
Abstract
Multiple sclerosis (MS) is a devastating autoimmune disorder of the central nervous system (CNS) for which there is no efficacious cure. Thanks to numerous preclinical and clinical studies, drugs able to mitigate the inexorable course of the disease have been made available recently. Still, there is a terrible need for compounds capable of reducing the severity of the autoimmune attack and of blocking progression of the disorder. Also, besides the classic immunosuppressive strategies, it is now appreciated that compounds directly targeting neuronal death can be of relevance to the treatment of MS patients. Acetylation homeostasis is a key regulator of both immune cell activation and neuronal survival. Of note, potent histone deacetylase inhibitors (HDACi) endowed with antiinflammatory and neuroprotective properties have been identified. Efficacy of HDACi in experimental models of MS has been reported consistently. In this review, we provide an appraisal of the literature on HDACi and MS, also discussing the mechanisms by which HDACi can suppress the autoimmune attack to the CNS.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS) is one of the most common inflammatory conditions of the central nervous system (CNS), and is characterized mainly as a neurodegenerative autoimmune disorder affecting the white matter tissue. Typically, the disease leads to the formation of plaques and lesions in various areas of the brain and the spinal cord. MS predominately affects people of temperate latitudes in the Western hemisphere, and its prevalence, depending on the different countries, ranges from 4 to 248 per 100,000 (1). About two million people worldwide suffer from MS, and it remains one of the leading causes of disability in young adults. MS onset typically occurs in subjects between the ages of 20 and 40 years. Women, in accordance with their increased incidence of autoimmune disorders, have a two-fold higher risk of developing MS. The very variable clinical manifestations of MS depend on number, localization and size of lesions.
Although the etiology of MS is yet to be defined, it is thought that the disease occurs in genetically predisposed individuals after an environmental or immunological stimulation triggers the autoimmune response. The central role of autoreactive CD4+ T cells in the pathogenesis of the disease is well established. Nevertheless, it has been demonstrated that the number of blood T cells reacting to specific myelin antigens does not differ between MS patients and healthy subjects. This notion clearly indicates that autoreactive T cells per se do not suffice to induce the disease (2,3).
It has been proposed repeatedly that MS stems from derangement of self tolerance. Recent research on the T-cell subtype Treg (regulatory T cells) has furthered our understanding of impairment of self tolerance significantly (4). Specifically, continuous Treg suppressing activity is required to prevent autoimmunity (5). As for MS, Treg depletion worsens neuropathology in mice with experimental autoimmune encephalomyelitis (EAE), the prototypical, experimental MS model (6), whereas significant protection from EAE is afforded by adoptive transfer of Treg cells (7).
Dendritic cells (DCs) emerged as key immune regulators involved in the pathogenesis of MS (8,9). In healthy subjects, immature DCs maintain peripheral tolerance by promoting differentiation of Treg. Strikingly, the number of immature DCs is decreased in MS patients, whereas that of fully mature (that is, activated) DCs is increased, with ensuing augmented production of proinflammatory cytokines and aberrant differentiation of naïve T lymphocytes toward autoreactive Th1 and Th17 subtypes (10,11). Once activated by DCs in the periphery, autoreactive T lymphocytes infiltrate the CNS, and trigger local inflammation and recruitment of innate immune cells such as microglia and macrophages. These cells contribute to myelin destruction, neuronal loss and lesion formation (12,13).
MS symptoms are a consequence of myelin sheath destruction and neuronal loss, and include vision dysfunction, dizziness, muscle-related symptoms, pain, fatigue and sensory deficit (14). As for clinical manifestations, MS most commonly (85% of cases) develops as a relapsing-remitting form (RRMS) that is characterized by attacks followed by total or partial recovery from symptoms. From a molecular point of view, it is thought that a central role in relapse occurrence is played by DCs. The latter process and present new antigens originating from tissue damage to autoreactive T cells, thereby triggering new waves of autoimmune attack. This DC-dependent broadening of the autoimmune response is called “epitope spreading,” and is proposed as responsible for MS relapses (8,15,16). Secondary progressive MS (SPMS) is the second, most common form of disease occurring in about 30% of cases. SPMS originates as RRMS but then evolves into irreversible, progressive disability owing to neuronal loss and axonal degeneration. The third form of MS is the primary progressive type (PPMS) that occurs in about 10% of patients and is characterized by a progressive disability without any remitting phase or recovery. Finally, the progressive relapsing form of MS (PRMS) is a rare subtype similar to PPMS but also displaying acutization and remissions phases.
Over the last two decades, several therapeutic strategies have been approved and shown to improve MS progression significantly. These drugs belong to the family of disease-modifying agents and include interferon-β (the first drug available for MS treatment since 1993), glatiramer acetate, mitoxantrone and the humanized monoclonal antibody natalizumab (10). Because of their safety profile and well-established effectiveness, interferon-β and glatiramer acetate are first-line drugs. The latter are able to modulate the immune response mainly by inhibiting lymphocyte proliferation, and skewing the T-cell response toward Th2 and Treg (17,18). Natalizumab is a monoclonal antibody directed against α4β1 integrin (VLA-4), reducing leukocyte infiltration into the CNS (19). Mitoxantrone is a chemoterapic agent that inhibits DNA and RNA synthesis, and is able to reduce proliferation of lymphocytes. Because of severe side effects, mitoxantrone and natalizumab are approved as second line therapy for treatment of aggressive MS only. Despite this therapeutic armamentarium, to date no cures are available for MS. Because of the autoimmune pathogenesis of the disease, immunosuppression and immunomodualtion have been the mainstays of experimental therapeutic strategies. Yet, MS treatment is still an unresolved therapeutic burden. Recently, Fingolimod, a sphingosine-1 phosphate agonist capable of sequestering lymphocytes in lymph nodes, received much attention in the MS field because of its therapeutic effects in patients as an oral drug, and is about to be approved in Europe and United States (20).
Effects of HDACi in Experimental Models of MS
At present, a wealth of knowledge indicates that aberrant regulation of acetylation homeostasis within neural cells might be a common pathogenetic mechanism underlying neurodegeneration in acute and chronic neurological diseases. Histone acetyl transferases (HATs) and histone deacetylases (HDACs) finely tune cellular acetylation, targeting not only histones but also numerous proteins with key roles in cell metabolism, signaling and death (21–23). Recently, we witnessed a real explosion of interest in the field of HDAC inhibition, and highly potent inhibitors are being evaluated currently in clinical trials for a disparate array of disorders. In this regard, preclinical evidence suggests that pharmacological inhibition of HDACs is a promising therapeutic strategy for the treatment of neurological disorders including MS (24).
In 2003, Pahan et al. reported the effects of the HDACi sodium phenylbutyrate (SPB) and its metabolite sodium phenylacetate (SPA) on adoptive transfer of EAE in SJL mice (25). The authors demonstrate that SPB (400 mg/kg, intraperitoneally [i.p.], daily from day 0) almost completely abrogates development of adoptive EAE. Similarly, its direct metabolite SPA (10 mg/mL in drinking water from day 0) suppresses neurological impairment in MBP-primed T-cell recipient mice dramatically. The authors demonstrate that both pretreatment of donor EAE mice in vivo and MBP-primed T cells in vitro with SPA are able to reduce adoptive EAE symptoms and neuropathology in recipient mice. Notably, because the relationship among butyrate, immune activation and HDAC was not well established in 2003, in their study, Pahan et al. do not mention the ability of SPA and SPB to inhibit HDACs, they refer only to their use in urea cycle disorders (25). Two years later, however, additional evidence that HDACs contribute to MS pathogenesis was provided. Camelo et al. (26) investigated the effects of HDAC inhibition in C57BL/6 mice immunized with the myelin oligodendrocyte glycoprotein peptide MOG35-55 an experimental model of chronic MS. The study reports that the potent HDACi trichostatin A (TSA) when injected in mice from 4 days to 40 after immunization, although not affecting disease onset, reduces neurological impairment significantly. Accordingly, neuropathology of mice with EAE is ameliorated by TSA treatment, with animals showing reduced spinal cord inflammatory infiltrates, demyelination and axonal loss. Of note, TSA treatment increases the number of motoneurons in the ventral horn, suggesting neuroprotection. Gene array analysis demonstrates that TSA treatment leads to changes in the expression of several genes in the spinal cord of EAE mice. Specifically, Camelo et al. report that TSA increases expression of neuroprotective proteins such as estrogen receptor-a, insulin growth factor-2 (IGF-2), glutamate transporter EAAT2 and glutathione peroxidase, and decreases that of proapoptotic Bax, Bid, caspase-2 and apoptosis-in-ducing factor (26). In keeping with these findings, increased histone acetylation levels in the spinal cord of TSA-treated EAE mice correlated with reduced levels of caspase 3 and 9. The same study shows that caspase downregulation by TSA also occurs in cultures of cortical neurons exposed to oxidative stress. Although the study by Camelo et al. focuses mainly on the effects of TSA on transcriptional regulation of neuroprotective and neurotoxic genes, a snapshot of the effects of the HDACi on the autoimmune response in EAE mice also is reported. Specifically, TSA reduces transcripts for IL-2 receptor, IL-8 receptor, IL-12 and the costimulatory molecule CD28 in the spleen of EAE mice. In cultured splenocytes from these mice, TSA also decreases expression of the chemokine macrophage inflammatory protein-2 (MIP-2). Further, splenocytes from TSA-treated EAE mice show reduced proliferation to MOG35-55 as well as to nonspecific T-cell activators such as concavalin-A and phytohemoagglutinin (26). These latter findings clearly indicate that TSA treatment during EAE severely affects development of the autoimmune response.
Possible Mechanisms Underlying the Therapeutic Effects of HDACi in Experimental Models of MS
As outlined above, experimental evidence showing that HDACi afford protection from EAE is based only on two independent reports (25,26). Nevertheless, this evidence is well in-line with the numerous studies indicating the potent antiinflammatory properties of different HDACi (27). Also, the therapeutic relevance of HDACi to MS is supported further by the pleiotypic effects of this class of drugs on immune cells considered of key relevance to the pathogenesis of the autoimmune attack to the CNS. Specifically, several lines of evidence demonstrate that immunological functions of DCs, T lymphocytes such as Th1, Th17 and Treg as well as glial cells are affected profoundly by HDACi.
In 2007, Nencioni et al. reported that exposure of in vitro-generated human DCs to the HDACi valproic acid or MS-275 impairs cell differentiation and immunogenicity (28). In particular, the two HDACi suppress LPS-triggered DC expression of the lineage marker CD1a as well as the costimulatory molecule CD80. The latter is upregulated in MS patients (29). Likewise, DC production of proinflammatory cytokines such as IL-6, -12 and TNFα is reduced by the two HDACi. Suppression of DC activity is not due to HDACi-dependent nonspecific cytotoxicity because expression of the costimulatory molecule CD86 is not affected by the compounds, whereas that of CD11a increased. Accordingly, no decrease of cell viability is observed. In keeping with these findings, the allostimulatory and migration activity of HDAC-challenged DCs are severely impaired. In this study, the authors do not investigate at the molecular level the mechanisms responsible for DC-suppression by HDACi. Yet, they report experiments showing that drug treatment impair the signaling pathways leading to NFκB and IRF-3 and -8 activation at a level downstream to MyD88 and IRAK-1 (28). Overall, these findings are in agreement with a subsequent paper by Reddy et al. reporting the suppressing effects of HDACi on mouse DCs in vitro and in vivo (30). The authors take advantage of two potent HDACi (SAHA and ITF2357) and demonstrate that both compounds used at nanomolar concentrations suppress IL-6, -12 and TNFα expression by mouse bone marrow-derived DCs (BM-DCs) challenged with different activators. The two HDACi also impaired costimulatory molecule expression by DCs as well as their T-cell stimulatory capacity. Of note, in a murine model of allogenic bone marrow transplantation set up to study graft versus host disease (GVHD), the authors show that preincubation of DCs with HDACi before transplantation severely impairs the severity of the allogenic reaction (30). In an attempt to understand the molecular mechanisms underpinning DC-immunosuppression by HDAC inhibition, the authors found that both drugs induced expression of indoleamine dioxygenase (IDO), the rate-limiting enzyme of tryptophan catabolism, through the kynurenine pathway. Recently, this enzyme received much attention because of its key immunoregulating functions (31). As for DCs, IDO has been reported to shift the immunostimulating activity of this cell type toward a tolerogenic phenotype (32). In keeping with this, impairment of DC function by HDACi vanishes when the drugs are tested on DCs from IDO KO mice or challenged with IDO siRNA. Likewise, GVHD is not ameliorated by HDACi when IDO null DCs are exposed to the drugs before transplantation (30). Collectively, these findings identify a key role of IDO induction in HDACi-dependent immunosuppression of DCs. Further work on HDACs and DC function demonstrates that LAQ824, a pan inhibitor of HDACs, alters a specific subset of immunoregulating genes in DCs and macrophages, selectively impairing DC-dependent Th1 but not Th2 proliferation and migration signals (33). The authors report that LAQ824 also inhibits DC and macrophage chemotaxis without altering that of neutrophils. As a whole, this study is in keeping with that of Jung et al. showing that the HDACi apicidin suppresses DC-dependent Th1 polarization (34). Several reports, therefore, confirm that HDACi impairs DC functions and the ensuing T-cell proliferation remarkably through mechanisms related to modulation of specific gene expression, and not because of an overall deregulation of gene transcription. In light of the emerging relevance of DCs to development of the immune attack on the CNS during MS, the ability of HDACi to suppress DC function certainly may provide the cellular basis for protection by these drugs in mice with EAE. HDACi-dependent DC suppression also might be of relevance to the therapy of MS patients during different phases of the disease.
A large body of information demonstrates that HDAC inhibition prompts T-cell apoptosis, an effect supporting the use of HDACi in lymphomas and leukemias (35). Yet, HDACi, when used at concentrations lower than those inducing T-cell death, significantly reduce T-cell proliferation and function (36). HDACi inhibit T-cell proliferation and the expression of IL-12 and IFN-γ (37), two cytokines fundamental for differentiation of naïve Th0 into Th1 CD4+ cells. HDACi also reduce T-cell expression of IL-2 and IL-2-dependent gene expression (38). As for MS, the ability of different HDACi to impair Th-1 and -17 actions, and to promote Treg immunosuppression is certainly of remarkable preclinical and clinical significance. Work by Bosisio et al. shows that the HDACi TSA and SAHA reduce the amount of IL-12 and -23 produced by human DCs in vitro or mouse DCs in vivo (39). Of note, as mentioned above, these cytokines are key agents necessary for DC-dependent polarization of T cells toward the Th1 and Th17 lineage, respectively, and play key roles in MS immunopathogenesis.
Further underscoring the therapeutic relevance of HDACi to MS therapy, several lines of evidence indicate that HDACs negatively regulate Treg generation and function (40 for review). As mentioned above, the Treg lineage is involved in suppression of effector T cells, induction of anergy toward specific antigens, thereby negatively regulating immune responses during homeostatic conditions or autoimmune disorders and tissue transplantation. Further, reduced number and functional deficiency of Treg has been reported in MS patients (6), whereas Treg transfer affords protection from EAE in rodents (7). The study by Hancock et al. (41) is of remarkable relevance to the therapeutic potential of HDACi in MS. The authors report that TSA assists proliferation of Treg as well as their suppressive function on effector T cells in vivo. Interestingly, these effects are due to inhibition of HDAC9-dependent Foxp3 transcription factor deacetylation, with ensuing Foxp3 hyperacetylation-dependent increased transactivation. Of note, these effects are confirmed in vivo by showing that TSA promotes Treg-dependent permanent cardiac and islet allograft survival (41). Further evidence indicates that functional enhancement also is prompted in freshly isolated and expanded human Treg by several HDACi with different molecular moieties (42). Apparently, promotion of Treg functions by HDACi is due to increased expression of the negative immune regulator CTLA-4 (CD152) (42). Overall, these findings suggest that the neuroprotective effects of HDACi in mice with EAE might be due, at least in part, to increased immune suppressing activities of Treg. Lastly, it is worth noting that HDAC inhibition by TSA also reduces the number of Th17 originating from Treg when stimulated by allogenic APCs (43). Whether this also occurs during the autoimmune response is not known. Yet, this finding further emphasizes the ability of HDACi to bias the acquired immune response toward anergy and tolerance.
Of course, the ability of HDACi to suppress innate immune responses of macrophages and glial cells can well participate to HDACi-dependent protection from EAE. Indeed, profound inhibition of proinflammatory cytokine production by mononuclear cells occurs when HDACi are used in in vitro and in vivo experimental settings (44,45). As for glial cells, both potentiating and suppressing effects by HDACi on neuroimmune activation have been reported (46–51). Also, expression of HDACs within the CNS has been questioned (52). Recent work from our group, however, clearly indicates that HDACs all are expressed within the mouse brain (53). Consistently, we also show that the HDACi SAHA and ITF2357 suppress the glial neuroinflammatory response in vitro and in vivo, and potentiate glia immunosuppression triggered by dexamethasone, a prototypical MS drug. Suppression of immune activation of glia inversely relates to the histone acetylation status, and is accompanied by direct alteration of AP1 and NFκB transcription factor subunit DNA binding in glial extracts (53). These data demonstrate that proinflammatory activation of glial cells, a well-known pathogenetic component during MS development and evolution, is suppressed by molecules inhibiting HDACs because of direct impairments of the transcriptional machinery.
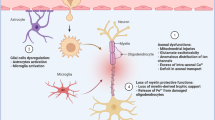
In light of EAE/MS pathogenesis, the ability of HDACi to suppress activation of the innate and acquired immune responses may well explain their ability to reduce the severity of the autoimmune attack on the CNS (Figure 1). Transcriptional therapy with HDACi, however, also is of potential relevance to neurodegeneration. As mentioned above, numerous studies recently have underscored the therapeutic potential of molecules able to increase acetylation of histones and various signaling proteins in preventing neuronal death in different models of neurodegenerative disorders. Molecular mechanisms underlying HDACi-dependent support of neuronal survival also are emerging (23). For instance, Ryu et al. show that acetylation of the transcription factor Sp1 protects neurons from oxidative stress-dependent death, and that HDACi increase Sp1 acetylation and provide protection in cultured neurons deprived of glutathione (54). These findings are in agreement with recent data showing that selective inhibition of HDAC6 protects neurons from oxidative stress, and promotes neurite/axonal growth on nonpermissive substrates such as myelin-associated glycoprotein (55). Interestingly, neuroprotection by HDAC6 inhibition seems to be mediated by hyperacetylation-dependent activation of the peroxiredoxin-1 and -2, whose main function is peroxide reduction (56). Further, in another model of inflammatory demyelination, the mechanism of impaired axonal transport appears dependent on HDAC1 activity (50). HDAC1 and -6 emerge therefore as molecular targets whose manipulation can protect myelin sheaths form the immune attack. Accumulating evidence indicates that neuronal loss occurs in the brain of MS patients and significantly contributes to neurological impairment (57,58). This knowledge widens the therapeutic potential of HDACi to MS therapy. That is, inhibitors of HDACs might exert protection from the autoimmune response within the nervous system via both their immunosuppressing effects and their promotion of neuronal survival (see Figure 1). Although this hypothesis is supported by experimental evidence, it is worth noting that the neurodegenerative component in MS patients is mainly present during disease progression. At present, however, there are no reports of the therapeutic efficacy of HDACi in experimental models of progressive EAE.

Effects of HDACi on the different cells involved in the pathogenesis of MS. The possible different sites of action of HDACi in the CNS and secondary lymphoid organs are shown. The two arrows from “HDACi” to either “Treg” and “Neuron” indicate promotion of activation (Treg) and survival (neuron). Each of the four lines with a bar on the end indicates inhibition.
These findings all support positive effects of HDACi on MS treatment. Yet, based on preclinical data, these drugs also might exert detrimental effects contributing to MS pathogenesis. Specifically, HDACi impair remyelination in cuprizone-treated mice. This feature might severely reduce the therapeutic potential of these compounds in MS in light of the emerging role of drug-induced remyelination in protection from EAE (59).
Conclusion and Future Perspectives
HDACi are pleiotypic agents that, based on preclinical and clinical evidence, show an expanding number of therapeutic indications, among which inflammatory and neurodegenerative disorders are well represented. It is not unexpected, therefore, that MS also is included in the list of disorders whose therapy can benefit from HDACi (24). Yet, preclinical studies carefully focusing on the therapeutic effects of HDACi in various MS models and animals species are limited. This is at odds with worldwide prevalence of MS as well as its morbidity and mortality. Hopefully, additional studies corroborating the therapeutic potential of HDACi in MS are forthcoming. Then, in light of the advanced clinical development of HDACi, clinical trials aimed at understanding the effects of these drugs in MS patients are expected.
Finally, it is worth noting that the various HDACi used in preclinical studies so far are pan HDAC inhibitors having offtarget effects (21–23). On the other hand, single HDACs might have specific roles in the immune response as well as in neuronal death. For instance, HDAC9 suppresses Treg expansion (41), whereas HDAC4 and -6 impair neuronal survival (60) and neurite outgrowth (55). It is hoped, therefore, that isoform-selective HDACi will be available shortly. These compounds could further our understanding of MS pathogenesis and also be of therapeutic relevance to CNS autoimmune disorders.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Rosati G, et al. (2001) Phase I study of a weekly schedule of oxaliplatin, high-dose leucovorin, and infusional fluorouracil in pretreated patients with advanced colorectal cancer. Ann. Oncol. 12:669–74.
Lovett-Racke AE, et al. (1998) Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. A marker of activated/memory T cells. J. Clin. Invest. 101:725–30.
Racke MK, et al. (2010) The mechanism of action of glatiramer acetate treatment in multiple sclerosis. Neurology. 74 Suppl 1:S25–30.
Venken K, et al. (2010) Disturbed regulatory T cell homeostasis in multiple sclerosis. Trends Mol. Med. 16:58–68.
Kim JM, et al. (2007) Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 8:191–7.
Zhang X, et al. (2004) IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int. Immunol. 16:249–56.
Kohm AP, et al. (2002) Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 169:4712–6.
Bailey SL, et al. (2007) CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat. Immunol. 8:172–80.
Miller SD, et al. (2007) Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann. N. Y. Acad. Sci. 1103:179–91.
Lopez-Diego RS and Weiner HL. (2008) Novel therapeutic strategies for multiple sclerosis—a multifaceted adversary. Nat. Rev. Drug. Discov. 7:909–25.
Gandhi R, et al. (2010) Role of the innate immune system in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 221:7–14.
Bar-Or A, et al. (1999) Molecular pathogenesis of multiple sclerosis. J. Neuroimmunol. 100:252–9.
Conlon P, et al. (1999) The immunobiology of multiple sclerosis: an autoimmune disease of the central nervous system. Neurobiol. Dis. 6:149–66.
Lublin FD and Reingold SC. (1996) Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 46:907–11.
Vanderlugt CL and Miller SD. (2002) Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2:85–95.
McMahon EJ, et al. (2005) Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 11:335–9.
Menge T, et al. (2008) Disease-modifying agents for multiple sclerosis: recent advances and future prospects. Drugs. 68:2445–68.
Liblau R. (2009) Glatiramer acetate for the treatment of multiple sclerosis: evidence for a dual antiinflammatory and neuroprotective role. J. Neurol. Sci. 287 Suppl 1:S17–23.
Polman CH and Killestein J. (2006) Anti-myelin antibodies in multiple sclerosis: clinically useful? J. Neurol. Neurosurg. Psychiatry. 77:712.
Fox EJ. (2010) Emerging oral agents for multiple sclerosis. Am. J. Manag. Care. 16:S219–26.
Langley B, et al. (2005) Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr. DrugTargets CNS Neurol. Disord. 4:41–50.
Saha RN and Pahan K. (2006) HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 13:539–50.
Kazantsev AG and Thompson LM. (2008) Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug. Discov. 7:854–68.
Gray SG and Dangond F. (2006) Rationale for the use of histone deacetylase inhibitors as a dual therapeutic modality in multiple sclerosis. Epigenetics. 1:67–75.
Dasgupta S, et al. (2003) Sodium phenylacetate inhibits adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice at multiple steps. J. Immunol. 170:3874–82.
Camelo S, et al. (2005) Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 164:10–21.
Blanchard F and Chipoy C. (2005) Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases? Drug Discov. Today. 10:197–204.
Nencioni A, et al. (2007) Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin. Cancer Res. 13:3933–41.
Genc K, et al. (1997) Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J. Clin. Invest. 99:2664–71.
Reddy P, et al. (2008) Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J. Clin. Invest. 118:2562–73.
Huang L, et al. (2010) Dendritic cells, indoleamine 2,3 dioxygenase and acquired immune privilege. Int. Rev. Immunol. 29:133–55.
Munn DH, et al. (2005) Dendritic cells have the option to express IDO-mediated suppression or not. Blood. 105:2618.
Brogdon JL, et al. (2007) Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood. 109:1123–30.
Jung ID, et al. (2009) Apicidin, the histone deacetylase inhibitor, suppresses Th1 polarization of murine bone marrow-derived dendritic cells. Int. J. Immunopathol Pharmacol. 22:501–15.
Minucci S and Pelicci PG. (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer. 6:38–51.
Moreira JM, et al. (2003) The histone deacetylase inhibitor Trichostatin A modulates CD4+ T cell responses. BMC Cancer. 3:30.
Saemann MD, et al. (2000) Anti-inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB J. 14:2380–2.
Koyama Y, et al. (2000) Histone deacetylase inhibitors suppress IL-2-mediated gene expression prior to induction of apoptosis. Blood. 96:1490–5.
Bosisio D, et al. (2008) Blocking TH17-polarizing cytokines by histone deacetylase inhibitors in vitro and in vivo. J. Leukoc. Biol. 84:1540–8.
Wang L, et al. (2009) Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells. Nat. Rev. Drug. Discov. 8:969–81.
Tao R, et al. (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 13:1299–307.
Akimova T, et al. (2010) Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3+ Tregs. Clin. Immunol. 136:348–63.
Koenen HJ, et al. (2008) Human CD25high-Foxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 112:2340–52.
Leoni F, et al. (2002) The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. U. S. A. 99:2995–3000.
Leoni F, et al. (2005) The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol. Med. 11:1–15.
Suuronen T, et al. (2003) Regulation of microglial inflammatory response by histone deacetylase inhibitors. J. Neurochem. 87:407–16.
Suuronen T, et al. (2005) Anti-inflammatory effect of selective estrogen receptor modulators (SERMs) in microglial cells. Inflamm. Res. 54:194–203.
Suuronen T, et al. (2006) Characterization of the pro-inflammatory signaling induced by protein acetylation in microglia. Neurochem. Int. 49:610–8.
Chen PS, et al. (2007) Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience. 149:203–12.
Kim HJ, et al. (2007) Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J. Pharmacol. Exp. Ther. 321:892–901.
Zhang B, et al. (2008) HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 1226:181–91.
Broide RS, et al. (2007) Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 31:47–58.
Faraco G, et al. (2009) Histone deacetylase (HDAC) inhibitors reduce the glial inflammatory response in vitro and in vivo. Neurobiol. Dis. 36:269–79.
Ryu H, et al. (2003) Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc. Natl. Acad. Sci. U. S. A. 100:4281–6.
Rivieccio MA, et al. (2009) HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. U. S. A. 106:19599–604.
Parmigiani RB, et al. (2008) HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. U. S. A. 105:9633–8.
Centonze D, et al. (2010) The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ. 17:1083–91.
Aktas O, et al. (2010) Neuroprotection, regeneration and immunomodulation: broadening the therapeutic repertoire in multiple sclerosis. Trends Neurosci. 33:140–52.
Taveggia C, et al. (2010) Signals to promote myelin formation and repair. Nat. Rev. Neurol. 6:276–87.
Bolger TA and Yao TP. (2005) Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 25:9544–53.
Acknowledgments
This work was supported by funds from Fondazione Italiana Sclerosi Multipla.
Author information
Authors and Affiliations
Corresponding author
Additional information
GF and LC contributed equally to this work.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Faraco, G., Cavone, L. & Chiarugi, A. The Therapeutic Potential of HDAC Inhibitors in the Treatment of Multiple Sclerosis. Mol Med 17, 442–447 (2011). https://doi.org/10.2119/molmed.2011.00077
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00077