
Abstract
Interleukin-1 is a prototypic pro-inflammatory cytokine that is elevated in cytokine storm syndromes (CSS), such as secondary hemophagocytic lymphohistiocytosis (sHLH) and macrophage activation syndrome (MAS). IL-1 has many pleotropic and redundant roles in both the innate and adaptive immune response. Blockade of IL-1 with recombinant human interleukin-1 receptor antagonist has shown efficacy in treating CSS. Recently, an IL-1 family member, IL-18, has been demonstrated to be contributory to CSS in autoinflammatory conditions, such as in inflammasomopathies (e.g., NLRC4 mutations). Anecdotally, recombinant IL-18 binding protein can be of benefit in treating IL-18 driven CSS. Lastly, another IL-1 family member, IL-33, has been postulated to contribute to CSS in an animal model of disease. Targeting of IL-1 and related cytokines holds promise in treating a variety of CSS.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Covid-19
- Anakinra
- Interleukin-1 (IL-1)
- Interleukin-18 (IL-18)
- Interleukin-33 (IL-33)
- IL-1 receptor antagonist
- Cytokine storm syndrome
- IL-18 binding protein
- Interleukin-1 receptor-like 1
- Macrophage activation syndrome
- Secondary hemophagocytic lymphohistiocytosis
Interleukin-1 (IL-1)
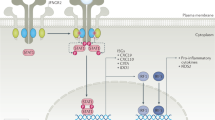
Cytokines are substances, such as growth factors, interferons, and interleukins, which are secreted by cells in the immune system to influence other cells. Interleukins, as the name implies, are glycoproteins that regulate immune response by communicating between white blood cells. Interleukin-1 (IL-1) was one of the first cytokines described in the immune system, over 40 years ago [1], as it plays a central host defense role against infection. IL-1, formerly known as endogenous pyrogen for its fever inducing effect, is a representative of the 11-member IL-1 family of cytokines [2]. In addition, there are ten unique IL-1 receptor (IL-1R) family members that can result in either pro- or anti-inflammatory functions upon binding IL-1 family members depending on the individual receptor and its co-receptor, making for a highly complex system of signaling. IL-1α and IL-1β are two distinct gene products located adjacent to one another on the long arm of chromosome 2. Their regulation differs, but both are able to bind IL-1R1 and the natural inhibitor of both, IL-1R antagonist (IL-1Ra) (Fig. 1) [2].

IL-1 family member biology. The producers of and responders to IL-1 family members are numerous and complex; this figure illustrates some of the main actors in IL-1 family member biology. IL-1β and IL-18 are released from myeloid cells after activation of the inflammasome. IL-33, normally retained in the nucleus, is released by endothelial or epithelial cells after necrotic cell death. Each of the cytokines are then sensed by their respective receptors on CD4+ and CD8+ T-cells resulting in activation of the transcription factor NFκB. Additionally, soluble inhibitors of each of these cytokines are produced which displace binding of the cytokine to its receptor: IL-1 receptor antagonist (IL1-RA) for IL-1β, IL-18 binding protein (IL-18BP) for IL-18, and soluble ST2 receptor (sST2) for IL-33. Figure courtesy of Dr. Ed Behrens, University of Pennsylvania
IL-1β has been termed a gatekeeper of inflammation and is involved in the pathophysiology of a variety of autoinflammatory diseases [3]. Monocytes/macrophages are a primary source of IL-1β, and IL-1β activity is tightly controlled and dependent on the conversion of an inactive precursor to an active cytokine by limited proteolysis. IL-1β can be processed intracellularly by caspase-1, which is activated by the inflammasome, a multiprotein complex that detects pathogenic organisms as well as sterile stressors to the cell [2]. The protein, NLRP3 (cryopyrin) is important for the assembly of this complex, and hyper-activating mutations in NLRP3 can lead to excess IL-1β activity, resulting in a family of autoinflammatory disorders ranging from familial cold urticaria to Muckle–Wells syndrome to the severest form, neonatal onset multisystem inflammatory disease (NOMID) [4]. NOMID can be quite devastating to infants primarily affecting the central nervous system, the bones/joints, and the skin. Fortunately, IL-1β blockade by monoclonal antibody, IL-1R fusion protein, or recombinant human IL-1Ra (rhIL-1Ra) has revolutionized the care of individuals with cryopyrinopathies [4].
Another more common autoinflammatory disorder of childhood responsive to IL-1 blockade is systemic juvenile idiopathic arthritis (sJIA). sJIA affects approximately 1 in 10,000 children worldwide and is characterized clinically by high spiking fever, evanescent salmon-colored rash, arthritis, adenopathy, and occasionally serositis [5]. This used to be a quite devastating disease of childhood until it was found that large amounts of IL-1β were released by sJIA patient peripheral blood mononuclear cells, and IL-1Ra could dramatically treat children with refractory sJIA [6]. Initial treatment of sJIA patients with rIL-1Ra was also shown to improve outcomes and reduce the requirement for corticosteroid use [7]. Eventually, randomized and blinded, placebo-controlled clinical trials with various IL-1 inhibitors bore out these initial anecdotal experiences [8,9,10]. IL-1 blockade, along with IL-6 blockade, have revolutionized the care of children with sJIA [11].
Children with sJIA are prone to develop a sometimes fatal cytokine storm syndrome (CSS) termed macrophage activation syndrome (MAS) or secondary hemophagocytic lymphohistiocytosis (sHLH) [12]. Seven to ten percent of sJIA patients will develop overt MAS, while another 30–40% will manifest with subclinical or occult MAS which can progress to multi-system organ failure [13, 14]. Those sJIA patients who are prone to MAS development frequently possess heterozygous mutations in known familial HLH genes involved in the perforin mediated cytolytic pathway (e.g., PRF1, UNC13D) employed by CD8 T cells and natural killer (NK) cells [15, 16]. This suggests some shared genetic risk factors for CSS/MAS in those with sJIA and other forms of sHLH/MAS [17]. Heterozygous defects in HLH genes have been shown to disrupt NK cell lytic activity by partial [17, 18] and complete dominant negative [19], and hypomorphic effects, including those with sJIA [20]. Defective cytotoxic killing of antigen presenting cells (APC) by lymphocytes has been shown to lead to prolonged interaction of the lytic lymphocyte and the APC resulting in a pro-inflammatory CSS [18, 21].
As CSS resembles MAS in sJIA patients, and sJIA patients were originally shown to be responsive to IL-1 blockade, rhIL-1Ra was employed as treatment for a severe CSS in a child with cytophagic histiocytic panniculitis [22]. This was the first explicit use of rhIL-1Ra reported to successfully treat CSS/MAS, and subsequently rhIL-1Ra was shown to effectively treat refractory MAS in another dozen patients with primarily rheumatic disorders, such as sJIA [23]. Since then, there have been numerous case reports and series of patients effectively treated with rhIL-1Ra for CSS/MAS/sHLH with etiologies ranging from sJIA to adult onset Still disease (Table 1), to systemic lupus erythematosus (Table 2), to autoinflammatory conditions (Table 3), to secondary infections in HIV/AIDS patients (Table 4). Moreover, a retrospective review of a large clinical trial in sepsis revealed that high dose rhIL-1Ra markedly improved survival in sepsis patients with features of MAS, namely hepatobiliary dysfunction and disseminated intravascular coagulopathy (Table 4) [24]. Thus, rhIL-1Ra therapy was anecdotally reported to effectively treat CSS in a variety of infectious and rheumatic disorders. Currently, a randomized, double-blind, placebo controlled clinical trial is underway to evaluate rhIL-1Ra treatment for children and adults with sHLH/CSS [ClinicalTrials.gov Identifier: NCT02780583].
Interleukin-18 (IL-18)
In 2014, activating mutations in the NLRC4 inflammasome component were demonstrated to lead to an autoinflammatory syndrome [25, 26], including an infant with recurrent MAS [27]. This child only partially responded to rhIL-1Ra treatment but was found to have very elevated serum IL-18 levels and was successfully treated with addition of recombinant human IL-18 binding protein (IL-18BP) [28]. rhIL-18BP is naturally occurring and analogous to rhIL-1Ra in blocking IL-18 and IL-1 function, respectively (Fig. 1). IL-18 is a member of the IL-1 superfamily and analogous to IL-1β is first synthesized as an inactive precursor and is activated following cleavage by caspase-1 [2]. Unlike, IL-1, however, IL-18 does not trigger fever [29]. Thus, despite overlapping features and functions, IL-18 has unique features from IL-1.
In the presence of IL-12 or IL-15, IL-18 induces interferon-gamma (IFNγ) in NK cells, CD4 T cells, and CD8 T cells [29]. IFNγ is believed to a major driver of CSS/HLH in animal models and in humans, and a recent case report depicted the benefit of anti-IFNγ (emapalumab) in treating refractory HLH [30]. A recent clinical trial of emapalumab in treating HLH has resulted in FDA approval for this indication [31]. IL-18 has also been shown to promote murine and human MAS demonstrating the pathogenicity of free (unbound) IL-18 [32, 33]. In addition, free IL-18 concentrations correlated with clinical status in sHLH/MAS patients [34], and IL-18 levels were predictive of MAS in children with sJIA [35]. Therefore, blockade of IL-18 may take on a more prominent role in treating a range of CSS.
Interleukin-33 (IL-33)
IL-33 is another IL-1 family member with close homology to IL-1, and it is considered an alarmin that is released as an active precursor upon cell damage [2]. IL-33 differs from IL-1 as it can act, depending on context, as either anti- or pro-inflammatory in nature [2]. In a pro-inflammatory setting, IL-33 binds the ST2 receptor which signals via MyD88, IL-1 receptor activated kinases (IRAKs), and the inflammatory transcription factor, NFκB (Fig. 1) [2]. A murine model of HLH showed a role for MyD88-dependent ST2 in disease, and demonstrated that blocking IL-33 signaling via monoclonal antibody directed against ST2 improved survival and the severity of multiple disease manifestations [36]. Moreover, in the long term ST2 blockage results in CD8 T cell exhaustion that does not alter mortality in the HLH murine model arguing for early use on ST2 blockade in CSS [37]. Thus, disruption of signaling of another IL-1 family member, IL-33, may be an option in treating CSS. Overall, these targeted (anti-cytokine) approaches to treating CSS are likely to be far less toxic then current chemotherapeutic approaches [38]. Identifying the correct target for individual patients remains the next challenge.
References
Dinarello, C. A., Renfer, L., & Wolff, S. M. (1977). Human leukocytic pyrogen: Purification and development of a radioimmunoassay. Proceedings of the National Academy of Sciences of the United States of America, 74(10), 4624–4627.
Dinarello, C. A. (2018). Overview of the IL-1 family in innate inflammation and acquired immunity. Immunological Reviews, 281(1), 8–27.
Dinarello, C. A. (2011). A clinical perspective of IL-1beta as the gatekeeper of inflammation. European Journal of Immunology, 41(5), 1203–1217.
Kuemmerle-Deschner, J. B. (2015). CAPS—pathogenesis, presentation and treatment of an autoinflammatory disease. Seminars in Immunopathology, 37(4), 377–385.
Lee, J. J. Y., & Schneider, R. (2018). Systemic juvenile idiopathic arthritis. Pediatric Clinics of North America, 65(4), 691–709.
Pascual, V., Allantaz, F., Arce, E., Punaro, M., & Banchereau, J. (2005). Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. The Journal of Experimental Medicine, 201(9), 1479–1486.
Nigrovic, P. A., Mannion, M., Prince, F. H., Zeft, A., Rabinovich, C. E., van Rossum, M. A., et al. (2011). Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: Report of forty-six patients from an international multicenter series. Arthritis and Rheumatism, 63(2), 545–555.
Ilowite, N. T., Prather, K., Lokhnygina, Y., Schanberg, L. E., Elder, M., Milojevic, D., et al. (2014). Randomized, double-blind, placebo-controlled trial of the efficacy and safety of rilonacept in the treatment of systemic juvenile idiopathic arthritis. Arthritis & Rhematology, 66(9), 2570–2579.
Quartier, P., Allantaz, F., Cimaz, R., Pillet, P., Messiaen, C., Bardin, C., et al. (2011). A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Annals of the Rheumatic Diseases, 70(5), 747–754.
Ruperto, N., Brunner, H. I., Quartier, P., Constantin, T., Wulffraat, N., Horneff, G., et al. (2012). Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. The New England Journal of Medicine, 367(25), 2396–2406.
Stoll, M. L., & Cron, R. Q. (2014). Treatment of juvenile idiopathic arthritis: A revolution in care. Pediatric Rheumatology Online Journal, 12, 13.
Ravelli, A., Grom, A. A., Behrens, E. M., & Cron, R. Q. (2012). Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: Diagnosis, genetics, pathophysiology and treatment. Genes and Immunity, 13(4), 289–298.
Behrens, E. M., Beukelman, T., Paessler, M., & Cron, R. Q. (2007). Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. The Journal of Rheumatology, 34(5), 1133–1138.
Bleesing, J., Prada, A., Siegel, D. M., Villanueva, J., Olson, J., Ilowite, N. T., et al. (2007). The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis and Rheumatism, 56(3), 965–971.
Kaufman, K. M., Linghu, B., Szustakowski, J. D., Husami, A., Yang, F., Zhang, K., et al. (2014). Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis & Rhematology, 66(12), 3486–3495.
Vastert, S. J., van Wijk, R., D'Urbano, L. E., de Vooght, K. M., de Jager, W., Ravelli, A., et al. (2010). Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford, England), 49(3), 441–449.
Zhang, M., Behrens, E. M., Atkinson, T. P., Shakoory, B., Grom, A. A., & Cron, R. Q. (2014). Genetic defects in cytolysis in macrophage activation syndrome. Current Rheumatology Reports, 16(9), 439–446.
Zhang, M., Bracaglia, C., Prencipe, G., Bemrich-Stolz, C. J., Beukelman, T., Dimmitt, R. A., et al. (2016). A heterozygous RAB27A mutation associated with delayed cytolytic granule polarization and hemophagocytic lymphohistiocytosis. Journal of Immunology, 196(6), 2492–2503.
Spessott, W. A., Sanmillan, M. L., McCormick, M. E., Patel, N., Villanueva, J., Zhang, K., et al. (2015). Hemophagocytic lymphohistiocytosis caused by dominant-negative mutations in STXBP2 that inhibit SNARE-mediated membrane fusion. Blood, 125(10), 1566–1577.
Schulert, G. S., Zhang, M., Husami, A., Fall, N., Brunner, H., Zhang, K., et al. (2018). Brief report: Novel UNC13D intronic variant disrupting an NF-kappaB enhancer in a patient with recurrent macrophage activation syndrome and systemic juvenile idiopathic arthritis. Arthritis & Rhematology, 70(6), 963–970.
Jenkins, M. R., Rudd-Schmidt, J. A., Lopez, J. A., Ramsbottom, K. M., Mannering, S. I., Andrews, D. M., et al. (2015). Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. The Journal of Experimental Medicine, 212(3), 307–317.
Behrens, E. M., Kreiger, P. A., Cherian, S., & Cron, R. Q. (2006). Interleukin 1 receptor antagonist to treat cytophagic histiocytic panniculitis with secondary hemophagocytic lymphohistiocytosis. The Journal of Rheumatology, 33(10), 2081–2084.
Miettunen, P. M., Narendran, A., Jayanthan, A., Behrens, E. M., & Cron, R. Q. (2011). Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: Case series with 12 patients. Rheumatology (Oxford, England), 50(2), 417–419.
Shakoory, B., Carcillo, J. A., Chatham, W. W., Amdur, R. L., Zhao, H., Dinarello, C. A., et al. (2016). Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: Reanalysis of a prior phase III trial. Critical Care Medicine, 44(2), 275–281.
Kitamura, A., Sasaki, Y., Abe, T., Kano, H., & Yasutomo, K. (2014). An inherited mutation in NLRC4 causes autoinflammation in human and mice. The Journal of Experimental Medicine, 211(12), 2385–2396.
Romberg, N., Al Moussawi, K., Nelson-Williams, C., Stiegler, A. L., Loring, E., Choi, M., et al. (2014). Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nature Genetics, 46(10), 1135–1139.
Canna, S. W., de Jesus, A. A., Gouni, S., Brooks, S. R., Marrero, B., Liu, Y., et al. (2014). An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nature Genetics, 46(10), 1140–1146.
Canna, S. W., Girard, C., Malle, L., de Jesus, A., Romberg, N., Kelsen, J., et al. (2017). Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. The Journal of Allergy and Clinical Immunology, 139(5), 1698–1701.
Kaplanski, G. (2018). Interleukin-18: Biological properties and role in disease pathogenesis. Immunological Reviews, 281(1), 138–153.
Lounder, D. T., Bin, Q., de Min, C., & Jordan, M. B. (2019). Treatment of refractory hemophagocytic lymphohistiocytosis with emapalumab despite severe concurrent infections. Blood Advances, 3(1), 47–50.
Al-Salama, Z. T. (2019). Emapalumab: First global approval. Drugs.
Girard-Guyonvarc'h, C., Palomo, J., Martin, P., Rodriguez, E., Troccaz, S., Palmer, G., et al. (2018). Unopposed IL-18 signaling leads to severe TLR9-induced macrophage activation syndrome in mice. Blood, 131(13), 1430–1441.
Weiss, E. S., Girard-Guyonvarc’h, C., Holzinger, D., de Jesus, A. A., Tariq, Z., Picarsic, J., et al. (2018). Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood, 131(13), 1442–1455.
Mazodier, K., Marin, V., Novick, D., Farnarier, C., Robitail, S., Schleinitz, N., et al. (2005). Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood, 106(10), 3483–3489.
Shimizu, M., Nakagishi, Y., Inoue, N., Mizuta, M., Ko, G., Saikawa, Y., et al. (2015). Interleukin-18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clinical Immunology, 160(2), 277–281.
Rood, J. E., Rao, S., Paessler, M., Kreiger, P. A., Chu, N., Stelekati, E., et al. (2016). ST2 contributes to T-cell hyperactivation and fatal hemophagocytic lymphohistiocytosis in mice. Blood, 127(4), 426–435.
Rood, J. E., Burn, T. N., Neal, V., Chu, N., & Behrens, E. M. (2018). Disruption of IL-33 signaling limits early CD8+ T cell effector function leading to exhaustion in murine hemophagocytic lymphohistiocytosis. Frontiers in Immunology, 9, 2642.
Ravelli, A., Davi, S., Minoia, F., Martini, A., & Cron, R. Q. (2015). Macrophage activation syndrome. Hematology/Oncology Clinics of North America, 29(5), 927–941.
Kelly, A., & Ramanan, A. V. (2008). A case of macrophage activation syndrome successfully treated with anakinra. Nature Clinical Practice. Rheumatology, 4(11), 615–620.
Durand, M., Troyanov, Y., Laflamme, P., & Gregoire, G. (2010). Macrophage activation syndrome treated with anakinra. The Journal of Rheumatology, 37(4), 879–880.
Bruck, N., Suttorp, M., Kabus, M., Heubner, G., Gahr, M., & Pessler, F. (2011). Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. Journal of Clinical Rheumatology, 17(1), 23–27.
Loh, N. K., Lucas, M., Fernandez, S., & Prentice, D. (2012). Successful treatment of macrophage activation syndrome complicating adult Still disease with anakinra. Internal Medicine Journal, 42(12), 1358–1362.
Kahn, P. J., & Cron, R. Q. (2013). Higher-dose Anakinra is effective in a case of medically refractory macrophage activation syndrome. The Journal of Rheumatology, 40(5), 743–744.
Barut, K., Yucel, G., Sinoplu, A. B., Sahin, S., Adrovic, A., & Kasapcopur, O. (2015). Evaluation of macrophage activation syndrome associated with systemic juvenile idiopathic arthritis: Single center experience over a one-year period. Turkish Archives of Pediatrics, 50(4), 206–210.
Mehta, M. V., Manson, D. K., Horn, E. M., & Haythe, J. (2016). An atypical presentation of adult-onset Still's disease complicated by pulmonary hypertension and macrophage activation syndrome treated with immunosuppression: A case-based review of the literature. Pulmonary Circulation, 6(1), 136–142.
Lenert, A., & Yao, Q. (2016). Macrophage activation syndrome complicating adult onset Still’s disease: A single center case series and comparison with literature. Seminars in Arthritis and Rheumatism, 45(6), 711–716.
Aytac, S., Batu, E. D., Unal, S., Bilginer, Y., Cetin, M., Tuncer, M., et al. (2016). Macrophage activation syndrome in children with systemic juvenile idiopathic arthritis and systemic lupus erythematosus. Rheumatology International, 36(10), 1421–1429.
Kumar, A., & Kato, H. (2016). Macrophage activation syndrome associated with adult-onset Still’s disease successfully treated with anakinra. Case Reports in Rheumatology, 2016, 3717392.
Agnihotri, A., Ruff, A., Gotterer, L., Walker, A., McKenney, A. H., & Brateanu, A. (2016). Adult onset Still’s disease associated with mycoplasma pneumoniae infection and hemophagocytic lymphohistiocytosis. Case Reports in Medicine, 2016, 2071815.
Schafer, E. J., Jung, W., & Korsten, P. (2016). Combination immunosuppressive therapy including rituximab for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adult-onset Still's disease. Case Reports in Rheumatology, 2016, 8605274.
Parisi, F., Paglionico, A., Varriano, V., Ferraccioli, G., & Gremese, E. (2017). Refractory adult-onset Still disease complicated by macrophage activation syndrome and acute myocarditis: A case report treated with high doses (8 mg/kg/d) of anakinra. Medicine (Baltimore), 96(24), e6656.
Sonmez, H. E., Demir, S., Bilginer, Y., & Ozen, S. (2018). Anakinra treatment in macrophage activation syndrome: A single center experience and systemic review of literature. Clinical Rheumatology, 37(12), 3329–3335.
Lilleby, V., Haydon, J., Sanner, H., Krossness, B. K., Ringstad, G., & Flato, B. (2014). Severe macrophage activation syndrome and central nervous system involvement in juvenile dermatomyositis. Scandinavian Journal of Rheumatology, 43(2), 171–173.
Shafferman, A., Birmingham, J. D., & Cron, R. Q. (2014). High dose anakinra for treatment of severe neonatal Kawasaki disease: A case report. Pediatric Rheumatology Online Journal, 12, 26.
Cron, R. Q., & Chatham, W. W. (2016). Development of spondyloarthropathy following episodes of macrophage activation syndrome in children with heterozygous mutations in haemophagocytic lymphohistiocytosis-associated genes. Clinical and Experimental Rheumatology, 34(5), 953.
Tayer-Shifman, O. E., & Ben-Chetrit, E. (2013). Refractory macrophage activation syndrome in a patient with SLE and APLA syndrome—Successful use of PET—CT and Anakinra in its diagnosis and treatment. Modern Rheumatology.
Mohr, V., Schulz, A., Lohse, P., Schumann, C., Debatin, K. M., & Schuetz, C. (2014). Urticaria, fever, and hypofibrinogenemia. Arthritis & Rhematology, 66(5), 1377.
Schulert, G. S., Bove, K., McMasters, R., Campbell, K., Leslie, N., & Grom, A. A. (2015). 11-month-old infant with periodic fevers, recurrent liver dysfunction, and perforin gene polymorphism. Arthritis Care and Research., 67(8), 1173–1179.
Barsalou, J., Blincoe, A., Fernandez, I., Dal-Soglio, D., Marchitto, L., Selleri, S., et al. (2018). Rapamycin as an adjunctive therapy for NLRC4 associated macrophage activation syndrome. Frontiers in Immunology, 9, 2162.
Butin, M., Mekki, Y., Phan, A., Billaud, G., Di Filippo, S., Javouhey, E., et al. (2013). Successful immunotherapy in life-threatening parvovirus B19 infection in a child. The Pediatric Infectious Disease Journal, 32(7), 789–792.
Rajasekaran, S., Kruse, K., Kovey, K., Davis, A. T., Hassan, N. E., Ndika, A. N., et al. (2014). Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children∗. Pediatric Critical Care Medicine, 15(5), 401–408.
Kumar, N., Goyal, J., Goel, A., Shakoory, B., & Chatham, W. (2014). Macrophage activation syndrome secondary to human monocytic ehrlichiosis. Indian Society of Hematology and Blood Transfusion, 30(Suppl 1), 145–147.
Divithotawela, C., Garrett, P., Westall, G., Bhaskar, B., Tol, M., & Chambers, D. C. (2016). Successful treatment of cytomegalovirus associated hemophagocytic lymphohistiocytosis with the interleukin 1 inhibitor—anakinra. Respirology Case Reports, 4(1), 4–6.
Wohlfarth, P., Agis, H., Gualdoni, G. A., Weber, J., Staudinger, T., Schellongowski, P., et al. (2017). Interleukin 1 receptor antagonist anakinra, intravenous immunoglobulin, and corticosteroids in the management of critically Ill adult patients with hemophagocytic lymphohistiocytosis. Journal of Intensive Care Medicine, 885066617711386.
Kumar, B., Aleem, S., Saleh, H., Petts, J., & Ballas, Z. K. (2017). A personalized diagnostic and treatment approach for macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in adults. Journal of Clinical Immunology, 37(7), 638–643.
Ocon, A. J., Bhatt, B. D., Miller, C., & Peredo, R. A. (2017). Safe usage of anakinra and dexamethasone to treat refractory hemophagocytic lymphohistiocytosis secondary to acute disseminated histoplasmosis in a patient with HIV/AIDS. BML Case Reports, 2017.
Nusshag, C., Morath, C., Zeier, M., Weigand, M. A., Merle, U., & Brenner, T. (2017). Hemophagocytic lymphohistiocytosis in an adult kidney transplant recipient successfully treated by plasmapheresis: A case report and review of the literature. Medicine (Baltimore), 96(50), e9283.
Summary
Although CSSs are frequently fatal, in part from the disease process but also secondary to broad immunosuppression used during treatment, novel approaches of targeting pro-inflammatory cytokines are being explored. Members of the IL-1 superfamily, including IL-1, IL-18, and IL-33 are being explored clinically and/or in murine models of CSS. IL-1 blockade seems like promising therapy for CSS, particularly in the setting of children with sJIA. Similarly, targeting IL-18 may be an important therapeutic option for certain genetic inflammasomopathies, such as activating NLRC4 mutations. Finally, murine models of CSS suggest disrupting IL-33 signaling dampens disease parameters and increases survival. Knowing which cytokine, or combinations of cytokines, to target for individual patients will keep physician-scientists busy for some time to come, yet cytokine blockade for frequently fatal CSS has shown some early promising results.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this chapter

Cite this chapter
Cron, R.Q. (2019). IL-1 Family Blockade in Cytokine Storm Syndromes. In: Cron, R., Behrens, E. (eds) Cytokine Storm Syndrome. Springer, Cham. https://doi.org/10.1007/978-3-030-22094-5_31
Download citation
DOI: https://doi.org/10.1007/978-3-030-22094-5_31
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-22093-8
Online ISBN: 978-3-030-22094-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)