
Abstract
Nonalcoholic fatty liver disease (NAFLD) is associated with the development of metabolic syndromes and hepatocellular carcinoma (HCC). Cholesterol accumulation is related to NAFLD, whereas its detailed mechanism is not fully understood. Previously, we reported that glycine N-methyltransferase (GNMT) knockout (Gnmt-/-) mice develop chronic hepatitis and HCC. In this study, we showed that Gnmt-/- mice had hyperlipidemia and steatohepatitis. Single photon emission computed tomography images of mice injected with 131I-labeled 6β-iodocholesterol demonstrated that Gnmt-/- mice had slower hepatic cholesterol uptake and excretion rates than wild-type mice. In addition, genes related to cholesterol uptake (scavenger receptor class B type 1 [SR-B1] and ATP-binding cassette A1 (ABCA1)), intracellular trafficking (Niemann-Pick type C1 protein [NPC1] and Niemann-Pick type C2 protein [NPC2]) and excretion (ATP-binding cassette G1 [ABCG1]) were downregulated in Gnmt-/- mice. Yeast two-hybrid screenings and coimmunoprecipitation assays elucidated that the C conserved region (81–105 amino acids) of NPC2 interacts with the carboxyl-terminal fragment (171–295 amino acids) of GNMT. Confocal microscopy demonstrated that when cells were treated with low-density lipoprotein, NPC2 was released from lysosomes and interacts with GNMT in the cytosol. Overexpression of GNMT doubled the half-lives of both NPC2 isoforms and reduced cholesterol accumulation in cells. Furthermore, GNMT was downregulated in the liver tissues from patients suffering with NAFLD as well as from mice fed a high-fat diet, high-cholesterol diet or methionine/choline-deficient diet. In conclusion, our study demonstrated that GNMT regulates the homeostasis of cholesterol metabolism, and hepatic cholesterol accumulation may result from downregulation of GNMT and instability of its interactive protein NPC2. Novel therapeutics for steatohepatitis and HCC may be developed by using this concept
Similar content being viewed by others


Introduction
Nonalcoholic fatty liver disease (NAFLD) includes illnesses ranging from hepatic steatosis to intermediate lesions, nonalcoholic steatohepatitis (NASH) and cirrhosis (1–3). Epidemiologists estimate that between 20% and 30% of all adults living in the U.S. and other developed countries have some form of NAFLD (4,5). NAFLD is considered a hepatic event in the metabolic syndrome and is associated with hepatocellular carcinoma (HCC) development (6,7). The liver plays a critical role in whole-body lipid metabolism (8,9). Deterioration in lipid uptake, transport, excretion, synthesis and catabolic mechanisms serves as the basis for NAFLD development (10). It is important to note that cholesterol content is increased in human fatty liver tissues (11–15), suggesting that cholesterol metabolism is dysregulated in NAFLD. However, the reason for the cholesterol accumulation is not fully understood.
Niemann-Pick type C2 (NPC2) protein is a small soluble glycoprotein and is mainly expressed in liver, kidney and testis (16,17). NPC2 plays an important role in regulating intracellular cholesterol trafficking and homeostasis through direct binding with free cholesterol (18,19). Deficiency of NPC2 in mice results in cholesterol accumulation in the liver tissues (20). Glycine N-methyltransferase (GNMT) is abundantly expressed in the liver cytosol (21) but is downregulated in human HCC tissues (22,23). According to genotypic analyses, the loss of heterozygosity at the GNMT locus in paired tumor and tumor-adjacent tissues from HCC patients ranges from 36% to 47% (24). Previously, we generated a GNMT knockout (Gnmt-/-) mice and reported on their tendencies toward chronic hepatitis, glycogen storage, hypercholesterolemia, fatty nodules and HCC (25,26). In this study, we showed that Gnmt-/- mice impaired cholesterol metabolism and developed steatohepatitis. We used full-length human GNMT as bait in a yeast two-hybrid screen system and identified NPC2 as a GNMT-interactive protein. Accordingly, this study aimed to investigate the role of GNMT-NPC2 interaction in the regulation of hepatic cholesterol homeostasis. We showed that GNMT was downregulated in the liver tissues from patients with NAFLD as well as from mice fed a high-fat diet, high-cholesterol diet or methionine/choline-deficient (MCD) diet. GNMT loss attenuates NPC2 protein stability and triggers cholesterol accumulation in hepatocytes.
Materials and Methods
Mice
Eleven-week-old and 9-month-old wild-type (WT) and Gnmt-/- mice on a C57BL/6 genetic background were used in this study. To induce fatty liver, for 11 wks, WT mice were fed an MCD diet (Research Diets, A02082002B), high-fat diet (TestDiet, 58V8) or high-cholesterol diet (Research Diets, D12336) for indicated periods. Both genders of ob/ob mice were purchased from the National Laboratory Animal Center in Taiwan. All animal protocols were approved by the Institutional Animal Care and Use Committee of National Yang-Ming University.
Serum and Hepatic Lipids Measurement
Serum cholesterol, low-density lipoprotein (LDL) and alanine amino-transferase were measured using a Kodak Vitros DT60 II (Kodak, Rochester, NY, USA). Nile red (N3013; Sigma, St. Louis, MO, USA) and filipin (F-9765; Sigma) fluorescence staining were used to detect the neutral lipids and free cholesterol, respectively. Tissue or cell cholesterol levels were extracted and quantified by colorimetric commercial kit (BioVision, Mountain View, CA, USA).
Western Blot, Real-Time Polymerase Chain Reaction Analysis and Immunohistochemical Staining
Hepatic proteins (50 µg) were separated by 8–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Primary antibodies for ATP-binding cassette A1 (ABCA1), ATP-binding cassette G1 (ABCG1), scavenger receptor class B type 1 (SR-B1), 3-hydroxy-3-methyl-glutarylcoenzyme A reductase (HMGCR), Niemann-Pick type C1 protein (NPC1), lysosome-associated membrane protein 1 (LAMP1) and cathepsin D were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Steroidogenic acute regulatory protein (StAR) monoclonal antibody was purchased from Abcam (Cambridge, UK). Sterol response element binding protein 2 (SREBP2) monoclonal antibody was purchased from BD Biosciences (Pharmingen, San Diego, CA, USA). Mouse anti-GNMT monoclonal antibody (catalog no. 14-1) and NPC2 (catalog no. 3-6B) monoclonal antibody were purchased from YMAC Biotech (Taipei, Taiwan). Immunoblotting signals were normalized to α-tubulin and quantified by densitometric scanning.
For real-time PCR, primer sequences are shown in Supplementary Table 1 and include ABCA1, SR-B1, SREBP2, NPC1, NPC2, HMGCR and StAR. The mRNA level was normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA level and subsequently expressed as fold changes relative to WT mice.
Immunohistochemical (IHC) staining of the GNMT and NPC2 protein was performed using monoclonal antibodies at a dilution of 1:200 (23). Paraffin-embedded liver sections were incubated with the antibody and detected using a Universal LSAB™2 kit (DakoCytomation, Carpinteria, CA, USA) according to the manufacturer’s instructions.
Single Photon Emission Computed Tomography Imaging and Immunoprecipitation
Detail procedurals are given in the supplementary materials.
LDL and Progesterone Treatments
The progesterone-related inhibition and restoration of lysosomal cholesterol trafficking is a useful experimental means of studying intracellular cholesterol transport (27). In the beginning, Chinese hamster ovary (CHO) cells were incubated in lipoprotein-deficient serum (LPDS) medium for 24 h and transfected with pNPC2-DsRed and pcDNA3-HA or pNPC2-DsRed and pGNMT-GFP for 24 h. Subsequently, these cells were subjected to 50 µg/mL LDL and progesterone (10 µg/mL). After 24 h, LDL and progesterone were washed out and incubated with LPDS medium for the indicated hours. For immunofluorescence staining, cells were fixed and imaged by confocal microscopy. The colocalization was quantified using the Metamorph program (Molecular Devices, Sunnyvale, CA, USA). For Western blot assay, SK-Hep1 cells were infected with 50 MOI (multiplicity of infection) of adenovirus-carrying GNMT cDNA (Ad-GNMT) for 24 h (28); cytosol and lysosome were isolated by a commercial lysosome isolation kit (LYSISO1; Sigma).
Cycloheximide and Pulse-Chase Analysis
For cycloheximide (CHX) treatment, HEK293T cells were transfected with either NPC2-HA or GNMT-Flag plus NPC2-HA for 24 h and then treated with CHX (50 µg/mL) for indicated hours. For pulse-chase analysis, HEK293T cells were transfected with either pNPC2-HA and pCMV-5a or pGNMT-Flag and pNPC2-HA. After 48 h, cells were incubated in methionine/cysteine-free Dulbecco’s modified Eagle’s medium + 10% dialyzed fetal calf serum for 30 min at 37°C. Labeling was performed for 20 min at 37°C with 0.5 mCi 35S-labeled cysteine and methionine. After 20 min of labeling, medium containing 1.5 mg/mL methionine and cysteine was added and incubated for the indicated hours.
Human Samples
Tissues from patients with fatty liver (n = 56; 29 men and 27 women) had been recruited between 2008 and 2010 from the Department of International Medicine, Taipei City Hospital, Ran-Ai Branch. The clinical features of the 56 fatty liver patients are shown in Supplementary Table 2. Diagnosis of fatty liver was according to abdominal ultrasonography by the gastroenterological physician and confirmed by histological examination. Fatty liver patients with other chronic liver disease or alcohol consumption >20 g/d were excluded. The Ethics Committee of the Taipei City Hospital, Ran-Ai Branch, approved the clinical investigations. The normal human liver tissue array (BN03011) was purchased from U.S. Biomax, Inc. (Rockville, MD, USA).
Glycosidase Digestion
Sixty micrograms of liver protein from 0- and 5-wk MCD diet-fed mice were incubated with or without peptide N-glycosidase F (PNGase F) according to the manufacturer’s instruction (New England Biolabs, Ipswich, MA, USA). The reaction products were subjected to Western blot analysis.
Statistical Analyses
Data are mean ± SD. Comparisons of mean values between groups were evaluated by two-tailed Student t test or non-parametric Wilcoxon signed-rank test. P < 0.05 was considered to be statistically significant.
All supplementary materials are available online at www.molmed.org .
Results
Gnmt-/- Mice Develop Hepatic Steatosis and Hyperlipidemia
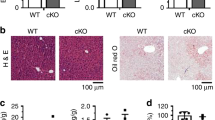
We used a Gnmt-/- mouse model to investigate the role of GNMT in lipid metabolism, specifically comparing hepatic steatosis and serum lipid levels between WT and Gnmt-/- mice. Liver sections taken from Gnmt-/- mice at 11 wks and 9 months of age showed macrovesicular and microvesicular intracytoplasmic lipid droplets and inflammatory infiltration (Figure 1A). Results from filipin fluorescence and Nile red staining indicate accumulations of free cholesterol and neutral lipids in Gnmt-/- mice livers (Figure 1B). In addition, analyses of total lipids in mouse liver tissues and serum samples showed that both male and female Gnmt-/- mice had significantly higher cholesterol and LDL levels than WT mice (Figures 1C, D; P < 0.05). Combined, the data suggest that GNMT loss results in hepatic steatosis and hyperlipidemia.

Gnmt-/- mice developed steatohepatitis and hyperlipidemia. (A) Hematoxylin and eosin staining of liver tissues from WT and Gnmt-/- mice at 11 wks and 9 months of age. Original magnification 200×. Inset: Higher magnification image of microvesicular intracytoplasmic lipid droplet. (B) Filipin and Nile red staining of liver tissues from 11-wk-old WT and Gnmt-/- mice. Arrow, free cholesterol; arrowhead, hydrophobic lipids. Scale bar, 20 µm. Hepatic (C) and serum cholesterol (D), free cholesterol and LDL levels in 11-wk-old WT and Gnmt-/- mice (n = 6 mice per group). *P < 0.05; **P < 0.01. ■, WT; □, Gnmt-/-.
Cholesterol Metabolism Is Impaired in Gnmt-/-Mouse Livers
To delineate the molecular mechanisms responsible for hepatic cholesterol accumulation in Gnmt-/- mice, we analyzed protein and mRNA expression levels for the following genes during different stages of cholesterol metabolism: (a) hepatic cholesterol uptake: ABCA1 and SR-B1; (b) cholesterol synthesis: HMGCR and SREBP2; (c) cholesterol trafficking: NPC1, NPC2 and StAR; and (d) hepatic cholesterol efflux: ABCG1 (Figures 2A, B). Proteins involved in cholesterol uptake (SR-B1 and ABCA1), efflux (ABCG1) and trafficking (NPC1) were significantly downregulated in both genders of Gnmt-/- mice compared with WT mice (P < 0.05). Although NPC2 protein levels were significantly downregulated in Gnmt-/- mice, no significant changes were noted in mRNA levels. There were no differences in HMGCR, SREBP2 and StAR expression between WT and Gnmt-/- mice.
To further evaluate and compare cholesterol uptake and excretion between WT and Gnmt-/- mice, we injected 131I-labeled 6β-iodocholesterol into mouse penis and normalized liver single photon emission computed tomography (SPECT) images to heart SPECT images at different time points (Figure 2C). Compared to WT mice, Gnmt-/- mice expressed much lower radioactivity and slower cholesterol uptake rates in their livers 10–30 min after injection (slope = 0.63 versus slope = 0.35) (Figure 2D). The excretion of 131I-labeled 6β-iodocholesterol from the liver was detected at 3 and 24 h after injection. Gnmt-/- mice excreted cholesterol at a much slower rate than WT mice (slope = −0.3 versus slope = −1.2) (see Figure 2D).

Cholesterol metabolism is impaired in the liver tissues of Gnmt-/- mice. (A) Western blot and (B) real-time PCR analyses of genes involved in hepatic cholesterol uptake (ABCA1 and SR-B1), cholesterol synthesis (HMGCR and SREBP2), cholesterol trafficking (NPC1, NPC2 and StAR) and hepatic cholesterol efflux (ABCG1) in 11-wk-old WT and Gnmt-/- mice (n = 5–6 mice per group). The expression of each gene is normalized to GAPDH. *P < 0.05; **P < 0.01. (C) 131I-labeled 6β-iodocholesterol injection site. (D) Representative SPECT images of WT and Gnmt-/- mice after injection of 131I-labeled 6β-iodocholesterol (n = 2 in each group). B: □, WT; ■, male Gnmt-/-; ▨, female Gnmt-/-; D: −□−, WT; −■−, Gnmt-/-.
Identification of NPC2 as a GNMT Interacting Protein
To identify proteins that interact with GNMT, we used full-length human GNMT as bait in a yeast two-hybrid screen system for a human liver cDNA library. After three rounds of screening, we selected a cDNA clone containing a nucleotide sequence encoding amino acid residues 43–151 of the NPC2 protein for further study. Specific GNMT-NPC2 interaction and colocalization were analyzed by coimmunoprecipitation, confocal microscopic examination and a combination of lysosomal/cytosol fractionation and Western blot analysis. As shown in Figure 3A, both GNMT and NPC2 proteins were capable of binding with their counterpart proteins in coimmunoprecipitation experiments using anti-HA or anti-Flag tag antibodies. Consistent with the above-described overexpression system, endogenous NPC2 was detected in immunoprecipitates prepared from the SK-Hep1 (hepatoma cell line) transfected with pGNMT-Flag (Figure 3B) and WT mouse liver lysates (Figure 3C). To map GNMT-NPC2 interactive domains, we constructed nine plasmids containing different regions of either GNMT or NPC2. The results indicate that the amino-terminal half of GNMT was not capable of pulling down NPC2 (lane 9, Supplementary Figure 1A), but the carboxyl-terminal half (containing amino acid residues 171–295) of GNMT was capable (lanes 10–12, Supplementary Figure 1A). On the other hand, the C region of NPC2 (containing amino acid residues 81–105) was the primary domain for GNMT interaction (lanes 9–10, Supplementary Figure 1B).

GNMT interacts and colocalizes with NPC2 in cytosol. 293T cells were cotransfected with GNMT-Flag and NPC2-HA for 24 h and harvested for coimmunoprecipitation. (A) GNMT was precipitated by anti-Flag beads and immunoblotted with anti-HA to detect NPC2 expression (arrowhead). NPC2 was precipitated by anti-HA beads and immunoblotted with anti-Flag to detect GNMT expression (arrowhead). (B) SK-Hep1 cells were transfected with GNMT-Flag for 24 h and harvested for coimmunoprecipitation. GNMT was precipitated by anti-Flag beads and immunoblotted with anti-NPC2 antibody to detect endogenous NPC2 expression (arrowhead). (C) Liver lysates from WT mice were precipitated with anti-GNMT antibodies and immunoblotted with anti-NPC2 antibodies to detect endogenous NPC2 expression (arrowhead). (D) Cytosol and lysosome fractions were isolated from the liver tissues of WT mice. Western blot analysis was used to detect subcellular NPC2 and GNMT localization. LAMP1 and cathepsin D are indicated as lysosome markers; α-tubulin is indicated as a cytosol marker. (E) Liver cytosol fractions from WT mice were precipitated with anti-GNMT antibodies and immunoblotted with anti-NPC2 antibodies to detect endogenous NPC2 expression (arrowhead). (F) HuH7 cells were transfected with GNMT-Flag and fixed for immunofluorescence assays using antibodies against Flag, NPC2 and Lysotracker to detect colocalization among GNMT, NPC2 and lysosomes. Colocalization percentage is shown in each merge panel. Scale bar, 20 µm. IB, immunoblot.
To prove GNMT colocalization and interaction with NPC2 in the cytosol, we isolated various lysosomal and cytosolic fractions from WT mouse liver for Western blot and coimmunoprecipitation analyses. Approximately 80% of the NPC2 was expressed in lysosomes (indicated by the lysosomal markers LAMP1 and cathepsin D), and 20% was expressed in the cytosol. In contrast, GNMT was expressed in the cytosol (Figure 3D). To confirm GNMT-NPC2 interaction within the cytosol, we immunoprecipitated GNMT from the cytosolic fraction of WT mouse liver and then detected NPC2 expression (Figure 3E). To gain further insight into GNMT-NPC2 interaction, we used immunofluorescence to assess GNMT and NPC2 colocalization. Our data indicate that 81.2% of endogenous NPC2 colocalized with Lysotracker in punctate structures, but part of the diffuse perinuclear compartment of NPC2 did not (Figure 3F, upper panel inset). GNMT-Flag did not colocalize with Lysotracker (Figure 3F, middle panel). It is important to note that a small amount (4.1%) of diffused NPC2 colocalized with GNMT-Flag in the cytosol of HuH7 cells (Figure 3F, lower panel inset), suggesting that GNMT-NPC2 interaction did not take place in the lysosome.
GNMT-NPC2 Influences Intracellular Cholesterol Homeostasis
To monitor dynamic distribution and colocalization between GNMT and NPC2, we used immunofluorescent staining, confocal microscopy and Western blot to detect NPC2-GNMT colocalization in cells treated with LDL and progesterone. Progesterone inhibits cholesteryl ester synthesis and blocks cholesterol trafficking pathways (27). When CHO cells were transfected with NPC2-DsRed and treated with LDL and progesterone for 24 h, NPC2 was accumulated in lysosomes (Figure 4A–4) because of the inhibitory effect of progesterone on cholesteryl ester synthesis in the endoplasmic reticulum (27). NPC2 subsequently appeared in the cytosol, peaking between 6 and 18 h after progesterone was removed from the medium (Figure 4A–5–7). When CHO cells overexpressed GNMT-GFP and NPC2-DsRed, colocalization signals between GNMT and NPC2 became prominent in the cytosol between 6 and 18 h after the removal of progesterone (Figure 4B–5–7). According to cytosol-lysosome isolation results, endogenous NPC2 was predominantly expressed in the lysosomal fractions of SK-Hep1 cells before LDL and progesterone treatment (Figure 4C). Endogenous NPC2 was also increased in cytosolic fractions after the removal of progesterone from the medium at 6 and 18 h. In contrast, GNMT remained in cytosolic fractions throughout the experiment (see Figure 4C).

GNMT coordinates with NPC2 and influences hepatic cholesterol homeostasis after LDL and progesterone treatment. CHO cells were cultured in normal medium (panel 1) and transfected with either NPC2-DsRed (A) or GNMT-GFP and NPC2-DsRed (B) for 24 h. After reseeding, CHO cells were cultured in LPDS (panel 2), LDL (panel 3) or LDL plus progesterone (panel 4) medium for 24 h. After washing out progesterone at 6, 12, 18 or 24 h (panels 5–8), confocal microscopy was used to detect colocalization between NPC2-DsRed (red) and GNMT-GFP (green). NPC2-GNMT colocalization percentages are shown in B. Scale bar, 20 µm. (C) SK-Hep1 cells were infected with Ad-GNMT for 24 h, followed by LDL or LDL plus progesterone treatment for 24 h. After washing out progesterone at 6 or 18 h, cells were harvested for cytosol-lysosome fractionation experiments. LAMP1 and α-tubulin are indicated as lysosome and cytosol markers, respectively. (D) Ad-GNMT and Ad-vector-infected SK-Hep1 cells were cultured in normal and LPDS media, followed by LDL or LDL plus progesterone treatment for 24 h. After washing out progesterone at 6 or 18 h, cells were harvested to determine cholesterol concentrations. *P < 0.05. −■−, Ad-GNMT; −□−, Ad-vector.

GNMT stabilizes NPC2 protein. (A) Analysis of NPC2 degradation followed by cycloheximide (CHX) treatment. The 293T cells were transfected with either NPC2-HA or GNMT-Flag plus NPC2-HA for 24 h before treatment with CHX for the indicated hours. NPC2 degradation was analyzed using Western blot. Single arrowheads indicate mono-glycosylated NPC2; double arrowheads indicate diglycosylated NPC2. (B) Pulse-chase analysis of NPC2. The 293T cells were transfected with either NPC2-HA or GNMT-Flag plus NPC2-HA for 24 h; 35S-Met/Cys incorporation measurements were used to determine NPC2 half-lives. −□−, pNPC2; −■−, pNPC2 + pGNMT.

GNMT is downregulated in fatty liver tissues. (A) IHC staining of human normal and fatty liver tissues using a monoclonal GNMT antibody. Western blot analyses of hepatic GNMT expression from WT mice fed with a high-cholesterol diet (B), high-fat diet (C) or MCD diet (D) are shown. (E) 8-wk-old ob/ob mice (n = 3–5 mice per group).

NPC2 is dysregulated in fatty liver tissues. (A) IHC staining of human normal and fatty liver using a monoclonal NPC2 antibody. Original magnification 200×. Western blot analysis of NPC2 expression from WT mice fed with high-cholesterol diet (B), high-fat diet (C) or MCD diet (E). (D) 8-wk-old ob/ob mice (n = 3–5 per group). (F) For endoglycanase digestion, liver lysates (60 µg) from 0- and 5-wk MCD diet-fed mice were incubated with PNGase F and subjected to Western blot analysis.
To evaluate whether GNMT coordinates with NPC2 in regulating intracellular homeostasis of cholesterol, SK-Hep1 cells were infected with Ad-GNMT for 24 h before treatment with LDL and progesterone. Compared to cells infected with a vector control, the cholesterol levels in GNMT-overexpressed SK-Hep1 cells decreased significantly 6–18 h after the removal of LDL and progesterone (Figure 4D, P < 0.05), indicating that the presence of GNMT prevents cholesterol accumulation in the cells.
GNMT Enhances NPC2 Protein Stability
Because the protein level of NPC2 decreased significantly in Gnmt-/- mice liver tissues while mRNA level remained unchanged (Figures 2A, B), we assumed that GNMT positively affects NPC2 stability at the posttranslational level. In cells transfected with pNPC2-HA, cycloheximide treatment degraded NPC2 isoforms in a time-dependent manner, with half-lives of approximately 1.4–1.5 h (Figure 5A). When cells were cotransfected with pNPC2-HA and pGNMT-Flag, the half-lives of mono-glycosylated and diglycosylated NPC2 isoforms increased to 3.7 and 3.9 h, respectively (see Figure 5A). Pulse-chase experiments were performed to confirm these findings. In the absence of GNMT, the half-lives of NPC2 isoforms were 1.1 and 1.3 h, respectively (Figure 5B). In the presence of GNMT, the half-lives of NPC2 isoforms more than doubled to 3.1 and 3.3 h, respectively (see Figure 5B). Taken together, GNMT overexpression doubled the half-lives of NPC2 isoforms in cells.
GNMT is Downregulated in Fatty Liver Tissues
According to our data, we therefore speculate that hepatic cholesterol accumulation may result from GNMT down-regulation-induced NPC2 protein instability. Previously, we reported that the expression of GNMT is diminished in human HCC using IHC staining (22,23). In this study, we used IHC staining to compare GNMT expression between normal human liver and fatty liver tissues. Compared with normal liver and steatosis-adjacent tissues, fatty liver tissues had much lower GNMT expression (Figure 6A). In 34 of 56 (61%) paired steatosis and steatosis-adjacent tissues, steatosis tissues had lower GNMT expression than steatosis-adjacent tissues (P = 0.04, Supplementary Table 3).
In mice, dietary models (such as MCD, high-cholesterol and high-fat diets) and genetic models (such as ob/ob mice) both mimic NAFLD in humans (10,29). We therefore fed WT mice a high-cholesterol diet, high-fat diet or MCD diet to prove the downregulation of GNMT in fatty liver tissues. As expected, hepatic steatosis and inflammatory infiltration were observed in 3 months of high-cholesterol diet and 20 wks of high-fat diet treatment (Supplementary Figure 2A). Eight-week-old ob/ob mice were obese and expressed hepatic steatosis (Figure 6E and Supplementary Figure 2A). On the other hand, MCD diet induced steatosis during the second week, and steatohepatitis appeared during the fifth week (Supplementary Figure 2B). We found that GNMT was downregulated in mice fed a high-cholesterol diet, high-fat diet or MCD diet but not in ob/ob mice (Figures 6B–E and Supplementary Figures 2C–F). No changes in GNMT expression were noted in mice fed a normal diet (data not shown).
Expression Pattern of NPC2 Is Dysregulated in Fatty Liver Tissues
To evaluate the expression of NPC2 in human fatty liver tissues, we used IHC staining to compare NPC2 expression between normal liver tissues and fatty liver tissues. Compared to normal liver, NPC2 displayed an enlargement of punctuate structure in steatosis tissues (Figure 7A). In 41 of 56 (73%) paired steatosis and steatosis-adjacent tissues, steatosis tissues had enlargement of punctuate structure of NPC2 expression compared with steatosis-adjacent tissues (P = 0.04, Supplementary Table 4).
NPC2 was downregulated in ob/ob mice as well as in mice fed a high-cholesterol diet or high-fat diet (Figures 7B–D). However, in the MCD diet-induced steatosis and steatohepatitis mouse model, a progressive increased glycosylated-NPC2 expression was observed in both genders of WT mice liver (Figure 7E). Because NPC2 is a glycoprotein, we used PNGase F digestion to confirm whether NPC2 protein heterogeneity is due to posttranslational glycosylation modification. After PNGase F treatment, NPC2 was visualized as a single immunoreactive band in the 0- and 5-wk MCD diet-fed WT mice liver (Figure 7F).
Discussion
GNMT is abundantly expressed in normal livers, but is downregulated in HCC tissues (22). GNMT deficiency in mice results in chronic hepatitis, fatty liver and HCC spontaneously (25,26,30). Recently, several putative metabolites identified in Gnmt-/- mice serum closely resemble those of NAFLD patients (31). Varela-Rey et al. (32) also showed that nicotinamide administration normalized hepatic Sadenosylmethionine (SAM) and fatty acid metabolism, as well as prevented fatty liver development in Gnmt-/- mice. However, the molecular mechanisms of GNMT deficiency-induced hepatic cholesterol accumulation remain unclear. Here, we provide a novel concept on GNMT and NPC2 interaction in the regulation of hepatic cholesterol homeostasis. NPC2 is a lysosomal glycoprotein involved in regulating intracellular cholesterol homeostasis through direct binding with free cholesterol (17,18). Besides, GNMT deficiency in mice results in phenotypes that resemble NPC disease, including hepatic free cholesterol accumulation, hepatomegaly, higher serum alanine aminotransferase and hypercholesterolemia (20,33,34). In this study, we also observed that NPC1 was decreased in Gnmt-/- mice. Because NPC1 coordinates with NPC2 and regulates intracellular cholesterol homeostasis (35), we cannot rule out the effect of NPC1 loss in hepatic cholesterol accumulation in Gnmt-/- mice. However, NPC1 was not identified as a GNMT interactive protein in our yeast two-hybrid screen system. Further experiments needed to be conducted to elucidate the mechanism associated with the downregulation of NPC1 in Gnmt-/- mice.
According to Harrison et al. (36), the Nogo-B receptor (NgBR) interacts with and stabilizes the NPC2 protein (36). However, we did not find appreciable differences in NgBR expression levels in Gnmt-/- mice, nor interaction between GNMT and NgBR (data not shown), suggesting that GNMT-NPC2 interaction occurs independently of NgBR. It was reported that the expression signal of vesicular lysosomal NPC2 diminishes as cholesterol is mobilized from lysosomes. However, NPC2 is still present in cells, indicating that NPC2 immunofluorescent signal loss is not due to protein degradation (37). Therefore, NPC2 may travel with cholesterol through the cell and perhaps even be secreted with it. Accordingly, we used confocal and biochemical techniques to show that NPC2 is released into the cytosol and colocalizes with GNMT after LDL and progesterone treatment (Figures 4A–C), and we found that GNMT overexpression decreases cholesterol accumulation in cells after progesterone is washed out (Figure 4D). Because NPC2 cholesterol binding occurs at both acidic and neutral pH levels (19), GNMT may physiologically interact with NPC2 and maintain NPC2 function in the cytosol before NPC2-NgBR interaction. In addition to supporting the idea of GNMT as an NPC2-interacting protein, these findings suggest avenues for enhancing NPC2 protein stability and preventing intracellular cholesterol accumulation.
N-glycosylation is important for the proper targeting and function of NPC2 (38). In this study, we showed that NPC2 was decreased in ob/ob mice and in high-cholesterol diet- and high-fat diet-induced fatty liver tissues (Figures 7B–D); however, MCD diet results in glycosylated NPC2 expression (Figures 7E, F). Because MCD diet impaired mitochondrial β-oxidation and reduced triglyceride and VLDL secretion (10,29), further study is required to address whether glycosylation of NPC2 causes a response to mitochondrial β-oxidation.
The most common form of chronic liver disease is NAFLD, which encompasses a clinicopathologic spectrum of illnesses ranging from isolated hepatic steatosis to NASH—a more aggressive form that can progress to cirrhosis and HCC (39,40). According to the two-hit hypothesis, lipid accumulation and subsequent inflammation and oxidative stress may trigger liver damage and HCC (41). Previously, we reported that downregulation of detoxification and antioxidant-related proteins may trigger oxidative stress and accelerate liver damage and HCC formation in Gnmt-/- mice (42). Several genetic deletion mouse models (including AOX, MAT1A, NRF-1, PTEN, SOD and NEMO) were reported with the spontaneous development of steatosis, steatohepatitis and HCC (43,44). In this study, we showed that Gnmt-/- mice had signs of hepatic steatosis with increased cholesterol deposition in liver tissues, as well as inflammatory infiltration before HCC formation (Figure 1). According to our previous report, 50% male and 100% female Gnmt-/- mice developed HCC spontaneously (26), suggesting that loss of GNMT is associated with gender disparity of liver cancer susceptibility. It is important to note that the percentages of female Gnmt-/- mice with fatty change were higher than in male Gnmt-/- mice at 11 wks and 9 months (Supplementary Table 5). In 18- to 24-month tumor-bearing mice, 100% of female Gnmt-/- mice had fatty change. In contrast, only 63% (10/16) of male Gnmt-/- mice had fatty change. This observation implies the possibility that deterioration of lipid metabolism in liver may affect the development of HCC, especially in female Gnmt-/- mice.
Hypercholesterolemia and cholesterol accumulation in the liver tissues causes development of NAFLD (11–13); however, the reason is still unclear. Our study demonstrated that increased cholesterol content in fatty liver tissues may due to the effects of GNMT deficiency-induced NPC2 protein instability and subsequent cholesterol accumulation. In addition to cholesterol, triglyceride and free fatty acid accumulation also contribute to the development of NAFLD (45). Indeed, accumulation of triglyceride and free fatty acids was also observed in Gnmt-/- mice liver (data not shown), indicating that loss of GNMT widely affects lipids metabolism. Because GNMT is abundantly expressed in normal liver cytosol, we cannot rule out other modulations such as GNMT loss-induced high SAM level (32), or other possibilities on GNMT and other lipid metabolism-related protein interactions.
Conclusion
In the present study, we showed that GNMT was downregulated in mice fed a high-cholesterol diet, high-fat diet or MCD diet but not in ob/ob mice (Figure 6), suggesting that GNMT may not be involved in the leptin mutated model. Thus, this discrepancy may because of the difference of experimental condition. Aberrant methylation of DNA is associated with liver tumorigenesis (46). In either human NAFLD or an MCD diet-fed mouse model, decreases of SAM and global DNA hypomethylation are observed in the liver tissues (46), suggesting that epigenetic modification may regulate the expression of GNMT. Indeed, GNMT promoter hypermethylation and silencing are found in various cancer cell lines, whereas 5-aza-deoxycitidine treatment increases GNMT expression (47). Further study is needed to clarify whether abnormal SAM and global DNA hypomethylation in fatty liver tissues contribute to downregulation of GNMT through promoter hypermethylation.
The GNMT protein has multiple functions. In addition to binding with folate (48), it also interacts with environmental carcinogens, such as benzo(a)pyrene and aflatoxin B1, and inhibits DNA adduct formation (28,49). To our knowledge, this is the first report demonstrating that GNMT plays an important role in regulating cholesterol homeostasis via interaction with NPC2. GNMT deficiency attenuates NPC2 protein stability and triggers cholesterol accumulation in the liver tissues. Because NASH is one of the key factors involved in cirrhosis and HCC development, our data have both physiological and pathological significance. These novel findings may provide important implications regarding the development of diagnostic or therapeutic strategies for fatty liver patients, and possibly for HCC as well.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Ong JP, Younossi ZM. (2007) Epidemiology and natural history of NAFLD and NASH. Clin. Liver Dis. 11:1–16.
Stickel F, Hellerbrand C. (2010) Non-alcoholic fatty liver disease as a risk factor for hepatocellular carcinoma: mechanisms and implications. Gut. 59:1303–7.
Day CP, James OF. (1998) plSteatohepatitis: a tale of two “hits”? Gastroenterology. 114:842–5.
Page JM, Harrison SA. (2009) NASH and HCC. Clin. Liver Dis. 13:631–47.
Bedogni G, et al. (2005) Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology. 42:44–52.
Bugianesi E. (2007) Non-alcoholic steatohepatitis and cancer. Clin. Liver Dis. 11:191–207.
Adams LA, Lindor KD. (2007) Nonalcoholic fatty liver disease. Ann. Epidemiol. 17:863–9.
Maxfield FR, Tabas I. (2005) Role of cholesterol and lipid organization in disease. Nature. 438:612–21.
Repa JJ, Mangelsdorf DJ. (2002) The liver X receptor gene team: potential new players in atherosclerosis. Nat. Med. 8:1243–8.
Anstee QM, Goldin RD. (2006) Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 87:1–16.
Simonen P, et al. (2011) Cholesterol synthesis is increased and absorption decreased in nonalcoholic fatty liver disease independent of obesity. J. Hepatol. 54:153–9.
Nakamuta M, et al. (2009) Impact of cholesterol metabolism and the LXRalpha-SREBP-1c pathway on nonalcoholic fatty liver disease. Int. J. Mol. Med. 23:603–8.
Caballero F, et al. (2009) Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 50:789–96.
Puri P, et al. (2007) A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 46:1081–90.
Reunanen A, Miettinen TA, Nikkila EA. (1969) Quantitative lipid analysis of human liver needle biopsy specimens. Acta. Med. Scand. 186:149–50.
Naureckiene S, et al. (2000) Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 290:2298–301.
Vanier MT, Millat G. (2004) Structure and function of the NPC2 protein. Biochim. Biophys. Acta. 1685:14–21.
Storch J, Xu Z. (2009) Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim. Biophys. Acta. 1791:671–8.
Ko DC, Binkley J, Sidow A, Scott MP. (2003) The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc. Natl. Acad. Sci. U. S. A. 100:2518–25.
Sleat DE, et al. (2004) Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. U. S. A. 101:5886–91.
Kerr SJ. (1972) Competing methyltransferase systems. J. Biol. Chem. 247:4248–52.
Chen YM, et al. (1998) Characterization of glycine-N-methyltransferase-gene expression in human hepatocellular carcinoma. Int. J. Cancer. 75:787–93.
Liu HH, et al. (2003) Characterization of reduced expression of glycine N-methyltransferase in cancerous hepatic tissues using two newly developed monoclonal antibodies. J. Biomed. Sci. 10:87–97.
Tseng TL, et al. (2003) Genotypic and phenotypic characterization of a putative tumor susceptibility gene, GNMT, in liver cancer. Cancer Res. 63:647–54.
Liu SP, et al. (2007) Glycine N-methyltransferase-/- mice develop chronic hepatitis and glycogen storage disease in the liver. Hepatology. 46:1413–25.
Liao YJ, et al. (2009) Characterization of a glycine N-methyltransferase gene knockout mouse model for hepatocellular carcinoma: Implications of the gender disparity in liver cancer susceptibility. Int. J. Cancer. 124:816–26.
Butler JD, et al. (1992) Progesterone blocks cholesterol translocation from lysosomes. J. Biol. Chem. 267:23797–805.
Chen SY, et al. (2004) Glycine N-methyltransferase tumor susceptibility gene in the benzo(a)pyrenedetoxification pathway. Cancer Res. 64:3617–23.
Hebbard L, George J. (2011) Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 8:35–44.
Martinez-Chantar ML, et al. (2008) Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 47:1191–9.
Barr J, et al. (2010) Liquid chromatography-mass spectrometry-based parallel metabolic profiling of human and mouse model serum reveals putative biomarkers associated with the progression of nonalcoholic fatty liver disease. J. Proteome Res. 9:4501–12.
Varela-Rey M, et al. (2010) Fatty liver and fibrosis in glycine N-methyltransferase knockout mice is prevented by nicotinamide. Hepatology. 52:105–14.
Beltroy EP, Richardson JA, Horton JD, Turley SD, Dietschy JM. (2005) Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology. 42:886–93.
Rimkunas VM, Graham MJ, Crooke RM, Liscum L. (2008) In vivo antisense oligonucleotide reduction of NPC1 expression as a novel mouse model for Niemann Pick type C-associated liver disease. Hepatology. 47:1504–12.
Liscum L, Sturley SL. (2004) Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochim. Biophys. Acta. 1685:22–7.
Harrison KD, et al. (2009) Nogo-B receptor stabilizes Niemann-Pick type C2 protein and regulates intracellular cholesterol trafficking. Cell. Metab. 10:208–18.
Zhang M, et al. (2003) Differential trafficking of the Niemann-Pick C1 and 2 proteins highlights distinct roles in late endocytic lipid trafficking. Acta. Paediatr. Suppl. 92:63–73.
Chikh K, Vey S, Simonot C, Vanier MT, Millat G. (2004) Niemann-Pick type C disease: importance of N-glycosylation sites for function and cellular location of the NPC2 protein. Mol. Genet. Metab. 83:220–30.
Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. (2008) Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med. 14:72–81.
Starley BQ, Calcagno CJ, Harrison SA. (2010) Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 51:1820–32.
Day CP. (2002) Pathogenesis of steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 16:663–78.
Liao YJ, et al. (2010) Deficiency of glycine N-methyltransferase results in deterioration of cellular defense to stress in mouse liver. Proteomics Clin. Appl. 4:394–406.
Varela-Rey M, et al. (2009) Non-alcoholic steatohepatitis and animal models: understanding the human disease. Int. J. Biochem. Cell. Biol. 41:969–76.
Fan JG, Qiao L. (2009) Commonly used animal models of non-alcoholic steatohepatitis. Hepatobiliary Pancreat. Dis. Int. 8:233–40.
Browning JD, Horton JD. (2004) Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114:147–52.
Davis CD, Uthus EO. (2004) DNA methylation, cancer susceptibility, and nutrient interactions. Exp. Biol. Med. (Maywood). 229:988–95.
Boumber YA, et al. (2004) Hypermethylation of glycine N-methyltransferase promoter in cancer cell lines and primary AML samples. In: Proceedings of AACR 2004 Annual Meeting; 2004 Mar 27–31; Orlando. Abstr nr 967-a. Available from: http://www.aacrmeetingabstracts.org/cgi/content/abstract/2004/1/967-a
Cook RJ, Wagner C. (1984) Glycine N-methyltransferase is a folate binding protein of rat liver cytosol. Proc. Natl. Acad. Sci. U. S. A. 81:3631–4.
Yen CH, et al. (2009) Glycine N-methyltransferase affects the metabolism of aflatoxin B1 and blocks its carcinogenic effect. Toxicol. Appl. Pharmacol. 235:296–304.
Acknowledgments
This study was supported by a grant from the National Science Council of the Republic of China (NSC99-2628-B-010-010-MY3 and NSC100-2325-B-010-008) and a grant from the Genomic Research Center of the National Yang-Ming University given by the Ministry of Education of the Republic of China (Top University and Center Grant). The authors thank Fu-Hui Wang (Molecular and Genetic Imaging Core), Chi-Hung Lin (National Research Program for Genomic Medicine) and Tung-Wei Chen and Chia-Yen Chen (Institute of Microbiology and Immunology, School of Life Science), all at National Yang-Ming University, for technical support.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
10020_2012_1803412_MOESM1_ESM.pdf
Glycine N-Methyltransferase Deficiency Affects Niemann-Pick Type C2 Protein Stability and Regulates Hepatic Cholesterol Homeostasis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Liao, YJ., Chen, TL., Lee, TS. et al. Glycine N-Methyltransferase Deficiency Affects Niemann-Pick Type C2 Protein Stability and Regulates Hepatic Cholesterol Homeostasis. Mol Med 18, 412–422 (2012). https://doi.org/10.2119/molmed.2011.00258
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00258