
Abstract
Network based prediction of interaction between drug compounds and target proteins is a core step in the drug discovery process. The availability of drug–target interaction data has boosted the development of machine learning methods for the in silico prediction of drug–target interactions. In this paper we focus on the crucial issue of data bias.
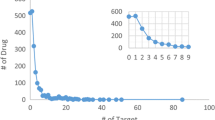
We show that four popular datasets contain a bias because of the way they have been constructed: all drug compounds and target proteins have at least one interaction and some of them have only a single interaction. We show that this bias can be exploited by prediction methods to achieve an optimistic generalization performance as estimated by cross-validation procedures, in particular leave-one-out cross validation. We discuss possible ways to mitigate the effect of this bias, in particular by adapting the validation procedure. In general, results indicate that the data bias should be taken into account when assessing the generalization performance of machine learning methods for the in silico prediction of drug–target interactions.
The datasets and source code for this article are available at
Chapter PDF
Similar content being viewed by others

References
Baumann, K., Rohrer, S.: Exploring benchmark dataset bias in ligand based virtual screening. Chemistry Central Journal 2(suppl. 1), P1 (2008)
Bleakley, K., Yamanishi, Y.: Supervised prediction of drug-target interactions using bipartite local models. Bioinformatics 25(18), 2397–2403 (2009)
Campillos, M., Kuhn, M., Gavin, A.-C., Jensen, L.J., Bork, P.: Drug target identification using side-effect similarity. Science 321(5886), 263–266 (2008)
Chen, X., Liu, M.-X., Yan, G.-Y.: Drug-target interaction prediction by random walk on the heterogeneous network. Mol. Biosyst. 8(7), 1970–1978 (2012)
Csermely, P., Korcsmáros, T., Kiss, H.J., London, G., Nussinov, R.: Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacology & Therapeutics 138(3), 333–408 (2013)
Davis, J., Goadrich, M.: The relationship between Precision-Recall and ROC curves. In: ICML 2006: Proceedings of the 23rd International Conference on Machine Learning, pp. 233–240. ACM (2006)
DeLong, E.R., DeLong, D.M., Clarke-Pearson, D.L.: Comparing the Areas under Two or More Correlated Receiver Operating Characteristic Curves: A Nonparametric Approach. Biometrics 44(3), 837–845 (1988)
Ding, H., Takigawa, I., Mamitsuka, H., Zhu, S.: Similarity-based machine learning methods for predicting drug–target interactions: a brief review. Briefings in Bioinformatics (2013)
Faulon, J.-L., Misra, M., Martin, S., Sale, K., Sapra, R.: Genome scale enzyme– metabolite and drug–target interaction predictions using the signature molecular descriptor. Bioinformatics 24(2), 225–233 (2008)
Fawcett, T.: An introduction to ROC analysis. Pattern Recognition Letters 27(8), 861–874 (2006)
Gönen, M.: Predicting drug-target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics 28(18), 2304–2310 (2012)
Günther, S., Kuhn, M., Dunkel, M., Campillos, M., Senger, C., Petsalaki, E., Ahmed, J., Urdiales, E.G.G., Gewiess, A., Jensen, L.J.J., Schneider, R., Skoblo, R., Russell, R.B., Bourne, P.E., Bork, P., Preissner, R.: SuperTarget and Matador: resources for exploring drug-target relationships. Nucleic Acids Res. 36(Database issue), D919–D922 (2008)
Hopkins, A.L., Groom, C.R.: The druggable genome. Nature reviews. Drug Discovery 1(9), 727–730 (2002)
Isaksson, A., Wallman, M., Göransson, H., Gustafsson, M.G.: Cross-validation and bootstrapping are unreliable in small sample classification. Pattern Recognition Letters 29(14), 1960–1965 (2008)
Jacob, L., Hoffmann, B., Stoven, B., Vert, J.-P.: Virtual screening of GPCRs: an in silico chemogenomics approach. BMC Bioinformatics 9, 363 (2008)
Kanehisa, M., Goto, S., Hattori, M., Aoki-Kinoshita, K.F., Itoh, M., Kawashima, S., Katayama, T., Araki, M., Hirakawa, M.: From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34(Database issue), D354–D357 (2006)
Keiser, M.J., Roth, B.L., Armbruster, B.N., Ernsberger, P., Irwin, J.J., Shoichet, B.K.: Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25(2), 197–206 (2007)
Keiser, M.J., Setola, V., Irwin, J.J., Laggner, C., Abbas, A.I., Hufeisen, S.J., Jensen, N.H., Kuijer, M.B., Matos, R.C., Tran, T.B., Whaley, R., Glennon, R.A., Hert, J., Thomas, K.L., Edwards, D.D., Shoichet, B.K., Roth, B.L.: Predicting new molecular targets for known drugs. Nature 462(7270), 175–181 (2009)
Kohavi, R.: A study of cross-validation and bootstrap for accuracy estimation and model selection. In: Proceedings of the 14th International Joint Conference on Artificial Intelligence, IJCAI 1995, vol. 2, pp. 1137–1143. Morgan Kaufmann Publishers Inc., Montreal (1995)
van Laarhoven, T., Marchiori, E.: Predicting Drug-Target Interactions for New Drug Compounds Using a Weighted Nearest Neighbor Profile. PLoS One 8(6), e66952 (2013)
van Laarhoven, T., Nabuurs, S.B., Marchiori, E.: Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 27(21), 3036–3043 (2011)
Mei, J.-P., Kwoh, C.-K., Yang, P., Li, X., Zheng, J.: Drug-target interaction prediction by learning from local information and neighbors. Bioinformatics 29(2), 238–245 (2013)
Okuno, Y., Tamon, A., Yabuuchi, H., Niijima, S., Minowa, Y., Tonomura, K., Kunimoto, R., Feng, C.: GLIDA: GPCR ligand database for chemical genomics drug discovery database and tools update. Nucleic Acids Research 36(suppl. 1), D907–D912 (2008)
Overington, J.: ChEMBL. An interview with John Overington, team leader, chemogenomics at the European Bioinformatics Institute Outstation of the European Molecular Biology Laboratory (EMBL-EBI). Interview by Wendy A. Warr. Journal of Computer-Aided Molecular Design 23(4), 195–198 (2009)
Rao, R.B., Fung, G.: On the Dangers of Cross-Validation. An Experimental Evaluation. In: SDM, pp. 588–596. SIAM (2008)
Schomburg, I., Chang, A., Ebeling, C., Gremse, M., Heldt, C., Huhn, G., Schomburg, D.: BRENDA, the enzyme database: updates and major new developments. Nucleic Acids Res. 32(suppl. 1), D431–D433 (2004)
Torralba, A., Efros, A.A.: Unbiased look at dataset bias. In: Proceedings of the 2011 IEEE Conference on Computer Vision and Pattern Recognition, CVPR 2011, pp. 1521–1528. IEEE Computer Society, Washington, DC (2011)
Wassermann, A.M., Geppert, H., Bajorath, J.: Ligand prediction for orphan targets using support vector machines and various target-ligand kernels is dominated by nearest neighbor effects. J. Chem. Inf. Model 49, 2155–2167 (2009)
Wishart, D.S., Knox, C., Guo, A.C.C., Cheng, D., Shrivastava, S., Tzur, D., Gautam, B., Hassanali, M.: DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 36(Database issue), D901–D906 (2008)
Yamanishi, Y., Araki, M., Gutteridge, A., Honda, W., Kanehisa, M.: Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 24, i232–i240 (2008)
Yamanishi, Y., Kotera, M., Kanehisa, M., Goto, S.: Drug-target interaction prediction from chemical, genomic and pharmacological data in an integrated framework. Bioinformatics 26(12), i246–i254 (2010)
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this paper
Cite this paper
van Laarhoven, T., Marchiori, E. (2014). Biases of Drug–Target Interaction Network Data. In: Comin, M., Käll, L., Marchiori, E., Ngom, A., Rajapakse, J. (eds) Pattern Recognition in Bioinformatics. PRIB 2014. Lecture Notes in Computer Science(), vol 8626. Springer, Cham. https://doi.org/10.1007/978-3-319-09192-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-09192-1_3
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09191-4
Online ISBN: 978-3-319-09192-1
eBook Packages: Computer ScienceComputer Science (R0)
