
Abstract
Apolipoprotein A-I (apoA-I) is the main protein of high-density lipoprotein (HDL). We investigated the involvement of apoA-I in diet-induced accumulation of triglycerides in hepatocytes and its potential role in the treatment of nonalcoholic fatty liver disease (NAFLD). ApoA-I-deficient (apoA-I−/−) mice showed increased diet-induced hepatic triglyceride deposition and disturbed hepatic histology while they exhibited reduced glucose tolerance and insulin sensitivity. Quantification of FASN (fatty acid synthase 1), DGAT-1 (diacylglycerol O-acyltransferase 1), and PPARγ (peroxisome proliferator-activated receptor γ) mRNA expression suggested that the increased hepatic triglyceride content of the apoA-I−/− mice was not due to de novo synthesis of triglycerides. Similarly, metabolic profiling did not reveal differences in the energy expenditure between the two mouse groups. However, apoA-I−/− mice exhibited enhanced intestinal absorption of dietary triglycerides (3.6 ± 0.5 mg/dL/min for apoA-I−/− versus 2.0 ± 0.7 mg/dL/min for C57BL/6 mice, P < 0.05), accelerated clearance of postprandial triglycerides and a reduced rate of hepatic very low density lipoprotein (VLDL) triglyceride secretion (9.8 ± 1.1 mg/dL/min for apoA-I−/− versus 12.5 ± 1.3 mg/dL/min for C57BL/6 mice, P < 0.05). In agreement with these findings, adenovirus-mediated gene transfer of apoA-IMilano in apoA-I−/− mice fed a Western-type diet for 12 wks resulted in a significant reduction in hepatic triglyceride content and an improvement of hepatic histology and architecture. Our data extend the current knowledge on the functions of apoA-I, indicating that in addition to its well-established properties in atheroprotection, it is also an important modulator of processes associated with diet-induced hepatic lipid deposition and NAFLD development in mice. Our findings raise the interesting possibility that expression of therapeutic forms of apoA-I by gene therapy approaches may have a beneficial effect on NAFLD.
Similar content being viewed by others

Introduction
Apolipoprotein A-I (apoA-I) is a single polypeptide of 243 amino acids with a molecular mass of 28.1 kDa, mainly present in plasma as a component of high-density lipoprotein (HDL) (1). ApoA-I is expressed primarily by the intestine and the liver, although other tissues also express it. Intestinally derived apoA-I enters the circulation associated with chylomicrons and then it is rapidly transferred to HDL during conversion of chylomicrons into chylomicron remnants by lipoprotein lipase (2,3), and hepatic apoA-I is secreted in the circulation in a lipid-free, or minimally lipidated form (2,4).
Lipid-free or minimally lipidated apoA-I plays an important role in the de novo biogenesis of HDL (1). In the early steps of this pathway, apoA-I acquires phospholipids and cholesterol via its interactions with the ATP-binding cassette A1 (ABCA1). Through a series of intermediate steps that are currently poorly understood, apoA-I is gradually lipidated and eventually forms discoidal particles that are then converted to spherical by the action of the plasma enzyme lecithin:cholesterol acyl transferase (LCAT). Both discoidal and spherical HDL particles interact functionally with scavenger receptor-class B type I (SR-BI) and this interaction is believed to be important for some of the atheroprotective functions of HDL. ApoA-I is a coactivator of LCAT in plasma (5). In vitro studies suggested that apoA-I is absolutely required for the activation of plasma LCAT (6–8). Similarly, in vivo studies showed that mice deficient in apoA-I (apoA-I−/−) (9) have a plasma LCAT activity level that corresponds to 20–25% of their wild-type (WT) counterparts (10), further confirming the important role of apoA-I in the activation of LCAT.
Additional functions of apoA-I were discovered as a result of studies of abnormal and/or diseased states. For example, some mutations in apoA-I have been implicated in pathological conditions, such as Tangier disease (11–13). In contrast, apoA-IMilano (14,15), a natural variant form of apoA-I, has been found to have a beneficial effect on atherosclerosis, although the precise mechanism has not been fully elucidated. In rare cases of combined hyperlipidemia, apoA-I can also be found as a component of triglyceride-rich lipoproteins, in which it interferes with their metabolism in plasma. Despite the key role that lipoproteins play in the trafficking of plasma lipids and the contribution of apoA-I in lipoprotein metabolism, to date very little is known on the effects of apoA-I on triglyceride deposition to the liver, a process that constitutes the first step in the development of diet-induced nonalcoholic fatty liver disease (NAFLD).
In humans, NAFLD ranges from a simple accumulation of triglycerides in the liver (hepatic steatosis) to hepatic steatosis with inflammation, fibrosis and cirrhosis (nonalcoholic steatohepatitis [NASH]) (16,17). Even though hepatic deposition of triglycerides is an important process, additional parameters such as disturbances in hepatic lipid homeostasis; increased generation of reactive oxygen species and, consequently, oxidative stress; changes in mitochondrial function; DNA damage; microbial infections; and release of various cytokines are also required for the establishment of NAFLD and its progression to NASH (17–20). Since NAFLD is also present in patients with metabolic syndrome, it has been proposed that it should be included as the hepatic component of metabolic syndrome (18). Even though aging, hormonal imbalance and genetic predisposition may contribute to NAFLD (19), a Western-type diet and sedentary lifestyle that result in excess body fat, physical inactivity and imbalance in caloric load are the most common contributors to the disease (20).
Here we studied the role of apoA-I in NAFLD development. Our data reveal a significant role of apoA-I in modulating diet-induced hepatic triglyceride accumulation, and raise the interesting possibility that therapeutic variants of apoA-I may find important applications in the treatment of NAFLD in future gene therapy approaches.
Materials and Methods
Animal Studies
The apoA-I−/− (9) and the C57BL/6 mice used in our studies were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Male mice 10–12 wks old were used in these studies. Mice in each group were caged individually (one mouse per cage) and were allowed unrestricted access to food and water under a 12-h light/dark cycle. To ensure similar average cholesterol, triglyceride and glucose levels and starting body weights, groups of 10 mice were formed after determining the fasting cholesterol, triglyceride and glucose levels and body weights of the individual mice. Mice were fed the standard Western-type diet (Mucedola SRL, Milan, Italy) for the indicated period, and body weight and fasting plasma cholesterol and triglyceride levels were determined at the indicated time points after diet initiation. At the end of each experiment, mice were killed and plasma and liver samples were collected. Carcasses were stored at −80°C. All animal studies were governed by the European Union directive for the protection and welfare of animals used for experimental and other scientific purposes (https://doi.org/eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:1986:358:0001:0028:EN:PDF). In our experiments we took into consideration the 3Rs (reduce, refine, replace) and we minimized the number of animal experiments to the absolute minimum. To date there is no in vitro system to mimic satisfactorily the lipid and lipoprotein transport system and the in vivo mechanisms leading to NAFLD, making the use of experimental animals mandatory. All procedures used in our studies involved minimal distress to the mice tested. The work was authorized by the appropriate committee of the Laboratory Animal Center of the University of Patras Medical School.
Indirect Calorimetry Studies
We performed indirect calorimetry studies using the Phenomaster small animal calorimetry system (TSE Systems, Bad Homburg, Germany). Groups of apoA-I−/− and C57BL/6 mice were acclimated in the calocages for 48 h and then energy expenditure (EE) was determined on the basis of the CO2 volume (VCO2) (mL/h) produced and VO2 (mL/h) consumed over an additional 24-h period. A total of 72 measurements of VCO2 and VO2 were collected. The EE per mouse (expressed in J/h) for each measurement was calculated by the formula EE = (15.818 × VO2) + (5.176 × VCO2) (21). Then, the average EE of the 72 different measurements was determined and plotted as a function of mouse body weight (g), and analysis of covariance (ANCOVA) was performed between the two groups of mice (21).
Generation and Large-Scale Production of the Recombinant Attenuated Adenovirus Expressing ApoA-IMilano
For the generation of the genomic DNA expressing apoA-IMilano, we used the QuickChange-XL (Stratagene, La Jolla, CA, USA) site-directed mutagenesis kit as described (22). The mutagenic primers used were: 5′-TTGGC CGCGC GCCTT GAG-3′ (forward) and 5′-CTGGC GCAGC TCGTC GCT-3′ (reverse) with the pCA13-AIgN vector (23), which contains the genomic human apoA-I sequence, as template. The recombinant adenoviruses were constructed as described previously (24). The titer of the virus preparation was 2 × 1010 pfu/mL. The recombinant attenuated adenovirus AdGFP-apoA-IMilano independently expresses green fluorescent protein (GFP) and apoA-IMilano under the control of two different CMV (cytomegalovirus) promoters.
Cell Culture Studies
HTB13 cells (SW1783, human astrocytoma) were grown to confluence in medium containing 10% fetal calf serum. Confluent cultures were washed twice with phosphate-buffered saline (PBS), switched to culture medium containing 2% heat-inactivated horse serum, and then infected with AdGFP-apoA-IMilano at a multiplicity of infection of 5. At 24 h postinfection, cells were washed twice with PBS, and fresh serum-free medium was added. Following an additional 24 h of incubation, medium was collected and analyzed by Western blotting as described below.
Western Blot Analysis
Western blot analysis for apoA-I was performed as described previously (24), using a goat anti-human apoA-I antibody (cat# K45252G; Biodesign International, Saco, ME, USA) as primary, and a rabbit anti-goat antibody (cat # sc-2768; Santa-Cruz Biotechnology, Sant Cruz, CA, USA) as secondary. For analysis of culture medium, 100 µL of medium was extensively dialyzed in H20 and then lyophilized. The dry powder was then reconstituted in 20 µL H2O and analyzed by Western blotting. For analysis of plasma samples, 10 µL of plasma was used. For peroxisome proliferator-activated receptor γ (PPARγ) detection, liver specimens were homogenized in radioimmunoprecipitation assay buffer with a minibead beater (Biospec, Bartlesville, OK, USA). Then 10 µg of protein extracts were resolved on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and Western blot analysis was performed with a PPARγ-specific antibody (cat # sc7273; Santa Cruz Biotechnology).
Plasma Lipid Determination
Following a 16-h fasting period, plasma samples were isolated from the experimental mice. Plasma cholesterol, triglyceride and free fatty acid levels were measured as described previously (24).
Fractionation of Plasma Lipoproteins by Density Gradient Ultracentrifugation
For the determination of plasma cholesterol and triglyceride levels in various plasma lipoproteins, 0.5 mL of pools of plasma from five apoA-I−/− and five C57BL/6 mice were fractionated by density gradient ultracentrifugation over a 10-mL KBr density gradient, as described previously (24). The cholesterol and triglyceride content of different density fractions were determined as described above.
Measurement of Total Hepatic Cholesterol and Triglyceride Content
Total hepatic cholesterol (free + esterified) and triglyceride determination was performed as described previously (25). Results are expressed as milligram of cholesterol or triglycerides per gram of tissue ± standard error of the mean.
Body Weight Determination and Body Mass Composition Analysis
At the indicated time points during the course of the experiments, mice in each group were briefly anesthetized with isofluorane and their body weight was determined by a Mettler® precision microscale. At the end of each experiment, at least six mice from each group were sacrificed. Body mass composition was determined as described previously (25,26).
Histological Analysis of Tissue Samples
At the end of each experiment, mice were killed and liver and visceral fat specimens were collected and either stored at −80°C or fixed in 10% formalin. We obtained 4-µm-thick sections from the formalin-fixed paraffin-embedded tissue for further histological analyses. Conventional hematoxylin and eosin (H&E) histological staining was performed to evaluate the microscopic morphology of the liver tissue samples. To assess the tissue structural integrity and architecture, the reticular fiber network was outlined with application of the reticulin stain according to the manufacturer’s instructions (Bioptica, Milan, Italy). Stained sections were visualized with an Olympus BX41 bright-field microscope. Histomorphometry was performed with Adobe Photoshop software. Specifically, five representative hepatic sections from each animal were used for histomorphometric measurements (number and average surface area of lipid-laden hepatocytes). From each section we photographed three different areas using a Nikon Eclipse 80i microscope with a Nikon DXM 1200C digital camera (original magnification 20x). The digital images were imported into Adobe Photoshop CS2 and a grid was added. For each area the number of lipid vacuoles that were intersected by the grid was determined and calculated independently by one pathologist (DJ Papachristou) and one investigator (KE Kypreos) in a blinded fashion. These data were then used to assess the total number of fat vacuoles accumulated within the hepatocytes of each section. Oil red O staining of hepatic sections was performed for the visualization of the unsaturated hydro-phobic lipids according to the manufacturer instructions (Oil Red O Stain kit, #RRSK14; Biostain Ready Reagents, Manchester, UK).
Determination of Daily Food Consumption
Food intake was assessed by determining the difference in food weight during a 7-d period to ensure reliable measurements, as described previously (26,27).
Determination of Postprandial Triglyceride Kinetics following Oral Administration of Olive Oil
Groups of five apoA-I−/− or C57BL/6 mice were tested. Determination of the postprandial triglyceride kinetics was performed as described previously (25). Values were expressed in milligrams per deciliter ± standard error of the mean.
Rate of Intestinal Secretion of Triglyceride-Rich Chylomicrons and Hepatic VLDL Triglyceride Production
To determine the rate of intestinal triglyceride secretion in the plasma of our experimental mice we measured the total rate of plasma triglyceride input (intestinal and hepatic) and then subtracted the rate of hepatic triglyceride secretion, as described previously (28).
Briefly, to determine the total rate of triglyceride input in the plasma of mice, groups of five apoA-I−/− or C57BL/6 mice were tested. Prior to the experiment, mice were fasted overnight for 16 h. The following day animals were gavaged with 0.3 mL of olive oil and placed back to their cages for 1 h (in our experimental setup dietary triglyceride absorption, measured as a postgavage increase in plasma triglyceride levels, becomes apparent at ~ 1 h following oral administration of olive oil). Immediately after, mice were injected with triton-WR1339 at a dose of 500 mg/kg body weight, as a 15% solution (weight/volume) in 0.9% NaCl, as described previously (22,24,29,30). Triton-WR1339 is known to inhibit the catabolism of triglyceride-rich lipoproteins by coating plasma lipoproteins and preventing their interactions with lipoprotein lipase and lipoprotein receptors (31). Then, serum samples were isolated at 30, 60, 90, 120, 150 and 180 min, after injection with triton-WR1339. The increase in plasma triglyceride levels reflects the total (intestinal plus hepatic) triglyceride input because triton-WR1339 prevents plasma triglyceride clearance. As a baseline control, serum samples were isolated approximately 1 min after injection with the detergent. Plasma triglyceride levels at each timepoint were determined as described above and linear graphs of triglyceride concentration versus time were generated. The rate of plasma triglyceride accumulation expressed in milligrams per deciliter per minute was calculated from the slope of the linear graphs. Then, slopes were reported as mean ± standard error of the mean. Total plasma triglyceride supply equaled the sum of intestinal and hepatic triglyceride secretion.
The rate of hepatic VLDL triglyceride secretion was measured as described previously (28). Groups of four to six apoA-I−/− and C57BL/6 mice were used.
Subtraction of the rate of hepatic triglyceride secretion from the total plasma triglyceride input yielded the rate of intestinal secretion of triglyceride-rich chylomicrons following an oral fat load, expressed as mean ± standard error of the mean.
Fasting Glucose Determination, Glucose Tolerance Test and Insulin Sensitivity Test
The glucose tolerance test (GTT) and insulin sensitivity test (IST) were performed as described previously (26).
Real-Time Polymerase Chain Reaction Analysis of Gene Expression
We extracted total RNA from fresh-frozen liver tissue using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions. Reverse transcription and real-time polymerase chain reaction (PCR) for the genes fatty acid synthase (FASN), PPARγ, diacylglycerol O-acyltransferase 1 (DGAT1) and the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was performed in a single tube using the Rotor-gene SYBR Green Real-Time One-Step RT-PCR kit (Qiagen, Valencia, CA, USA), in a Rotor-Gene RG-3000 cycler by CORBETT, Research (Mortlake, Sydney, Australia). Primers for FASN were forward: AGG TGG TGA TAG CCG GTA TGT and reverse: TGG GTA ATC CAT AGA GCC CAG; for PPARγ were forward: GGA AGA CCA CTC GCA TTC CTT and reverse: GTA ATC AGC AAC CAT TGG GTC A, for DGAT1 were forward: GTG CCA TCG TCT GCA AGA TTC and reverse: GCA TCA CCA CAC ACC AAT TCA G and for GAPDH were forward: AGG TCG GTG TGA ACG GAT TTG and reverse: GGG GTC GTT GAT GGC AAC A.
Primers were synthesized by Lab Supplies EPE (Athens, Greece) and their sequences were found in PrimerBank (https://doi.org/pga.mgh.harvard.edu/primerbank). Results were analyzed with the REST 2009 Software. Data were normalized for GAPDH expression and reported as relative (%) induction ± standard error of the mean.
Statistical Analysis
Data are reported as mean ± standard error of the mean. * indicates P < 0.05 and ** indicates P < 0.005. Comparison of data from two groups of mice was performed with the Student t test. Analysis of the metabolic data was performed by ANCOVA. Tests were performed with SPSS software.
Results
ApoA-I−/− Deficiency Enhances Hepatic Triglyceride Accumulation and the Development of NAFLD in Mice
To test the effects of apoA-I on hepatic triglyceride accumulation, groups of 10–12-wk-old male apoA-I−/− and WT C57BL/6 mice were placed on a Western-type diet for 24 wks. H&E staining of liver sections from WT C57BL/6 and apoA-I−/− mice at the beginning of the experiment (wk 0) did not reveal any significant accumulation of hepatic lipids in the form of lipid droplets within the hepatocytes of these mice (data not shown). However, at wk 24, both C57BL/6 and apoA-I−/− mice showed accumulation of hepatic lipids (Figures 1A, B). Statistical analysis following histomorphometric evaluation of the H&E sections revealed that the number of lipid droplets within hepatocytes was significantly elevated in the apoA-I−/− mice than in the C57BL/6 mice (P = 0.0001) (Figures 1A, B, E, F). The observed steatosis was diffuse and of the mixed (macro- and microvesicular) type. In agreement with these data, staining of hepatic sections with reticulin showed that in apoA-I−/− mice fed a Western-type diet for 24 wks, NAFLD was much more progressed and resulted in a significant disruption of the normal architecture of the liver extracellular reticulin fibrils (Figure 1D), compared with C57BL/6 mice (Figure 1C).

Histological analyses of liver sections from apoA-I−/− and C57BL/6 mice. Panels (A) and (B) are representative pictures of H&E-stained hepatic sections from C57BL/6 (A) and apoA-I−/−mice (B) at wk 24 on a Western-type diet. (C, D) Representative pictures of reticulin-stained hepatic sections from C57BL/6 (C) and apoA-I−/−mice (D) at wk 24 of the experiment. All pictures were taken under original magnification, 20x. (E, F) Percentage (E) and average surface area (F) of lipid loaded cells in the livers of apoA-I−/− and C57BL/6 mice fed a Western-type diet for 24 wks.
Biochemical measurement of total hepatic cholesterol (free + esterified) and triglyceride content showed that apoA-I−/− mice fed a Western-type diet for 24 wks had a total hepatic cholesterol content of 4.0 ± 3.9 mg per gram of liver, whereas C57BL/6 mice had 10.5 ± 0.9 mg/g of liver (Figure 2A) (P < 0.05). In contrast, apoA-I−/− mice had a triglyceride content of 295.5 ± 15.8 mg/g of hepatic tissue, whereas C57BL/6 mice had a significantly lower hepatic triglyceride content (155.7 ± 10 mg/g of hepatic tissue, P < 0.005) (Figure 2A).

Biochemical parameters of apoA-I−/− and C57BL/6 mice fed a Western-type diet for a period of 24 wks. (A) Changes in hepatic triglycerides (TG) and cholesterol (Chol); (B) body weight; (C) plasma cholesterol; and (D) plasma triglycerides of mice fed a Western-type diet for 24 wks. (E, F) Cholesterol and triglyceride content, respectively, of the different density lipoprotein fractions following separation of plasma lipoproteins by density gradient ultracentrifugation. The different lipoprotein fractions are indicated. (G) Average food consumption of apoA-I−/− and C57BL/6 mice at wk 0 of the experiment. (H) Fasting plasma glucose levels of the mice at wk 0 and 24 of the experiment.
Body Weight, Body Fat Content and Plasma Lipid Levels of Mice Fed a Western-Type Diet for 24 wks
To test the effects of apoA-I deficiency on body weight gain, the weights of apoA-I−/− and C57BL/6 mice fed a Western-type diet were monitored every 6 wks for a total period of 24 wks.
As expected, C57BL/6 mice showed an increase in their body weight during the course of the experiment (27). At wk 6, C57BL/6 mice had an average body weight of 31.8 ± 1.7 g (23.5% ± 3.9% increase compared with their starting weight of 25.8 ± 1 g at wk 0, P < 0.05). At wk 12 their body weight was already 35.3 ± 0.6 g, and at wk 24 it increased further to 42.8 ± 1.7 g (66.7% ± 5.6% increase compared with their starting weight at wk 0, P < 0.05) (Figure 2B).
ApoA-I−/− mice showed a similar increase in their body weights during the course of the experiment (Figure 2B). Specifically, at wk 6 of the experiment, the apoA-I−/− mouse group had an average body weight of 26.9 ± 1.3 grams (24.8% ± 2.6% increase compared with their starting weight of 21.53 ± 0.741g at wk 0, P < 0.05). At wk 12, their average body weight was 31.9 ± 1.3 g, and at wk 24 their body weight further increased to 41.5 ± 2.2 g (98.1% ± 3.2% increase, compared with their starting weight at wk 0, P < 0.05) (Figure 2B).
To our surprise, apoA-I-deficient mice were found to consume significantly less food than the control C57BL/6 mice. Average daily food consumption, measured daily during a 7-d period, showed that apoA-I−/− mice consumed 3.2 ± 0.1 g/d per mouse while C57BL/6 mice consumed 3.8 ± 0.1 g/d per mouse (P < 0.001).(Figure 2G). At wk 24 apoA-I−/− mice had a mean body weight that was similar to that of C57BL/6 mice (P > 0.05) (Figure 2B), although they had a higher body fat content than the control mice (26 ± 2 g fat for apoA-I−/− mice and 22 ± 2 g fat for C57BL/6 mice, P < 0.05).
C57BL/6 mice on a high-fat diet for 24 wks had significantly elevated fasting cholesterol levels (224.6 ± 21 mg/dL) compared with their starting cholesterol levels at wk 0 (91.9 ± 10 mg/dL) (Figure 2C), while their plasma triglyceride levels remained normal (34.1 ± 3.0 mg/dL at wk 24 versus 8.2 ± 1.1 mg/dL at wk 0) (Figure 2D). ApoA-I−/− mice also showed a modest increase in their plasma cholesterol levels during the course of the experiment. At wk 24 of the experiment plasma cholesterol levels of the apoA-I−/− mice were elevated (76.9 ± 1.4 mg/dL at wk 24 versus 22.4 ± 7.3 mg/dL at wk 0) (Figure 2C), while their plasma triglyceride levels remained within the normal range (15.8 ± 2.8 mg/dL at wk 24 versus 4.0 ± 1.4 mg/dL at wk 0, P < 0.001) (Figure 2D).
Fractionation of plasma lipoproteins by density gradient ultracentrifugation followed by determination of total cholesterol and triglycerides in the different density fractions revealed that deficiency in apoA-I had a profound effect on the plasma lipoproteins of mice fed a Western-type diet for 24 wks. Specifically, in apoA-I−/− mice HDL cholesterol was significantly reduced (Figure 2E). In contrast, in C57BL/6 mice the vast majority of cholesterol was distributed in the HDL density fractions (Figure 2E). Both apoA-I−/− and C57BL/6 mice also had low levels of triglyceride-rich chylomicrons and VLDL (Figure 2F).
Expression of Lipogenic Genes and Indirect Calorimetry Analysis
To determine if the increased hepatic triglyceride levels of the apoA-I−/− mice was due to increased de novo fatty acid and triglyceride synthesis in the liver of these mice, we measured the mRNA expression of key lipogenic enzymes such as FASN, DGAT-1 and PPARγ by realtime PCR. As shown in Figure 3A, we did not observe any significant differences in the expression of these lipogenic markers between apoA-I−/− and C57BL/6 mice. In agreement with these results, Western blot analysis for PPARγ (Figure 3B) followed by scanning densitometry did not reveal any significant differences in the average PPARγ protein abundance between C57BL/6 and apoA-I−/− mice fed a Western-type diet for 24 wks.

Expression levels of lipogenic genes and indirect calorimetry analysis. (A) Relative mRNA expression of FASN, PPARγ and DGAT-1 genes. (B) Protein levels of hepatic PPARγ in the livers of C57BL/6 and apoA-I−/− mice fed a Western-type diet for 24 wks. (C) Relative mRNA expression of FASN, PPARγ and DGAT-1 in untreated apoA-I−/− mice or apoA-I−/− mice treated with the apoA-IMilano-expressing or a control AdGFP adenovirus. Error bars indicate the standard error of the mean. (D) Linear regression plot of EE (J/h) versus mouse body weight (g) for the C57BL/6 and apoA-I−/− mouse groups.
In an effort to determine whether differences in the EE may be a causal factor for the differences in adipose and liver lipid levels observed between the two mouse groups, indirect calorimetry analysis was performed. We chose to characterize our mice at the beginning of the experiment (wk 0) on the basis of the recommendations of Tschop and coworkers (22) that such metabolic phenotyping should ideally be performed early in an experiment when body weight and body fat content are identical between mouse groups. Average EE was 2030 ± 164 J/h for apoA-I−/− mice and 1930 ± 65 J/h for C57BL/6 mice (P > 0.05). ANCOVA analysis indicated that when controlled for body weight, apoA-I−/− and C57BL/6 mouse groups exhibited similar EE (F = 0.368, P = 0.556, n = 8) (Figure 3D).
Increased Hepatic Deposition of Triglycerides in the Livers of ApoA-I−/− Mice Correlates with Reduced Glucose Tolerance and Insulin Sensitivity
To determine if the increased hepatic triglyceride content of the apoA-I−/− mice correlates with disturbances in plasma glucose homeostasis we performed the standard tests, GTT and IST. At wk 0 both mouse groups had similar fasting plasma glucose levels (84.6 ± 9.5 mg/dL for apoA-I−/− mice and 69.5 ± 1.9 mg/dL for C57BL/6 mice, P > 0.05) (Figure 2H) and normal responses to intraperitoneal administration of glucose and insulin (Figures 4A, C). Similarly, at wk 24 of the experiment, the apoA-I−/− mice had fasting glucose levels of 93.8 ± 6.5 mg/dL while the C57BL/6 mice had fasting plasma glucose of 107.6 ± 3.7 mg/dL (P = 0,054) (Figure 2H). However, apoA-I−/− mice showed a significant deterioration in their ability to clear plasma glucose in a GTT (Figure 4B) and to respond to intraperitoneal administration of insulin in an IST, compared with C57BL/6 mice (Figure 4D).

Glucose tolerance test (A, B) and insulin sensitivity test (C, D) of apoA-I−/− and C57BL/6 mice at wk 0 (A, C) and 24 (B, D) of the experiment.
Rate of Hepatic Triglyceride Secretion in ApoA-I−/− and C57BL/6 Mice
One mechanism that could affect hepatic triglyceride content is the secretion of VLDL triglycerides in the circulation. Thus, to determine the effects of apoA-I deficiency on the secretion of hepatic triglycerides, we next performed a classical VLDL triglyceride secretion assay in apoA-I−/− and C57BL/6 mice. We found that apoA-I−/− mice had a reduced rate of hepatic VLDL-triglyceride secretion when compared with C57BL/6 mice. Specifically, secretion rates were 7.43 ± 0.81 mg/dL/min versus 12.52 ± 1.33 mg/dL/min for apoA-I−/− and C57BL/6 mice respectively (P < 0.005) (Figure 5A).

Kinetic parameters of apoA-I−/− and C57BL/6 mice at the initiation of the experiment (wk 0). (A) Rate of hepatic VLDL triglyceride secretion in apoA-I−/− and C57BL/6 mice at wk 0 of the experiment. The bar graph represents the mean ± standard deviation of the individual rates of VLDL triglyceride secretion per mouse group. (B) Total rate of triglyceride input in the plasma of apoA-I−/− and C57BL/6 mice. (C) Rate of intestinal triglyceride secretion of apoA-I−/− and C57BL/6 mice that results from subtracting the rate of hepatic triglyceride secretion, determined in (A), from the total rate of plasma triglyceride supply, determined in (B). (D) Kinetics of postprandial triglyceride clearance in apoA-I−/− and C57BL/6 mice. Values were expressed in milligrams per deciliter ± standard error of the mean.
Rate of Intestinal Triglyceride Secretion in ApoA-I−/− and C57BL/6 Mice
One additional mechanism that could explain the increased sensitivity of apoA-I−/− mice to diet-induced NAFLD could be increased intestinal secretion of triglyceride-rich lipoproteins to the plasma of these mice. To determine the effects of apoA-I deficiency on the rate of intestinal triglyceride secretion we first determined the total rate (intestinal and hepatic) of the plasma triglyceride supply in apoA-I−/− and C57BL/6 mice fed a Western-type diet, following an oral fat load. Groups of five apoA-I−/− and C57BL/6 mice each were fasted for 16 h, and then administered an oral fat load of 300 µL olive oil, as described in Materials and Methods. At 1 h postgavage, mice were injected with triton WR1339, and then plasma triglyceride levels were measured as a function of time and a linear graph of plasma triglycerides versus time was created. As shown in Figure 5B, both mouse strains exhibited comparable rates of plasma triglyceride increase (14.5 ± 1.2 mg/dL/min for C57BL/6 versus 17.11 ± 1.1 mg/dL/min for apoA-I−/− mice, P > 0.05) as determined from the slopes of the graphs.
Then, by subtracting the rate of hepatic triglyceride secretion (determined above) from the total rate of plasma triglyceride supply, the rate of intestinal triglyceride secretion was determined as 9.83 ± 1.51 mg/dL/min for the apoA-I−/− mice and 1.93 ± 1.12 mg/dL/min for the C57BL/6 mice (P < 0.05) (Figure 5C).
Kinetics of Postprandial Triglyceride Clearance in ApoA-I−/− and C57BL/6 Mice
Another potential mechanism that could explain the increased sensitivity of apoA-I−/− mice to diet-induced NAFLD is increased clearance of plasma triglycerides in these mice. To determine the effects of apoA-I deficiency on the kinetics of postprandial triglyceride clearance, groups of five apoA-I−/− and five C57BL/6 mice were fasted for 16 h, then administered an oral fat load of 300 µL of olive oil. Plasma samples were then isolated at 30, 60, 120, 180, 240 and 360 min postgavage. As shown in Figure 5D, plasma triglyceride levels in apoA-I−/− mice started increasing at 120 min (31.4 ± 6.8 mg/dL), reached a modest peak at 240 min (37.4 ± 5.4 mg/dL) and rapidly declined back to baseline by 360 min (28.4 ± 5.15 mg/dL). In contrast, control C57BL/6 mice showed a much slower kinetics of catabolism of postprandial triglycerides. Specifically, plasma triglyceride levels in C57BL/6 started increasing as early as 60 min (61.6 ± 6.6 mg/dL) postgavage. At 120 and 180 min, plasma triglyceride levels were 177.4 ± 50.8 mg/dL and 159.7 ± 18.3 mg/dL, respectively, and then started declining slowly, reaching a concentration of 67.7 ± 19.5 mg/dL at 180 min of the experiment (Figure 5D).
Ectopic Expression of ApoA-I by a Recombinant Attenuated Adenovirus Significantly Reduces Hepatic Triglyceride Content and Improves Hepatic Histology and Architecture
To evaluate the potential of ectopic apoA-I expression in reducing hepatic triglyceride load, as a proof of principle we used adenovirus-mediated gene transfer of apoA-IMilano in male apoA-I−/− mice fed a Western-type diet for a period of 12 wks
We generated a recombinant attenuated adenovirus expressing apoA-IMilano, designated as AdGFP-apoA-IMilano. The capacity of the adenovirus to infect cultured cells and produce apoA-IMilano protein was confirmed by Western blot analysis of media from infected HTB-13 (human astrocytoma) cell cultures (Figure 6A). We next proceeded with the administration of adenoviruses to apoA-I−/− mice that were fed a Western-type diet for 12 wks. Specifically, five mice were treated with the AdGFP-apoA-IMilano adenovirus, five mice were treated with the control AdGFP adenovirus, and five mice remained untreated. We chose to treat mice with a low dose of 8 × 108 pfu of the adenoviruses to avoid infection-induced inflammation and liver damage that could affect the outcome of our experiment. Following infection, mice were switched to chow diet for 10 d, then they were killed and plasma and livers were collected for further analyses.

Effects of ectopic apoA-IMilano expression on hepatic lipid content of mice fed a Western-type diet for 24 wks. (A) Western Blot analysis of media from infected HTB-13 (human astrocytoma) cells, confirming expression of apoA-IMilano. (B) Western blot analysis of plasma of infected apoA-I−/− mice, confirming apoA-IMilano expression. ultracentrifugation fraction (UCF) cholesterol (C) and triglyceride (D) profiles of uninfected, AdGFP-control infected and AdGFP-apoA-IMilano-infected mice. The different lipoprotein fractions are indicated.
The expression of apoA-IMilano in the plasma of the AdGFP-apoA-IMilano infected mice was confirmed by Western blot analysis (Figure 6B). Furthermore, expression of apoA-IMilano resulted in a slight increase in the HDL cholesterol of these mice (Figure 6C) in agreement with previously published results (32), whereas no significant difference was observed in their triglyceride levels compared with their AdGFP-infected and noninfected counterparts (Figure 6D).
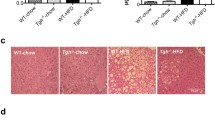
As shown in Figure 7A, H&E staining of livers from uninfected apoA-I−/− mice fed a Western-type diet for 12 wks revealed significant accumulation of lipids in the hepatocytes of these mice. Similarly apoA-I−/− mice infected with 8 × 108 pfu of the control adenovirus AdGFP showed a similar accumulation of hepatic triglycerides and histological signs of inflammation (Figure 7A). In contrast, apoA-I−/− mice infected with 8 × 108 pfu of the AdGFP-apoA-IMilano adenovirus had a profoundly reduced deposition of lipid droplets within the hepatocytes (Figure 7B), reflecting a significant reduction of the hepatic triglycerides, and exhibited a significantly improved liver histology. Histomorphometry followed by statistical analysis showed that the number of lipid droplets and their average surface area were significantly reduced in the livers of apoA-I−/− mice infected with the apoA-IMilano-expressing adenovirus compared with the livers of the uninfected or the control AdGFP adenovirus-infected apoA-I−/− mice (P = 0.0001) (Figures 7G, H). Oil red O staining (Figures 7C, D) further confirmed that apoA-I−/− mice treated with the AdGFP-apoA-IMilano adenovirus (Figure 7D) had significantly reduced hepatic lipid content compared with the apoA-I−/− mice treated with the control adenovirus AdGFP (Figure 7C). Staining with reticulin stain showed that treatment of apoA-I−/− mice with AdGFP-apoA-IMilano also resulted in a significant improvement in the hepatic architecture of these mice (Figures 7E, F). These differences could not be attributed to reduced de novo lipogenesis following infection with the apoA-IMilano-expressing adenovirus because no changes were observed in the hepatic mRNA expression of FASN, DGAT-1 and PPARγ between mouse groups (Figure 3C).

Histological analysis of liver sections from mice infected with an AdGFP-apoA-IMilano-expressing or a control adenovirus. (A, B) Representative pictures of H&E-stained hepatic sections from apoA-I−/− mice that were fed a Western-type diet for 12 wks and then infected with the control AdGFP adenovirus (A) or the AdGFP-apoA-IMilano adenovirus (B). (C, D) are representative pictures of oil red O-stained hepatic sections from apoA-I−/− mice that were fed a Western-type diet for 12 wks, and then infected with the control AdGFP (C) or the AdGFP-apoA-IMilano (D) adenovirus. (E, F) Representative pictures of reticulin-stained hepatic sections from control AdGFP-infected (E) or AdGFP-apoA-IMilano-infected (F) adenoviruses. All pictures were taken under original magnification of 20x. Panels (G) and (H) indicate the percentage of fat cells and the average surface area of the lipid droplets in the livers of mice infected with AdGFP and AdGFP-apoA-IMilano adenoviruses.
Taken together, the findings establish that ectopic expression of apoA-IMilano results in a significant reduction of hepatic triglyceride content and an improvement of hepatic histology and architecture in apoA-I−/− mice fed a Western-type diet for 12 wks.
Discussion
To date, the vast majority of published studies have focused almost exclusively on the effects of apoA-I expression on plasma lipid levels and atherosclerosis, whereas the involvement of apoA-I in hepatic triglyceride deposition and NAFLD has not been investigated thus far. To address this question, in the present study we tested the sensitivity of apoA-I−/− mice toward diet-induced hepatic lipid deposition and NAFLD.
Histological evaluation of liver samples from C57BL/6 and apoA-I−/−mice fed a Western-type diet for 24 wks revealed increased levels of steatosis in the apoA-I−/− mice. Biochemical analyses showed that apoA-I−/− mice had a much higher hepatic triglyceride content and significantly reduced hepatic cholesterol content than C57BL/6 mice, suggesting that the increased steatosis of the apoA-I−/− mice was due to hepatic accumulation of triglycerides. In an attempt to provide a mechanistic interpretation to our histological findings, we measured the mRNA expression levels of key lipogenic enzymes such as FASN, DGAT-1 and PPARγ. This analysis did not reveal significant differences between apoA-I−/− and C57BL/6 mice in the expression of these lipogenic markers. Because PPARγ is the master regulator of lipogenesis we also determined hepatic PPARγ protein expression levels. Again, no significant difference in PPARγ protein levels was obtained between the two mouse groups. The findings suggest that the increased deposition of triglycerides in the livers of apoA-I−/− mice is not due to increased de novo biogenesis of fatty acids and triglycerides in these mice. Similarly, indirect calorimetry analysis did not reveal any significant differences in the EE between the two mouse groups, suggesting that the observed phenotypic differences cannot be explained by differences in the EE of the mice. Taken together, these results suggest that the effects of apoA-I deficiency on hepatic triglyceride deposition may be mediated by its functions as a modulator of lipoprotein metabolism in plasma.
Therefore we performed kinetic analyses, which indicated that deficiency in apoA-I enhances intestinal absorption and secretion of dietary triglycerides in the plasma while it promotes a more efficient catabolism of postprandial triglyceride-rich lipoproteins as determined by the rate of clearance of postprandial triglycerides from the circulation (Figures 5A–D). It is possible that apoA-I, as a structural component of intestinally secreted chylomicrons, modulates their rate of assembly and subsequent secretion in the circulation. Likewise, because excess apoA-I is a known inhibitor of lipoprotein lipase (33), it is possible that deficiency in apoA-I accelerates lipoprotein lipase-mediated lipolysis of chylomicrons, thus promoting their catabolism. These possibilities warrant further investigation in future studies.
Our results indicate that mice deficient in apoA-I had significantly reduced hepatic secretion of triglyceride-rich VLDL compared with the control group. In line with our data, in a recently reported study Gambino and coworkers (34) identified a single-nucleotide polymorphism in microsomal triglyceride transfer protein (MTTP), which is associated with increased NAFLD and reduced apoA-I and HDL cholesterol levels in carriers of this polymorphism. It would be very interesting to determine in the future if this polymorphism reduces MTTP activity.
Surprisingly, in our experimental setup apoA-I−/− mice had higher body fat content than control C57BL/6 mice, though the average body weights of the two groups were similar during the course of the experiment. Histological analysis of visceral adipocytes did not reveal any size difference between the two groups (not shown). Apparently, the slightly reduced food consumption of the apoA-I−/− mice is counteracted by an enhanced intestinal absorption and post-prandial lipid deposition in these mice, eventually leading to higher hepatic lipid content and fat tissue hypertrophy.
As a proof of principle for the potential therapeutic role of apoA-I in NAFLD, ectopic expression of apoA-IMilano resulted in significantly reduced triglyceride content in the livers of these mice and in an improved hepatic histology compared with mice infected with the control AdGFP adenovirus or the noninfected mice. We administered apoA-IMilano because of its improved biological properties compared with WT apoA-I (32,35,36). The inclusion of the AdGFP-infected group was necessary to correct for any nonspecific effects due to the infection process. Interestingly, only 10 d of apoA-IMilano expression were sufficient to improve the histopathological condition of the livers of these mice. We hypothesize that the natural ability of the liver to regenerate could be a factor in the improvement of hepatic histology following ectopic expression of apoA-IMilano.
We have recent data showing that LCAT deficiency also promotes deposition of dietary triglycerides in the liver and results in significant histological and architectural alteration associated with NAFLD in response to feeding a Western-type diet (28). Previous studies in mice (37) and in humans (38,39) indicated that apoA-I is a strong activator of LCAT activity in plasma, whereas deficiency in apoA-I or mutations that affect its function result in a significant reduction in LCAT activity (6,7,10,40,41). In particular, it is well established in the literature that the apoA-I−/− mice that we used in our studies (9) have significantly reduced plasma LCAT activity (20–25% of their WT C57BL/6 counterparts) (10). Thus, it is possible that the enhanced deposition of hepatic triglycerides and the deteriorated hepatic pathology and architecture observed in the apoA-I−/− mice in response to a high-fat diet could be due, at least in part, to the reduced LCAT activity of these mice.
In a recent report (42), it was found that in H-ras12V transgenic mice with steatosis, apoA-I was elevated and accumulated around fatty vacuoles, leading these investigators to propose that apoA-I promotes steatosis. Our data allow for an alternative interpretation of those results, suggesting that the increased apoA-I presence around fat vacuoles may instead reflect a protective mechanism aimed at reducing the already elevated hepatic lipid content of H-ras12V transgenic mice.
Conclusion
Although NAFLD and reduced HDL cholesterol levels coexist in individuals with metabolic syndrome (16,17,43–45), no clear mechanistic link between these two conditions has been established in the literature. Because expression of apoA-I is absolutely essential for the formation of mature functional HDL, our data establish that the HDL metabolic pathway is a central contributor to the deposition of dietary triglycerides to the liver and to the development of NAFLD and other related metabolic dysfunctions associated with metabolic syndrome. Our data further support that the coexistence of reduced HDL levels and NAFLD in an individual with metabolic syndrome may not be a mere coincidence, rather it underlies a strong causative relationship between these two conditions. This observation extends the role of HDL beyond atherosclerosis, to the development of NAFLD, a pathological component found in patients with metabolic syndrome. Importantly, our findings raise the interesting possibility that expression of beneficial forms of apoA-I by gene therapy approaches may find therapeutic applications for the treatment of NAFLD in the future.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Zannis VI, Kypreos KE, Chroni A, Kardassis D, Zanni EE. (2004) Lipoproteins and atherogenesis. In: Molecular Mechanisms of Atherosclerosis. Loscalzo J (ed). Taylor & Francis, New York, NY, pp. 111–74.
Herbert PN, Assmann G, Gotto AM Jr, Fredrickson DS. (1982) Familial lipoprotein deficiency: alpha beta lipoproteinemia, hypobetalipoproteinemia, and Tangier disease. In: The Metabolic Basis of Inherited Disease. Stanbury JB, Wyngaarden JB, Fredrickson DS, Goldstein JL, and Brown MS (eds.). McGraw-Hill, New York, pp. 589–651.
Brunham LR, et al. (2006) Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Invest. 116:1052–62.
Timmins JM, et al. (2005) Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J. Clin. Invest. 115:1333–42
Fielding CJ, Shore VG, Fielding PE. (1972) A protein cofactor of lecithin:cholesterol acyltransferase. Biochem. Biophys. Res. Commun. 46:1493–8.
Utermann G, et al. (1984) Apolipoprotein A-IGiessen (Pro143—Arg). A mutant that is defective in activating lecithin:cholesterol acyltransferase. Eur. J. Biochem. 144:325–31
Rall SC Jr, et al. (1984) Abnormal lecithin:cholesterol acyltransferase activation by a human apolipoprotein A-I variant in which a single lysine residue is deleted. J. Biol. Chem. 259:10063–70.
Sparrow JT, Gotto AM Jr. (1982) Apolipoprotein/lipid interactions: studies with synthetic polypeptides. CRC Crit. Rev. Biochem. 13:87–107.
Williamson R, Lee D, Hagaman J, Maeda N. (1992) Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Proc. Natl. Acad. Sci. U. S. A. 89:7134–8.
Parks JS, Li H, Gebre AK, Smith TL, Maeda N. (1995) Effect of apolipoprotein A-I deficiency on lecithin:cholesterol acyltransferase activation in mouse plasma. J. Lipid Res. 36:349–55.
Gordon JI, et al. (1983) Proteolytic processing of human preproapolipoprotein A-I. A proposed defect in the conversion of pro A-I to A-I in Tangier’s disease. J. Biol. Chem. 258:4037–44.
Schaefer EJ, Kay LL, Zech LA, Brewer HB Jr. (1982) Tangier disease. High density lipoprotein deficiency due to defective metabolism of an abnormal apolipoprotein A-i (ApoA-ITangier). J. Clin. Invest. 70:934–45.
Zannis VI, Lees AM, Lees RS, Breslow JL. (1982) Abnormal apoprotein A-I isoprotein composition in patients with Tangier disease. J. Biol. Chem. 257:4978–86.
Weisgraber KH, et al. (1983) Apolipoprotein A-IMilano. Detection of normal A-I in affected subjects and evidence for a cysteine for arginine substitution in the variant A-I. J. Biol. Chem. 258:2508–13.
Franceschini G, Sirtori CR, Capurso A, Weisgraber KH, Mahley RW. (1980) A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J. Clin. Invest. 66:892–900.
Preiss D, Sattar N. (2008) Non-alcoholic fatty liver disease: an overview of prevalence, diagnosis, pathogenesis and treatment considerations. Clin. Sci. 115:141–50.
Angulo P. (2002) Nonalcoholic fatty liver disease. N. Engl. J. Med. 346:1221–31.
Marchesini G, et al. (2001) Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 50:1844–50.
Chitturi S, Farrell GC. (2001) Etiopathogenesis of nonalcoholic steatohepatitis. Semin. Liver Dis. 21:27–41.
Browning JD, Horton JD. (2004) Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114:147–52.
Tschop MH, et al. (2012) A guide to analysis of mouse energy metabolism. Nat. Methods. 9:57–63.
Kypreos KE, Van Dijk KW, Havekes LM, Zannis VI. (2005) Generation of a recombinant apolipoprotein E variant with improved biological functions: hydrophobic residues (LEU-261, TRP-264, PHE-265, LEU-268, VAL-269) of apoE can account for the apoE-induced hypertriglyceridemia. J. Biol. Chem. 280:6276–84.
Chroni A, et al. (2004) Substitutions of glutamate 110 and 111 in the middle helix 4 of human apolipoprotein A-I (apoA-I) by alanine affect the structure and in vitro functions of apoA-I and induce severe hypertriglyceridemia in apoA-I-deficient mice. Biochemistry. 43:10442–57.
Kypreos KE. (2008) ABCA1 Promotes the de novo biogenesis of apolipoprotein CIII-containing HDL particles in vivo and modulates the severity of apolipoprotein CIII-induced hypertriglyceridemia. Biochemistry. 47:10491–502.
Karavia EA, Papachristou DJ, Kotsikogianni I, Giopanou I, Kypreos KE. (2011) Deficiency in apolipoprotein E has a protective effect on diet-induced nonalcoholic fatty liver disease in mice. FEBS J. 278:3119–29.
Karagiannides I, Abdou R, Tzortzopoulou A, Voshol PJ, Kypreos KE. (2008) Apolipoprotein E predisposes to obesity and related metabolic dysfunctions in mice. FEBS J. 275:4796–809.
Duivenvoorden I, et al. (2005) Apolipoprotein C3 deficiency results in diet-induced obesity and aggravated insulin resistance in mice. Diabetes. 54:664–71.
Karavia EA, Papachristou DJ, Kotsikogianni I, Triantafyllidou IE, Kypreos KE. (2012) Lecithin/cholesterol acyltransferase modulates diet-induced hepatic deposition of triglycerides in mice. J. Nutr. Biochem. 2012, Jul 23 [Epub ahead of print].
Kypreos KE, Teusink B, Van Dijk KW, Havekes LM, Zannis VI. (2001) Analysis of the structure and function relationship of the human apolipoprotein E in vivo, using adenovirus-mediated gene transfer. FASEB J. 15:1598–600.
Kypreos KE, Van Dijk KW, van Der Zee A, Havekes LM, Zannis VI. (2001) Domains of apolipoprotein E contributing to triglyceride and cholesterol homeostasis in vivo. Carboxylterminal region 203–299 promotes hepatic very low density lipoprotein-triglyceride secretion. J. Biol. Chem. 276:19778–86.
Schotz MC, Scanu A, Page IH. (1957) Effect of triton on lipoprotein lipase of rat plasma. Am. J. Physiol. 188:399–402.
Shah PK, et al. (1998) Effects of recombinant apolipoprotein A-I(Milano) on aortic atherosclerosis in apolipoprotein E-deficient mice. Circulation. 97:780–5.
Yamamoto M, et al. (2003) Effects of plasma apolipoproteins on lipoprotein lipase-mediated lipolysis of small and large lipid emulsions. Biochim. Biophys. Acta. 1632:31–9.
Gambino R, Cassader M, Pagano G, Durazzo M, Musso G. (2007) Polymorphism in microsomal triglyceride transfer protein: a link between liver disease and atherogenic postprandial lipid profile in NASH? Hepatology. 45:1097–107.
Nissen SE, et al. (2003) Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 290:2292–300.
Ameli S, et al. (1994) Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation. 90:1935–41.
Plump AS, et al. (1997) ApoA-I knockout mice: characterization of HDL metabolism in homozygotes and identification of a post-RNA mechanism of apoA-I up-regulation in heterozygotes. J. Lipid Res. 38:1033–47.
Soutar AK, et al. (1975) Effect of the human plasma apolipoproteins and phosphatidylcholine acyl donor on the activity of lecithin: cholesterol acyltransferase. Biochemistry. 14:3057–64.
Santos RD, et al. (2008) Characterization of high density lipoprotein particles in familial apolipoprotein A-I deficiency. J. Lipid Res. 49:349–57.
Martin-Campos JM, et al. (2002) ApoA-I(MAL-LORCA) impairs LCAT activation and induces dominant familial hypoalphalipoproteinemia. J. Lipid Res. 43:115–23
Funke H, et al. (1991) A frameshift mutation in the human apolipoprotein A-I gene causes high density lipoprotein deficiency, partial lecithin: cholesterol- acyltransferase deficiency, and corneal opacities. J. Clin. Invest. 87:371–6.
Wang AG, et al. (2011) Steatosis induced by the accumulation of apolipoprotein A-I and elevated ROS levels in H-ras12V transgenic mice contributes to hepatic lesions. Biochem. Biophys. Res. Commun. 409:532–8.
Hamaguchi M, et al. (2005) The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann. Intern. Med. 143:722–8.
Fan JG, et al. (2005) Fatty liver and the metabolic syndrome among Shanghai adults. J. Gastroenterol. Hepatol. 20:1825–32.
Browning JD, et al. (2004) Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 40:1387–95.
Acknowledgments
This work was supported by the European Community’s Seventh Framework Programme [FP7/2007–2013] grant agreements PIRG02-GA-2007-219129 and PIRG02-GA-2009-256402, and the University of Patras Karatheodoris research grants C566 and D155. This work was part of the activities of the intramural research network MetSNet of the University of Patras.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Karavia, E.A., Papachristou, D.J., Liopeta, K. et al. Apolipoprotein A-I Modulates Processes Associated with Diet-Induced Nonalcoholic Fatty Liver Disease in Mice. Mol Med 18, 901–912 (2012). https://doi.org/10.2119/molmed.2012.00113
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2012.00113