in 85% yield (Scheme 9.15a). Heating, either conventional or using microwave, gave lower yields due to the premature cleavage of Fmoc protecting group and the instability of the Boc group to high temperatures. Conversely, lipidation of Nα-Ac cysteine 98 appeared to be straightforward under all conditions tested giving good to excellent yields of the expected Nα-Ac and S-palmitoylated product 99. However the most effective conversion was when CH2Cl2 and AIBN were used under microwave heating (100 W, 70 °C) for 80 min leading to quantitative formation of desired product 99 (Scheme 9.15b) (Yang et al. 2016).

(a) Lipidation of Fmoc-Cys-OH (90) and Boc-Cys-OH (96) with vinyl palmitate (91) using optimized conditions for CLipPA; (b) Lipidation of Ac-Cys-OH (98) with vinyl palmitate (91) using optimized conditions for CLipPA; (c) On-resin lipidation of antigen 100 using Nα-protected S-palmitoylated cysteine building blocks 92 and 99 (Wright et al. 2013a; Wright et al. 2013b; Yang et al. 2016). Reagents and conditions: (i) 91 (1.5 equiv), DMPA (1 equiv), CH2Cl2, 1 h, hν 365 nm, 85%; (ii) 91 (1.5 equiv), AIBN (1 equiv), CH2Cl2, 80 min, MW, 100 W, 70 °C, 99%; (iii) 92 or 99, PyBOP, 2,4,6-trimethylcollidine, CH2Cl2/DMF (1:1), 1 h, rt; (iv) (for building block 92): 20% piperidine in DMF, then 20% Ac2O in DMF; (v) TFA/3,6-dioxa-1,8-octane-dithiol (DODT)/H2O/iPr3SiH (94:2.5:2.5:1, v/v/v/v)
The choice of Nα-protecting group may influence the degree of racemization during the coupling step when SPPS is performed (Zhang et al. 2012). Therefore, the coupling of S-palmitoylated, Nα–protected building blocks, 92 or 99 to a model peptide sequence was evaluated (Kopycinski et al. 2010). The Met residue of NLVPMVATV was substituted with Cys(tBu) to demonstrate applicability of the ‘thiol-ene ’ reaction conditions to a suitably protected cysteine thiol. The resin-bound and side-chain protected peptide H2N-S(tBu)K(Boc)K(Boc)K(Boc)K(Boc)-N(Trt)LVPC(tBu)VAT(tBu)V-resin (100) was prepared using Fmoc SPPS at room temperature with HATU/iPr2NEt and piperidine as coupling and Fmoc deprotection reagents and acylation with the lipidated building blocks, 92 or 99 was undertaken using racemization-suppressing conditions (PyBOP, 2,4,6-collidine, room temperature) (Zhang et al. 2012; Carpino et al. 1994; Carpino and El-Faham 1994). In the case of Nα-Fmoc-protected 92, the Fmoc protecting group was removed after coupling and subsequently exchanged for an acetyl group before TFA-mediated peptide cleavage was performed affording 101 (Scheme 9.15c). This allowed for a direct comparison of RP HPLC profiles to assess the degree of racemization. The RP HPLC chromatogram investigation of crude 101, obtained by using either 92 or 99 building block revealed that 1:1 ratio of epimers was formed when acetamide protecting group was used for lipidated cysteine incorporation. No detectable epimerization was however observed when Nα-Fmoc-protected 92 was used for lipid incorporation. The type of Nα-protecting group clearly influenced the degree of racemization during the study indicating the preferred choice of Fmoc-protected building block 92 for Fmoc SPPS-mediated peptide lipidation.
We then focused on reaction conditions that would allow direct lipidation of a thiol-containing peptide affording an S-palmitoylated construct 101 in a convergent-like approach.
The construct 102, derived from resin-bound 100, incorporated two cysteine residues; an N-terminal Cys with a sulfhydryl group ready for ‘thiol-ene ’ conjugation and the side chain of the second, internally located cysteine was masked with tBu. Subsequent photoinitiated lipidation at 365 nm of 102 using vinyl palmitate 91 (7 equiv) and previously reported conditions [DMPA (0.5 equiv), DTT (3 equiv) in NMP for 60 min] afforded S-palmitoylated peptide 101 albeit in variable yields (Scheme 9.16a) (Wright et al. 2013a; Wright et al. 2013b). A careful examination of LC-MS profiles of the ‘thiol-ene ’ reaction leading to desired conjugate 101 identified formation of unwanted by-products such as DTT-adducts and bis-palmitoylated peptide 104. The competitive formation of 104 by-product was found to increase with increasing levels of vinyl palmitate in the reaction mixture. Substitution of DTT with the more bulky mercaptan tert-butyl thiol (tBuSH) proved superior in suppressing an unwanted addition of the thiol scavenger to the carbon-centered radical 103. Formation of undesired bis-palmitoylated adduct 104 was also diminished by including an organosilane-based coreductant (iPr3SiH) that facilitated hydrogen transfer to the radical intermediate 103. Furthermore, decreasing the pH of reaction mixture with TFA led to a cleaner reaction profile, presumably a result of protonation of electron-rich amine residues. Moreover, a large excess of vinyl palmitate (91), tert-butyl mercaptan and iPr3SiH were also needed to maximise conversion of 102 to the desired 101. Although a large excess of vinyl palmitate was used in the optimized, photoinitiated (hν 365 nm) conditions [91 (70 equiv), DMPA (0.5 equiv), tBuSH (80 equiv), iPr3SiH (80 equiv), TFA (5% v/v) in NMP for 30 min], a now quantitative conversion of peptide 102 to the S-monopalmitoylated construct 101 (95%, based on the corresponding peak integration on the RP HPLC profile) was observed with negligible levels of bis-adduct 104 formed (Scheme 9.16b).

CLipPA direct conjugation of vinyl palmitate (91) and semiprotected peptide 102 under unoptimised conditions (a) and optimized conditions (b) (Yang et al. 2016). Reagents and conditions: (i) 91 (7 equiv), DMPA (0.5 equiv), DTT (3 equiv) NMP, 1 h, hν 365 nm; (ii) 91 (70 equiv), DMPA (0.5 equiv), tBuSH (80 equiv), iPr3SiH (80 equiv), TFA/NMP (5:95, v/v), 30 min, hν 365 nm
The optimized CLipPA technology could be used to effect direct S-monopalmitoylation of complex, unprotected peptide substrates as demonstrated for long peptides including Ac-CSKKKK-GARGPESRLLEFYLAMPFATPMEAELARRSLAQDAPPL-OH and H2N-CSKKKK-VPGVLLKEFTVSGNILTIRLTAADHR-OH, derived from NY-ESO-1(79-116) and NY-ESO-1(118-143), respectively. An excellent conversion to the desired lipidated peptide 105 (81%) and good 46% conversion to 106, based on RP HPLC profiles, demonstrated the power of this new strategy (Fig. 9.14) (Yang et al. 2016).

S-Palmitoylated long peptide products accessed using CLipPA technology (Yang et al. 2016)
The CLipPA technology offers a feasible one-step approach to lipidated peptide constructs containing all-natural bonds. We believe that this technique has strong potential to play a key role in self-adjuvanting peptide-based vaccine development in the future. The use of CLipPA eliminates the need for complex, multi-step and timeconsuming solution-phase synthesis of lipidated building blocks that are not readily available in all research laboratories. Depending on the vaccine construct requirements, either a stepwise SPPS approach, or a direct, convergent-like substrate lipidation can be executed using the ‘thiol-ene’ reaction and the optimized CLipPA conditions to afford S-palmitoylated assemblies in excellent yields with high selectivity.
9.2 Conclusions
Lipidation of peptides and proteins plays an important role in improving pharmacokinetic and pharmacodynamic profiles of peptides which may lead to potent analogues with clinical potential. Lipidated peptides activating TLR2 are crucial for peptide-based self-adjuvanting vaccine development. A simple, efficient and low-cost synthetic approach for incorporation of lipid motifs into peptides for subsequent bioevaluation is required. Synthesis of lipidated peptides via a standard SPPS technique using orthogonal protecting group strategy poses a challenge due to decreased solubility of lipopeptides. Novel synthetic advances such as the atom economical and functional group compatible CLipPA technique provides a useful approach to access S-palmitoylated peptides with a range of applications including vaccine design.
References
Abdel-Aal AB, Lakshminarayanan V, Thompson P, Supekar N, Bradley JM, Wolfert MA, Cohen PA, Gendler SJ, Boons GJ (2014) Immune and anticancer responses elicited by fully synthetic aberrantly glycosylated MUC1 tripartite vaccines modified by a TLR2 or TLR9 agonist. ChemBioChem 15(10):1508–1513
Agnihotri G, Crall BM, Lewis TC, Day TP, Balakrishna R, Warshakoon HJ, Malladi SS, David SA (2011) Structure-activity relationships in toll-like receptor 2-agonists leading to simplified monoacyl lipopeptides. J Med Chem 54(23):8148–8160
Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13(1):11–26
Anderson RJ, Tang CW, Daniels NJ, Compton BJ, Hayman CM, Johnston KA, Knight DA, Gasser O, Poyntz HC, Ferguson PM, Larsen DS, Ronchese F, Painter GF, Hermans IF (2014) A self-adjuvanting vaccine induces cytotoxic T lymphocytes that suppress allergy. Nat Chem Biol 10(11):943–949
Anderson RJ, Compton BJ, Tang C-W, Authier-Hall A, Hayman CM, Swinerd GW, Kowalczyk R, Harris P, Brimble MA, Larsen DS, Gasser O, Weinkove R, Hermans IF, Painter GF (2015) NKT cell-dependent glycolipid-peptide vaccines with potent anti-tumour activity. Chem Sci 6(9):5120–5127
Asada H, Douen T, Mizokoshi Y, Fujita T, Murakami M, Yamamoto A, Muranishi S (1994) Stability of acyl derivatives of insulin in the small intestine: relative importance of insulin association characteristics in aqueous solution. Pharm Res 11(8):1115–1120
Asada H, Douen T, Waki M, Adachi S, Fujita T, Yamamoto A, Muranishi S (1995) Absorption characteristics of chemically modified-insulin derivatives with various fatty acids in the small and large intestine. J Pharm Sci 84(6):682–687
Avadisian M, Gunning PT (2013) Extolling the benefits of molecular therapeutic lipidation. Mol Biosyst 9(9):2179–2188
Avadisian M, Fletcher S, Liu B, Zhao W, Yue P, Badali D, Xu W, Schimmer AD, Turkson J, Gradinaru CC, Gunning PT (2011) Artificially induced protein–membrane anchorage with cholesterol-based recognition agents as a new therapeutic concept. Angew Chem Int Ed Engl 50(28):6248–6253
Backes BJ, Ellman JA (1999) An alkanesulfonamide “safety-catch” linker for solid-phase synthesis. J Org Chem 64(7):2322–2330
Basto AP, Leitao A (2014) Targeting TLR2 for vaccine development. J Immunol Res 2014:619410
Bednarek MA, Feighner SD, Pong SS, McKee KK, Hreniuk DL, Silva MV, Warren VA, Howard AD, Van Der Ploeg LH, Heck JV (2000) Structure-function studies on the new growth hormone-releasing peptide, ghrelin: minimal sequence of ghrelin necessary for activation of growth hormone secretagogue receptor 1a. J Med Chem 43(23):4370–4376
Benedict RG, Langlykke AF (1947) Antibiotic activity of Bacillus polymyxa. J Bacteriol 54(1):24
Berndt N, Hamilton AD, Sebti SM (2011) Targeting protein prenylation for cancer therapy. Nat Rev Cancer 11(11):775–791
Braun V (1975) Covalent lipoprotein from the outer membrane of escherichia coli. Biochim Biophys Acta Rev Biomemr 415(3):335–377
Bray GA (2015) Obesity: liraglutide[mdash]another weapon in the war against obesity? Nat Rev Endocrinol 11(10):569–570
Brimble MA, Wright TH, Dunbar RP, Williams GM (2014) Amino acid and peptide conjugates and conjugation process. WO2014207708 A3
Brimble MA, Edwards PJ, Harris PW, Norris GE, Patchett ML, Wright TH, Yang SH, Carley SE (2015) Synthesis of the antimicrobial S-linked glycopeptide, glycocin F. Chemistry 21(9):3556–3561
Brink AJ, Richards GA, Colombo G, Bortolotti F, Colombo P, Jehl F (2014) Multicomponent antibiotic substances produced by fermentation: implications for regulatory authorities, critically ill patients and generics. Int J Antimicrob Agents 43(1):1–6
Brown LE, Jackson DC (2005) Lipid-based self-adjuvanting vaccines. Curr Drug Deliv 2(4):383–393
Buskas T, Li Y, Boons GJ (2004) The immunogenicity of the tumor-associated antigen Lewis(y) may be suppressed by a bifunctional cross-linker required for coupling to a carrier protein. Chemistry 10(14):3517–3524
Buskas T, Ingale S, Boons GJ (2005) Towards a fully synthetic carbohydrate-based anticancer vaccine: synthesis and Immunological evaluation of a lipidated glycopeptide containing the tumor-associated Tn antigen. Angew Chem Int Ed Engl 44(37):5985–5988
Buwitt-Beckmann U, Heine H, Wiesmuller KH, Jung G, Brock R, Akira S, Ulmer AJ (2005a) Toll-like receptor 6-independent signaling by diacylated lipopeptides. Eur J Immunol 35(1):282–289
Buwitt-Beckmann U, Heine H, Wiesmuller KH, Jung G, Brock R, Ulmer AJ (2005b) Lipopeptide structure determines TLR2 dependent cell activation level. FEBS J 272(24):6354–6364
Cai H, Huang ZH, Shi L, Zhao YF, Kunz H, Li YM (2011) Towards a fully synthetic MUC1-based anticancer vaccine: efficient conjugation of glycopeptides with mono-, di-, and tetravalent lipopeptides using click chemistry. Chemistry 17(23):6396–6406
Cai H, Sun ZY, Huang ZH, Shi L, Zhao YF, Kunz H, Li YM (2013) Fully synthetic self-adjuvanting thioether-conjugated glycopeptide-lipopeptide antitumor vaccines for the induction of complement-dependent cytotoxicity against tumor cells. Chemistry 19(6):1962–1970
Cai H, Sun ZY, Chen MS, Zhao YF, Kunz H, Li YM (2014) Synthetic multivalent glycopeptide-lipopeptide antitumor vaccines: impact of the cluster effect on the killing of tumor cells. Angew Chem Int Ed Engl 53(6):1699–1703
Carpino LA, El-Faham A (1994) Effect of tertiary bases on O-benzotriazolyluronium salt-induced peptide segment coupling. J Org Chem 59(4):695–698
Carpino LA, El-Faham A, Albericio F (1994) Racemization studies during solid-phase peptide synthesis using azabenzotriazole-based coupling reagents. Tetrahedron Lett 35(15):2279–2282
Cavallari M, Stallforth P, Kalinichenko A, Rathwell DC, Gronewold TM, Adibekian A, Mori L, Landmann R, Seeberger PH, De Libero G (2014) A semisynthetic carbohydrate-lipid vaccine that protects against pneumoniae in mice. Nat Chem Biol 10(11):950–956
Chamberlain LH, Shipston MJ (2015) The physiology of protein S-acylation. Physiol Rev 95(2):341–376
Cheng C, Jain P, Bettahi I, Pal S, Tifrea D, de la Maza LM (2011) A TLR2 agonist is a more effective adjuvant for a Chlamydia major outer membrane protein vaccine than ligands to other TLR and NOD receptors. Vaccine 29(38):6641–6649
Chua BY, Zeng W, Lau YF, Jackson DC (2007) Comparison of lipopeptide-based immunocontraceptive vaccines containing different lipid groups. Vaccine 25(1):92–101
Chua BY, Eriksson EM, Brown LE, Zeng W, Gowans EJ, Torresi J, Jackson DC (2008) A self-adjuvanting lipopeptide-based vaccine candidate for the treatment of hepatitis C virus infection. Vaccine 26(37):4866–4875
Chua BY, Johnson D, Tan A, Earnest-Silveira L, Sekiya T, Chin R, Torresi J, Jackson DC (2012) Hepatitis C VLPs delivered to dendritic cells by a TLR2 targeting lipopeptide results in enhanced antibody and cell-mediated responses. PLoS One 7(10):e47492
Chua BY, Olson MR, Bedoui S, Sekiya T, Wong CY, Turner SJ, Jackson DC (2014) The use of a TLR2 agonist-based adjuvant for enhancing effector and memory CD8 T-cell responses. Immunol Cell Biol 92(4):377–383
Chua BY, Wong CY, Mifsud EJ, Edenborough KM, Sekiya T, Tan AC, Mercuri F, Rockman S, Chen W, Turner SJ, Doherty PC, Kelso A, Brown LE, Jackson DC (2015) Inactivated influenza vaccine that provides rapid, innate-immune-system-mediated protection and subsequent long-term adaptive immunity. MBio 6(6):e01024–e01015
Clement S, Still JG, Kosutic G, McAllister RG (2002) Oral insulin product hexyl-insulin monoconjugate 2 (HIM2) in type 1 diabetes mellitus: the glucose stabilization effects of HIM2. Diabetes Technol Ther 4(4):459–466
Clement S, Dandona P, Still JG, Kosutic G (2004) Oral modified insulin (HIM2) in patients with type 1 diabetes mellitus: results from a phase I/II clinical trial. Metabolism 53(1):54–58
Cochrane SA, Vederas JC (2016) Lipopeptides from Bacillus and Paenibacillus spp.: a gold mine of antibiotic candidates. Med Res Rev 36(1):4–31
Cordy JM, Hooper NM, Turner AJ (2006) The involvement of lipid rafts in Alzheimer’s disease. Mol Membr Biol 23(1):111–122
Cory D, Choong I, Glenn JS (2015) Treatment of hepatitis delta virus (HDV) infection with combination of lonafarnib and ritonavir. WO 2015168648:A1
Davda D, Martin BR (2014) Acyl protein thioesterase inhibitors as probes of dynamic S-palmitoylation. MedChemComm 5(3):268–276
Dawson P, Muir T, Clark-Lewis I, Kent S (1994) Synthesis of proteins by native chemical ligation. Science 266(5186):776–779
Debono M, Barnhart M, Carrell CB, Hoffmann JA, Occolowitz JL, Abbott BJ, Fukuda DS, Hamill RL, Biemann K, Herlihy WC (1987) A21978C, a complex of new acidic peptide antibiotics: isolation, chemistry, and mass spectral structure elucidation. J Antibiot 40(6):761–777
Deeg MA, Humphrey DR, Yang SH, Ferguson TR, Reinhold VN, Rosenberry TL (1992) Glycan components in the glycoinositol phospholipid anchor of human erythrocyte acetylcholinesterase. Novel fragments produced by trifluoroacetic acid. J Biol Chem 267(26):18573–18580
Demotz S, Lanzavecchia A, Eisel U, Niemann H, Widmann C, Corradin G (1989) Delineation of several DR-restricted tetanus toxin T cell epitopes. J Immunol 142(2):394–402
Dey G, Bharti R, Sen R, Mandal M (2015) Microbial amphiphiles: a class of promising new-generation anticancer agents. Drug Discov Today 20(1):136–146
Di L (2015) Strategic approaches to optimizing peptide ADME properties. AAPS J 17(1):134–143
Dondoni A, Marra A (2012) Recent applications of thiol-ene coupling as a click process for glycoconjugation. Chem Soc Rev 41(2):573–586
Ekrami HM, Kennedy AR, Shen WC (1995) Water-soluble fatty acid derivatives as acylating agents for reversible lipidization of polypeptides. FEBS Lett 371(3):283–286
Elbrond B, Jakobsen G, Larsen S, Agerso H, Jensen LB, Rolan P, Sturis J, Hatorp V, Zdravkovic M (2002) Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects. Diabetes care 25(8):1398–1404
Eriksson EM, Jackson DC (2007) Recent advances with TLR2-targeting lipopeptide-based vaccines. Curr Protein Pept Sci 8(4):412–417
Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24(1):24–41
Ferguson MAJ, Kinoshita T, Hart GW (2009) Glycosylphosphatidylinositol anchors. In: Varki A, Cummings RD, Esko JD, Freeze HH, Bertozzi CR, Hart GW, Etzler ME (eds) Essentials of glycobiology, 2nd edn. Cold Spring Harbor Laboratory Press, New York, pp 143–161
Flora DB 2002. Fatty acid-acylated insulin analogs. US6444641 B1.
Fosgerau K, Hoffmann T (2015) Peptide therapeutics: current status and future directions. Drug Discov Today 20(1):122–128
Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM (2003) Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med 198(2):267–279
Fujita T, Kawahara I, Y-s Q, Hattori K, Takenaka K, Muranishi S, Yamamoto A (1998) Permeability characteristics of tetragastrins across intestinal membranes using the Caco-2 monolayer system: comparison between acylation and application of protease inhibitors. Pharm Res 15(9):1387–1392
Gao S, Yu R, Zhou X (2016) The role of geranylgeranyltransferase I-mediated protein prenylation in the brain. Mol Neurobiol 53(10):6925–6937
Gay NJ, Gangloff M (2007) Structure and function of Toll receptors and their ligands. Annu Rev Biochem 76:141–165
Goddard-Borger ED, Stick RV (2007) An efficient, inexpensive, and shelf-stable diazotransfer reagent: imidazole-1-sulfonyl azide hydrochloride. Org Lett 9(19):3797–3800
Godfrey DI, Kronenberg M (2004) Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest 114(10):1379–1388
Gordon LB, Rothman FG, López-Otín C, Misteli T (2014) Progeria: a paradigm for translational medicine. Cell 156(3):400–407
Green BR, White KL, McDougle DR, Zhang L, Klein B, Scholl EA, Pruess TH, White HS, Bulaj G (2010) Introduction of lipidization-cationization motifs affords systemically bioavailable neuropeptide Y and neurotensin analogs with anticonvulsant activities. J Pept Sci 16(9):486–495
Green BR, Smith M, White KL, White HS, Bulaj G (2011) Analgesic neuropeptide W suppresses seizures in the brain revealed by rational repositioning and peptide engineering. ACS Chem Neurosci 2(1):51–56
Grgurina I, Barca A, Cervigni S, Gallo M, Scaloni A, Pucci P (1994) Relevance of chlorine-substituent for the antifungal activity of syringomycin and syringotoxin, metabolites of the phytopathogenic bacterium Pseudomonas syringae pv. syringae. Experientia 50(2):130–133
Gupta S, Takebe N, LoRusso P (2010) Review: targeting the hedgehog pathway in cancer. Ther Adv Med Oncol 2(4):237–250
Gutierrez JA, Solenberg PJ, Perkins DR, Willency JA, Knierman MD, Jin Z, Witcher DR, Luo S, Onyia JE, Hale JE (2008) Ghrelin octanoylation mediated by an orphan lipid transferase. Proc Natl Acad Sci U S A 105(17):6320–6325
Hamley IW (2015) Lipopeptides: from self-assembly to bioactivity. Chem. Commun 51(41):8574–8583
Hamman JH, Enslin GM, Kotze AF (2005) Oral delivery of peptide drugs: barriers and developments. BioDrugs 19(3):165–177
Han Y, Bontems SL, Hegyes P, Munson MC, Minor CA, Kates SA, Albericio F, Barany G (1996) Preparation and applications of xanthenylamide (XAL) handles for solid-phase synthesis of C-terminal peptide amides under particularly mild conditions. J Org Chem 61(18):6326–6339
Hannoush RN (2015) Synthetic protein lipidation. Curr Opin Chem Biol 28:39–46
Harris PW, Brimble MA (2010) Toward the total chemical synthesis of the cancer protein NY-ESO-1. Biopolymers 94(4):542–550
Harris PW, Brimble MA (2013) A comparison of Boc and Fmoc SPPS strategies for the preparation of C-terminal peptide alpha-thiolesters: NY-ESO-1 (3)(9)Cys-(6)(8)Ala-COSR. Biopolymers 100(4):356–365
Harris PW, Brimble MA (2015) Chemical synthesis of a polypeptide backbone derived from the primary sequence of the cancer protein NY-ESO-1 enabled by kinetically controlled ligation and pseudoprolines. Biopolymers 104(2):116–127
Harris PWR, Brimble MA, Dunbar R, Kent SBH (2007) Synthesis of a C-terminal thioester derivative of the lipopeptide Pam2CSKKKKG using Fmoc SPPS. Synlett 2007(5):0713–0716
Harris PW, Squire C, Young PG, Brimble MA (2015) Chemical synthesis of gamma-secretase activating protein using pseudoglutamines as ligation sites. Biopolymers 104(1):37–45
Hashimoto M, Takada K, Kiso Y, Muranishi S (1989) Synthesis of palmitoyl derivatives of insulin and their biological activities. Pharm Res 6(2):171–176
Hashizume M, Douen T, Murakami M, Yamamoto A, Takada K, Muranishi S (1992) Improvement of large intestinal absorption of insulin by chemical modification with palmitic acid in rats. J Pharm Pharmacol 44(7):555–559
Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V (2003) NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol 171(10):5140–5147
Hicks DA, Nalivaeva NN, Turner AJ (2012) Lipid rafts and Alzheimer’s disease: protein-lipid interactions and perturbation of signaling. Front Physiol 3:189
Hida T, Hayashi K, Yukishige K, Tanida S, Kawamura N, Harada S (1995) Synthesis and biological activities of TAN-1511 analogues. J Antibiot 48(7):589–603
Hofmann K (2000) A superfamily of membrane-bound O-acyltransferases with implications for Wnt signaling. Trends Biochem Sci 25(3):111–112
Home P, Kurtzhals P (2006) Insulin detemir: from concept to clinical experience. Expert Opin Pharmacother 7(3):325–343
Honeycutt L, Wang J, Ekrami H, Shen WC (1996) Comparison of pharmacokinetic parameters of a polypeptide, the Bowman-Birk protease inhibitor (BBI), and its palmitic acid conjugate. Pharm Res 13(9):1373–1377
Hoyle CE, Bowman CN (2010) Thiol-ene click chemistry. Angew Chem Int Ed Engl 49(9):1540–1573
Hutchinson JA, Burholt S, Hamley IW (2017) Peptide hormones and lipopeptides: from self-assembly to therapeutic applications. J Pept Sci 23(2):82–94
Iepsen EW, Torekov SS, Holst JJ (2015) Liraglutide for Type 2 diabetes and obesity: a 2015 update. Expert Rev Cardiovasc Ther 13(7):753–767
Ingale S, Buskas T, Boons GJ (2006) Synthesis of glyco(lipo)peptides by liposome-mediated native chemical ligation. Org Lett 8(25):5785–5788
Ingale S, Wolfert MA, Gaekwad J, Buskas T, Boons GJ (2007) Robust immune responses elicited by a fully synthetic three-component vaccine. Nat Chem Biol 3(10):663–667
Ingale S, Wolfert MA, Buskas T, Boons GJ (2009) Increasing the antigenicity of synthetic tumor-associated carbohydrate antigens by targeting Toll-like receptors. ChemBioChem 10(3):455–463
Ingenito R, Bianchi E, Fattori D, Pessi A (1999) Solid phase synthesis of peptide C-terminal thioesters by Fmoc/t-Bu chemistry. J Am Chem Soc 121(49):11369–11374
Jackson DC, Lau YF, Le T, Suhrbier A, Deliyannis G, Cheers C, Smith C, Zeng W, Brown LE (2004) A totally synthetic vaccine of generic structure that targets Toll-like receptor 2 on dendritic cells and promotes antibody or cytotoxic T cell responses. Proc Natl Acad Sci U S A 101(43):15440–15445
Jackson SH, Martin TS, Jones JD, Seal D, Emanuel F (2010) Liraglutide (Victoza): the first once-daily incretin mimetic injection for type-2 diabetes. P T 35(9):498–529
Jacques P (2011) Surfactin and other lipopeptides from Bacillus spp. In: Soberón-Chávez G (ed) Biosurfactants, Microbiology monographs, vol 20. Springer, Berlin/Heidelberg, pp 57–91
Johannessen L, Remsberg J, Gaponenko V, Adams KM, Barchi JJ, Tarasov SG, Jiang S, Tarasova NI (2011) Peptide structure stabilization by membrane anchoring and its general applicability to the development of potent cell-permeable inhibitors. ChemBioChem 12(6):914–921
Jones AR (1975) The metabolism of 3-chloro,- 3-bromo- and 3-iodoprpan-1,2-diol in rats and mice. Xenobiotica 5(3):155–165
Kaiser A, Gaidzik N, Becker T, Menge C, Groh K, Cai H, Li YM, Gerlitzki B, Schmitt E, Kunz H (2010) Fully synthetic vaccines consisting of tumor-associated MUC1 glycopeptides and a lipopeptide ligand of the Toll-like receptor 2. Angew Chem Int Ed Engl 49(21):3688–3692
Kakugawa S, Langton PF, Zebisch M, Howell S, Chang T-H, Liu Y, Feizi T, Bineva G, O’Reilly N, Snijders AP, Jones EY, Vincent J-P (2015) Notum deacylates Wnts to suppress signalling activity. Nature 519(7542):187–192
Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, Han SH, Lee H, Paik SG, Lee JO (2009) Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31(6):873–884
Karsdal MA, Henriksen K, Bay-Jensen AC, Molloy B, Arnold M, John MR, Byrjalsen I, Azria M, Riis BJ, Qvist P, Christiansen C (2011) Lessons learned from the development of oral calcitonin: the first tablet formulation of a protein in phase III clinical trials. J Clin Pharmacol 51(4):460–471
Karsdal MA, Riis BJ, Mehta N, Stern W, Arbit E, Christiansen C, Henriksen K (2015) Lessons learned from the clinical development of oral peptides. Br J Clin Pharmacol 79(5):720–732
Kaspar AA, Reichert JM (2013) Future directions for peptide therapeutics development. Drug Discov Today 18(17–18):807–817
Khong H, Overwijk WW (2016) Adjuvants for peptide-based cancer vaccines. J Immunother Cancer 4:1–11
Kipnes M, Dandona P, Tripathy D, Still JG, Kosutic G (2003) Control of postprandial plasma glucose by an oral insulin product (HIM2) in patients with type 2 diabetes. Diabetes care 26(2):421–426
Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thogersen H, Wilken M, Agerso H (2000) Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem 43(9):1664–1669
Koh C, Canini L, Dahari H, Zhao X, Uprichard SL, Haynes-Williams V, Winters MA, Subramanya G, Cooper SL, Pinto P, Wolff EF, Bishop R, Ai Thanda Han M, Cotler SJ, Kleiner DE, Keskin O, Idilman R, Yurdaydin C, Glenn JS, Heller T (2015) Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: a proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect Dis 15(10):1167–1174
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K (1999) Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402(6762):656–660
Konitsiotis AD, Jovanović B, Ciepla P, Spitaler M, Lanyon-Hogg T, Tate EW, Magee AI (2015) Topological analysis of hedgehog acyltransferase, a multipalmitoylated transmembrane protein. J Biol Chem 290(6):3293–3307
Kopycinski J, Osman M, Griffiths PD, Emery VC (2010) Sequence flexibility of the immunodominant HLA A*0201 restricted ppUL83 CD8 T-cell epitope of human cytomegalovirus. J Med Virol 82(1):94-103
Krall N, da Cruz FP, Boutureira O, Bernardes GJL (2016) Site-selective protein-modification chemistry for basic biology and drug development. Nat Chem 8(2):103–113
Kudryashov V, Glunz PW, Williams LJ, Hintermann S, Danishefsky SJ, Lloyd KO (2001) Toward optimized carbohydrate-based anticancer vaccines: epitope clustering, carrier structure, and adjuvant all influence antibody responses to Lewis(y) conjugates in mice. Proc Natl Acad Sci U S A 98(6):3264–3269
Kurtzhals P (2007) Pharmacology of insulin detemir. Endocrinol Metab Clin North Am 36(1):14–20
Lakshminarayanan V, Thompson P, Wolfert MA, Buskas T, Bradley JM, Pathangey LB, Madsen CS, Cohen PA, Gendler SJ, Boons GJ (2012) Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc Natl Acad Sci U S A 109(1):261–266
Lau J, Bloch P, Schaffer L, Pettersson I, Spetzler J, Kofoed J, Madsen K, Knudsen LB, McGuire J, Steensgaard DB, Strauss HM, Gram DX, Knudsen SM, Nielsen FS, Thygesen P, Reedtz-Runge S, Kruse T (2015) Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem 58(18):7370–7380
Le Floch J-P (2010) Critical appraisal of the safety and efficacy of insulin detemir in glycemic control and cardiovascular risk management in diabetics. Diabetes Metab Syndr Obes 3:197–213
Leclerc C, Deriaud E, Mimic V, van der Werf S (1991) Identification of a T-cell epitope adjacent to neutralization antigenic site 1 of poliovirus type 1. J Virol 65(2):711–718
Lee DJ, Harris PW, Brimble MA (2011) Synthesis of MUC1 Neoglycopeptides using efficient microwave-enhanced chaotrope-assisted click chemistry. Org Biomol Chem 9(5):1621–1626
Lee RTH, Zhao Z, Ingham PW (2016) Hedgehog signalling. Development 143(3):367–372
Lewis AL, Richard J (2015) Challenges in the delivery of peptide drugs: an industry perspective. Ther Deliv 6(2):149–163
Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK (2014) Peptide vaccine: progress and challenges. Vaccines 2(3):515–536
Lin David TS, Conibear E (2015) Enzymatic protein depalmitoylation by acyl protein thioesterases. Biochem Soc Trans 43(2):193
Liu C-F, Li F (2012) Method for modification of organic molecules. WO 2012/158122 A1
Lowe AB (2010) Thiol-ene “click” reactions and recent applications in polymer and materials synthesis. Polym Chem 1(1):17–36
Lowe AB (2014) Thiol-ene “click” reactions and recent applications in polymer and materials synthesis: a first update. Polym Chem 5(17):4820–4870
Madder A, Gunnoo SB (2016) Chemical protein modification via cysteine. ChemBioChem 17(7):529–553
Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thøgersen H, Wilken M, Johansen NL (2007) Structure−activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness. J Med Chem 50(24):6126–6132
Makovitzki A, Avrahami D, Shai Y (2006) Ultrashort antibacterial and antifungal lipopeptides. Proc Natl Acad Sci U S A 103(43):15997–16002
Maletínskâ L, Neugebauer W, Parê M-C, Pêrodin J, Pham D, Escher E (1996) Lipid masking and reactivation of angiotensin analogues. Helv Chim Acta 79(7):2023–2034
Maletinska L, Neugebauer W, Perodin J, Lefebvre M, Escher E (1997) Angiotensin analogues palmitoylated in positions 1 and 4. J Med Chem 40(20):3271–3279
Malm-Erjefalt M, Bjornsdottir I, Vanggaard J, Helleberg H, Larsen U, Oosterhuis B, van Lier JJ, Zdravkovic M, Olsen AK (2010) Metabolism and excretion of the once-daily human glucagon-like peptide-1 analog liraglutide in healthy male subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase. Drug Metab Dispos 38(11):1944–1953
Martinez-Saez N, Supekar NT, Wolfert MA, Bermejo IA, Hurtado-Guerrero R, Asensio JL, Jimenez-Barbero J, Busto JH, Avenoza A, Boons GJ, Peregrina JM, Corzana F (2016) Mucin architecture behind the immune response: design, evaluation and conformational analysis of an antitumor vaccine derived from an unnatural MUC1 fragment. Chem Sci 7(3):2294–2301
Matevossian A, Resh MD (2015) Membrane topology of hedgehog acyltransferase. J Biol Chem 290(4):2235–2243
McDonald DM, Wilkinson BL, Corcilius L, Thaysen-Andersen M, Byrne SN, Payne RJ (2014) Synthesis and immunological evaluation of self-adjuvanting MUC1-macrophage activating lipopeptide 2 conjugate vaccine candidates. Chem Commun 50(71):10273–10276
McDonald DM, Byrne SN, Payne RJ (2015) Synthetic self-adjuvanting glycopeptide cancer vaccines. Front Chem 3:60
Medini K, Harris PW, Hards K, Dingley AJ, Cook GM, Brimble MA (2015) Chemical synthesis of a pore-forming antimicrobial protein, caenopore-5, by using native chemical ligation at a Glu-Cys site. ChemBioChem 16(2):328–336
Medini K, Harris PWR, Menorca A, Hards K, Cook GM, Brimble MA (2016) Synthesis and activity of a diselenide bond mimetic of the antimicrobial protein caenopore-5. Chem Sci 7(3):2005–2010
Mejuch T, Waldmann H (2016) Synthesis of lipidated proteins. Bioconjug Chem 27(8):1771–1783
Metzger JW, Wiesmuller KH, Jung G (1991) Synthesis of N alpha-Fmoc protected derivatives of S-(2,3-dihydroxypropyl)-cysteine and their application in peptide synthesis. Int J Pept Protein Res 38(6):545–554
Metzger JW, Beck-Sickinger AG, Loleit M, Eckert M, Bessler WG, Jung G (1995) Synthetic S-(2,3-dihydroxypropyl)-cysteinyl peptides derived from the N-terminus of the cytochrome subunit of the photoreaction centre of Rhodopseudomonas viridis enhance murine splenocyte proliferation. J Pept Sci 1(3):184–190
Mifsud EJ, Tan AC, Jackson DC (2014) TLR agonists as modulators of the innate immune response and their potential as agents against infectious disease. Front Immunol 5:79
Mnif I, Ghribi D (2015) Review lipopeptides biosurfactants: mean classes and new insights for industrial, biomedical, and environmental applications. Biopolymers 104(3):129–147
Monji T, Pious D (1997) Exogenously provided peptides fail to complex with intracellular class II molecules for presentation by antigen-presenting cells. J Immunol 158(7):3155–3164
Moyle PM, Toth I (2008) Self-adjuvanting lipopeptide vaccines. Curr Med Chem 15(5):506–516
Muhlradt PF, Kiess M, Meyer H, Sussmuth R, Jung G (1997) Isolation, structure elucidation, and synthesis of a macrophage stimulatory lipopeptide from Mycoplasma fermentans acting at picomolar concentration. J Exp Med 185(11):1951–1958
Muhlradt PF, Kiess M, Meyer H, Sussmuth R, Jung G (1998) Structure and specific activity of macrophage-stimulating lipopeptides from Mycoplasma hyorhinis. Infect Immun 66(10):4804–4810
Müller TD, Nogueiras R, Andermann ML, Andrews ZB, Anker SD, Argente J, Batterham RL, Benoit SC, Bowers CY, Broglio F, Casanueva FF, D’Alessio D, Depoortere I, Geliebter A, Ghigo E, Cole PA, Cowley M, Cummings DE, Dagher A, Diano S, Dickson SL, Diéguez C, Granata R, Grill HJ, Grove K, Habegger KM, Heppner K, Heiman ML, Holsen L, Holst B, Inui A, Jansson JO, Kirchner H, Korbonits M, Laferrère B, LeRoux CW, Lopez M, Morin S, Nakazato M, Nass R, Perez-Tilve D, Pfluger PT, Schwartz TW, Seeley RJ, Sleeman M, Sun Y, Sussel L, Tong J, Thorner MO, van der Lely AJ, van der Ploeg LHT, Zigman JM, Kojima M, Kangawa K, Smith RG, Horvath T, Tschöp MH (2015) Ghrelin. Mol Metab 4(6):437–460
Muranishi S, Sakai A, Yamada K, Murakami M, Takada K, Kiso Y (1991) Lipophilic peptides: synthesis of lauroyl thyrotropin-releasing hormone and its biological activity. Pharm Res 8(5):649–652
Nauck MA, Petrie JR, Sesti G, Mannucci E, Courrèges J-P, Lindegaard ML, Jensen CB, Atkin SL (2016) A phase 2, randomized, dose-finding study of the novel once-weekly human GLP-1 analog, semaglutide, compared with placebo and open-label liraglutide in patients with type 2 diabetes. Diabetes care 39(2):231–241
Nile AH, Hannoush RN (2016) Fatty acylation of Wnt proteins. Nat Chem Biol 12(2):60–69
Orwa JA, Govaerts C, Busson R, Roets E, Van Schepdael A, Hoogmartens J (2001) Isolation and structural characterization of polymyxin B components. J Chromatogr A 912(2):369–373
Otvos L, Wade JD (2014) Current challenges in peptide-based drug discovery. Front Chem 2:62
Park K, Kwon IC, Park K (2011) Oral protein delivery: current status and future prospect. React Funct Polym 71(3):280–287
Pattabiraman VR, McKinnie SM, Vederas JC (2008) Solid-supported synthesis and biological evaluation of the lantibiotic peptide bis(desmethyl) lacticin 3147 A2. Angew Chem Int Ed Engl 47(49):9472–9475
Paulick MG, Bertozzi CR (2008) The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry 47(27):6991–7000
Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, Bixler SA, Ambrose CM, Garber EA, Miatkowski K, Taylor FR, Wang EA, Galdes A (1998) Identification of a palmitic acid-modified form of human sonic hedgehog. J Biol Chem 273(22):14037–14045
Porotto M, Yokoyama CC, Palermo LM, Mungall B, Aljofan M, Cortese R, Pessi A, Moscona A (2010) Viral entry inhibitors targeted to the membrane site of action. J Virol 84(13):6760–6768
Raaijmakers JM, de Bruijn I, de Kock MJD (2006) Cyclic lipopeptide production by plant-associated Pseudomonas spp.: diversity, activity, biosynthesis, and regulation. Mol Plant Microbe Interact 19(7):699–710
Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knölker H-J, Simons K (2008a) Efficient inhibition of the Alzheimer’s disease β-secretase by membrane targeting. Science 320(5875):520–523
Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knölker H-J, Simons K (2008b) Corrections and clarifications. Science 321(5891):912–912
Rajendran L, Udayar V, Goodger ZV (2012) Lipid-anchored drugs for delivery into subcellular compartments. Trends Pharmacol Sci 33(4):215–222
Ramesan RM, Sharma CP (2014) Recent advances in the oral delivery of insulin. Recent Pat Drug Deliv Formul 8(2):155–159
Remsberg JR, Lou H, Tarasov SG, Dean M, Tarasova NI (2007) Structural analogues of smoothened intracellular loops as potent inhibitors of hedgehog pathway and cancer cell growth. J Med Chem 50(18):4534–4538
Renukuntla J, Vadlapudi AD, Patel A, Boddu SHS, Mitra AK (2013) Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm 447(0):75-93
Resh MD (2012) Targeting protein lipidation in disease. Trends Mol Med 18(4):206–214
Resh MD (2013) Covalent lipid modifications of proteins. Curr Biol 23(10):R431–R435
Resh MD (2016) Fatty acylation of proteins: the long and the short of it. Prog Lipid Res 63:120-131
Rigato M, Fadini GP (2014) Comparative effectiveness of liraglutide in the treatment of type 2 diabetes. Diabetes Metab Syndr Obes 7:107–120
Roberts WL, Myher JJ, Kuksis A, Low MG, Rosenberry TL (1988a) Lipid analysis of the glycoinositol phospholipid membrane anchor of human erythrocyte acetylcholinesterase. Palmitoylation of inositol results in resistance to phosphatidylinositol-specific phospholipase C. J Biol Chem 263(35):18766–18775
Roberts WL, Santikarn S, Reinhold VN, Rosenberry TL (1988b) Structural characterization of the glycoinositol phospholipid membrane anchor of human erythrocyte acetylcholinesterase by fast atom bombardment mass spectrometry. J Biol Chem 263(35):18776–18784
Robertson CR, Scholl EA, Pruess TH, Green BR, White HS, Bulaj G (2010) Engineering galanin analogues that discriminate between GalR1 and GalR2 receptor subtypes and exhibit anticonvulsant activity following systemic delivery. J Med Chem 53(4):1871–1875
Rockenfeller P, Koska M, Pietrocola F, Minois N, Knittelfelder O, Sica V, Franz J, Carmona-Gutierrez D, Kroemer G, Madeo F (2015) Phosphatidylethanolamine positively regulates autophagy and longevity. Cell Death Differ 22(3):499–508
Rostovtsev VV, Green LG, Fokin VV, Sharpless KB (2002) A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl 41(14):2596–2599
Ryan GJ, Hardy Y (2011) Liraglutide: once-daily GLP-1 agonist for the treatment of type 2 diabetes. J Clin Pharm Ther 36(3):260–274
Saar I, Lahe J, Langel K, Runesson J, Webling K, Järv J, Rytkönen J, Närvänen A, Bartfai T, Kurrikoff K, Langel Ü (2013) Novel systemically active galanin receptor 2 ligands in depression-like behavior. J Neurochem 127(1):114–123
Salunke DB, Shukla NM, Yoo E, Crall BM, Balakrishna R, Malladi SS, David SA (2012) Structure–activity relationships in human Toll-like Receptor 2-specific monoacyl lipopeptides. J Med Chem 55(7):3353–3363
Sarkar S, Salyer AC, Wall KA, Sucheck SJ (2013) Synthesis and immunological evaluation of a MUC1 glycopeptide incorporated into l-rhamnose displaying liposomes. Bioconjug Chem 24(3):363–375
Sato T, Oishi K, Ida T, Kojima M (2015) Physiological functions and pathology of ghrelin. Am J Life Sci 3(3-2):8–16
Setoh K, Murakami M, Araki N, Fujita T, Yamamoto A, Muranishi S (1995) Improvement of transdermal delivery of tetragastrin by lipophilic modification with fatty acids. J Pharm Pharmacol 47(10):808–811
Shi L, Cai H, Huang ZH, Sun ZY, Chen YX, Zhao YF, Kunz H, Li YM (2016) Synthetic MUC1 antitumor vaccine candidates with varied glycosylation pattern bearing R/S-configured Pam3CysSerLys4. ChemBioChem 17(15):1412–1415
Shindou H, Eto M, Morimoto R, Shimizu T (2009) Identification of membrane O-acyltransferase family motifs. Biochem Biophys Res Commun 383(3):320–325
Simerska P, Moyle PM, Toth I (2011) Modern lipid-, carbohydrate-, and peptide-based delivery systems for peptide, vaccine, and gene products. Med Res Rev 31(4):520–547
Son SJ, Harris PW, Squire CJ, Baker EN, Kent SB, Brimble MA (2014) Total chemical synthesis of an orf virus protein, ORFV002, an inhibitor of the master gene regulator NF-kappaB. Biopolymers 102(2):137–144
Spohn R, Buwitt-Beckmann U, Brock R, Jung G, Ulmer AJ, Wiesmuller KH (2004) Synthetic lipopeptide adjuvants and Toll-like receptor 2-structure-activity relationships. Vaccine 22(19):2494–2499
Stansly PG, Schlosser ME (1947) Studies on polymyxin: isolation and identification of Bacillus polymyxa and differentiation of polymyxin from certain known antibiotics. J Bacteriol 54(5):549–556
Steinhagen F, Kinjo T, Bode C, Klinman DM (2011) TLR-based immune adjuvants. Vaccine 29(17):3341–3355
Stevenson FT, Bursten SL, Locksley RM, Lovett DH (1992) Myristyl acylation of the tumor necrosis factor alpha precursor on specific lysine residues. J Exp Med 176(4):1053–1062
Stevenson FT, Bursten SL, Fanton C, Locksley RM, Lovett DH (1993) The 31-kDa precursor of interleukin 1 alpha is myristoylated on specific lysines within the 16-kDa N-terminal propiece. Proc Natl Acad Sci U S A 90(15):7245–7249
Still GJ (2002) Development of oral insulin: progress and current status. Diabetes Metab Res Rev 18(S1):S29–S37
Takeuchi O, Kaufmann A, Grote K, Kawai T, Hoshino K, Morr M, Muhlradt PF, Akira S (2000) Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent signaling pathway. J Immunol 164(2):554–557
Tan AC, Mifsud EJ, Zeng W, Edenborough K, McVernon J, Brown LE, Jackson DC (2012) Intranasal administration of the TLR2 agonist Pam2Cys provides rapid protection against influenza in mice. Mol Pharm 9(9):2710–2718
Tanaka K, Fujita T, Yamamoto Y, Murakami M, Yamamoto A, Muranishi S (1996) Enhancement of intestinal transport of thyrotropin-releasing hormone via a carrier-mediated transport system by chemical modification with lauric acid. Biochim Biophys Acta 1283(1):119–126
Thompson P, Lakshminarayanan V, Supekar NT, Bradley JM, Cohen PA, Wolfert MA, Gendler SJ, Boons GJ (2015) Linear synthesis and immunological properties of a fully synthetic vaccine candidate containing a sialylated MUC1 glycopeptide. Chem Commun 51(50):10214–10217
Tomlinson B, Hu M, Zhang Y, Chan P, Liu Z-M (2016) Investigational glucagon-like peptide-1 agonists for the treatment of obesity. Expert Opin Investig Drugs 25(10):1167–1179
Tornoe CW, Christensen C, Meldal M (2002) Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 67(9):3057–3064
Toth I, Flinn N, Hillery A, Gibbons WA, Artursson P (1994) Lipidic conjugates of luteinizing hormone releasing hormone (LHRH)+ and thyrotropin releasing hormone (TRH)+ that release and protect the native hormones in homogenates of human intestinal epithelial (Caco-2) cells. Int J Pharm 105(3):241–247
Trabocchi A, Guarna A (2014) The basics of peptidomimetics. In: Peptidomimetics in organic and medicinal chemistry. Wiley, Chichester, pp 1–17
Velkov T, Roberts KD, Thompson PE, Li J (2016) Polymyxins: a new hope in combating Gram-negative superbugs? Future Med Chem 8(10):1017–1025
Vilhena C, Bettencourt A (2012) Daptomycin: a review of properties, clinical use, drug delivery and resistance. Mini Rev Med Chem 12(3):202–209
Wang M, Casey PJ (2016) Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol 17(2):110–122
Wang J, Shen D, Shen WC (1999) Preparation, purification, and characterization of a reversibly lipidized desmopressin with potentiated anti-diuretic activity. Pharm Res 16(11):1674–1679
Wang J, Wu D, Shen WC (2002) Structure-activity relationship of reversibly lipidized peptides: studies of fatty acid-desmopressin conjugates. Pharm Res 19(5):609–614
Wang J, Chow D, Heiati H, Shen W-C (2003) Reversible lipidization for the oral delivery of salmon calcitonin. J Control Release 88(3):369–380
Wang J, Hogenkamp DJ, Tran M, Li W-Y, Yoshimura RF, Johnstone TBC, Shen W-C, Gee KW (2006) Reversible lipidization for the oral delivery of leu-enkephalin. J Drug Target 14(3):127–136
Ward BP, Ottaway NL, Perez-Tilve D, Ma D, Gelfanov VM, Tschöp MH, DiMarchi RD (2013) Peptide lipidation stabilizes structure to enhance biological function. Mol Metab 2(4):468–479
Watson E, Jonker DM, Jacobsen LV, Ingwersen SH (2010) Population pharmacokinetics of liraglutide, a once-daily human glucagon-like peptide-1 analog, in healthy volunteers and subjects with type 2 diabetes, and comparison to twice-daily exenatide. J Clin Pharmacol 50(8):886–894
Wexler-Cohen Y, Shai Y (2009) Membrane-anchored HIV-1 N-heptad repeat peptides are highly potent cell fusion inhibitors via an altered mode of action. PLOS Pathog 5(7):e1000509
Wiertz EJ, van Gaans-van den Brink JA, Gausepohl H, Prochnicka-Chalufour A, Hoogerhout P, Poolman JT (1992) Identification of T cell epitopes occurring in a meningococcal class 1 outer membrane protein using overlapping peptides assembled with simultaneous multiple peptide synthesis. J Exp Med 176(1):79–88
Wilkinson BL, Malins LR, Chun CK, Payne RJ (2010) Synthesis of MUC1-lipopeptide chimeras. Chem Commun 46(34):6249–6251
Wilkinson BL, Day S, Malins LR, Apostolopoulos V, Payne RJ (2011) Self-adjuvanting multicomponent cancer vaccine candidates combining per-glycosylated MUC1 glycopeptides and the Toll-like receptor 2 agonist Pam3CysSer. Angew Chem Int Ed Engl 50(7):1635–1639
Wilkinson BL, Day S, Chapman R, Perrier S, Apostolopoulos V, Payne RJ (2012) Synthesis and immunological evaluation of self-assembling and self-adjuvanting tricomponent glycopeptide cancer-vaccine candidates. Chemistry 18(51):16540–16548
Willems MMJHP, Zom GG, Khan S, Meeuwenoord N, Melief CJM, van der Stelt M, Overkleeft HS, Codée JDC, van der Marel GA, Ossendorp F, Filippov DV (2014) N-Tetradecylcarbamyl lipopeptides as novel agonists for Toll-like Receptor 2. J Med Chem 57(15):6873–6878
Wong CY, Martinez J, Dass CR (2016) Oral delivery of insulin for treatment of diabetes: status quo, challenges and opportunities. J Pharm Pharmacol 68(9):1093–1108
Wright MH, Heal WP, Mann DJ, Tate EW (2010) Protein myristoylation in health and disease. J Chem Biol 3(1):19–35
Wright TH, Brooks AES, Didsbury AJ, Williams GM, Harris PWR, Dunbar PR, Brimble MA (2013a) Direct peptide lipidation through thiol–ene coupling enables rapid synthesis and evaluation of self-adjuvanting vaccine candidates. Angew Chem Int Ed Engl 52(40):10616–10619
Wright TH, Brooks AES, Didsbury AJ, Williams GM, Harris PWR, Dunbar PR, Brimble MA (2013b) Corrigendum: direct peptide lipidation through thiol–ene coupling enables rapid synthesis and evaluation of self-adjuvanting vaccine candidates. Angew Chem Int Ed Engl 52(45):11686–11686
Wright TH, Brooks AES, Didsbury AJ, McIntosh JD, Burkert K, Yeung H, Williams GM, Dunbar PR, Brimble MA (2013c) An improved method for the synthesis of lipopeptide TLR2-agonists using click chemistry. Synlett 24(14):1835–1841
Wu W, Li R, Malladi SS, Warshakoon HJ, Kimbrell MR, Amolins MW, Ukani R, Datta A, David SA (2010) Structure-activity relationships in toll-like receptor-2 agonistic diacylthioglycerol lipopeptides. J Med Chem 53(8):3198–3213
Yang J, Brown MS, Liang G, Grishin NV, Goldstein JL (2008) Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132(3):387–396
Yang SH, Wojnar JM, Harris PW, DeVries AL, Evans CW, Brimble MA (2013) Chemical synthesis of a masked analogue of the fish antifreeze potentiating protein (AFPP). Org Biomol Chem 11(30):4935–4942
Yang SH, Harris PWR, Williams GM, Brimble MA (2016) Lipidation of cysteine or cysteine-containing peptides using the thiol-ene reaction (CLipPA). Eur J Org Chem 2016(15):2608–2616
Ye J, Wang C, Sumpter R, Brown MS, Goldstein JL, Gale M (2003) Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci U S A 100(26):15865–15870
Yeung H, Lee DJ, Williams GM, Harris PWR, Dunbar RP, Brimble MA (2012) A method for the generation of Pam2Cys-based lipopeptide mimics via CuAAC click chemistry. Synlett 23(11):1617–1620
Yodoya E, Uemura K, Tenma T, Fujita T, Murakami M, Yamamoto A, Muranishi S (1994) Enhanced permeability of tetragastrin across the rat intestinal membrane and its reduced degradation by acylation with various fatty acids. J Pharmacol Exp Ther 271(3):1509–1513
Young SG, Yang SH, Davies BSJ, Jung H-J, Fong LG (2013) Targeting protein prenylation in progeria. Sci Transl Med 5(171):171ps173-171ps173
Yuan L, Wang J, Shen WC (2005) Reversible lipidization prolongs the pharmacological effect, plasma duration, and liver retention of octreotide. Pharm Res 22(2):220–227
Zaman M, Toth I (2013) Immunostimulation by synthetic lipopeptide-based vaccine candidates: structure-activity relationships. Front Immunol 4:318
Zeng W, Jackson DC, Rose K (1996) Synthesis of a new template with a built-in adjuvant and its use in constructing peptide vaccine candidates through polyoxime chemistry. J Pept Sci 2(1):66–72
Zeng W, Ghosh S, Macris M, Pagnon J, Jackson DC (2001) Assembly of synthetic peptide vaccines by chemoselective ligation of epitopes: influence of different chemical linkages and epitope orientations on biological activity. Vaccine 19(28-29):3843–3852
Zeng W, Ghosh S, Lau YF, Brown LE, Jackson DC (2002) Highly immunogenic and totally synthetic lipopeptides as self-adjuvanting immunocontraceptive vaccines. J Immunol 169(9):4905–4912
Zeng W, Horrocks KJ, Robevska G, Wong CY, Azzopardi K, Tauschek M, Robins-Browne RM, Jackson DC (2011) A modular approach to assembly of totally synthetic self-adjuvanting lipopeptide-based vaccines allows conformational epitope building. J Biol Chem 286(15):12944–12951
Zhang L, Bulaj G (2012) Converting peptides into drug Leads by lipidation. Curr Med Chem 19(11):1602–1618
Zhang L, Robertson CR, Green BR, Pruess TH, White HS, Bulaj G (2009) Structural requirements for a lipoamino acid in modulating the anticonvulsant activities of systemically active galanin analogues. J Med Chem 52(5):1310–1316
Zhang Y, Muthana SM, Farnsworth D, Ludek O, Adams K, Barchi JJ Jr, Gildersleeve JC (2012) Enhanced epimerization of glycosylated amino acids during solid-phase peptide synthesis. J Am Chem Soc 134(14):6316–6325
Zhang X, Cheong S-M, Amado NG, Reis AH, MacDonald BT, Zebisch M, Jones EY, Abreu JG, He X (2015) Notum is required for neural and head induction via Wnt deacylation, oxidation, and inactivation. Dev Cell 32(6):719–730
Zhao L, Tong P, Chen Y-X, Hu Z-W, Wang K, Zhang Y-N, Zhao D-S, Cai L-F, Liu K-L, Zhao Y-F, Li Y-M (2012) A multi-functional peptide as an HIV-1 entry inhibitor based on self-concentration, recognition, and covalent attachment. Org Biomol Chem 10(32):6512–6520
Zom GG, Khan S, Britten CM, Sommandas V, Camps MG, Loof NM, Budden CF, Meeuwenoord NJ, Filippov DV, van der Marel GA, Overkleeft HS, Melief CJ, Ossendorp F (2014) Efficient induction of antitumor immunity by synthetic toll-like receptor ligand-peptide conjugates. Cancer Immunol Res 2(8):756–764
Zom GG, Welters MJ, Loof NM, Goedemans R, Lougheed S, Valentijn RR, Zandvliet ML, Meeuwenoord NJ, Melief CJ, de Gruijl TD, Van der Marel GA, Filippov DV, Ossendorp F, Van der Burg SH (2016) TLR2 ligand-synthetic long peptide conjugates effectively stimulate tumor-draining lymph node T cells of cervical cancer patients. Oncotarget 7(41):67087–67100
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Kowalczyk, R., Harris, P.W.R., Williams, G.M., Yang, SH., Brimble, M.A. (2017). Peptide Lipidation – A Synthetic Strategy to Afford Peptide Based Therapeutics. In: Sunna, A., Care, A., Bergquist, P. (eds) Peptides and Peptide-based Biomaterials and their Biomedical Applications. Advances in Experimental Medicine and Biology, vol 1030. Springer, Cham. https://doi.org/10.1007/978-3-319-66095-0_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-66095-0_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-66094-3
Online ISBN: 978-3-319-66095-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)