
Abstract
Systemic lupus erythematosus (SLE) is a prototypic autoimmune inflammatory disease characterized by the production of autoantibodies directed against nuclear antigens such as nucleosomes, DNA and histone proteins found within the body’s cells and plasma. Autoantibodies may induce disease by forming immune complexes that lodge in target organs or by crossreacting with targeted antigens and damaging tissue. In addition to autoantibody production, apoptotic defects and impaired removal of apoptotic cells contribute to an overload of autoantigens that initiate an autoimmune response. Besides the well-recognized genetic susceptibility to SLE, environmental and epigenetic factors play a crucial role in disease pathogenesis as evidenced by monozygotic twins typically being discordant for disease. Changes in DNA methylation and histone acetylation alter gene expression and are thought to contribute to the epigenetic deregulation in disease. In SLE, global and gene-specific DNA methylation changes have been demonstrated to occur. Additionally, aberrant histone acetylation is evident in individuals with SLE. Moreover, histone deacetylase inhibitors (HDACi) have been shown to reverse the skewed expression of multiple genes involved in SLE. In this review, we discuss the implications of epigenetic alterations in the development and progression of SLE, and how therapeutics designed to alter histone acetylation status may constitute a promising avenue to target disease.
Similar content being viewed by others

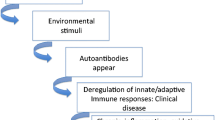
Introduction
There are approximately 80 different known autoimmune diseases affecting at least 5% of the population in the Western world (1). SLE is a prototypic autoimmune disease of unknown etiology in which a genetic predisposition coupled with environmental agents trigger disease. SLE is characterized by the production of antibodies against its own cells and various components within the cell. SLE targets many organs, including the joints, skin, kidneys, heart, lungs, blood vessels and brain. People with SLE have many different symptoms; the most common ones include extreme fatigue, painful or swollen joints (arthritis), unexplained fever, skin rashes and kidney dysfunction. The severity of these problems increases with age and disease duration (2–5). In SLE, the major cause of morbidity and mortality is lupus nephritis (LN), which affects over half of all SLE patients. Traditional treatment for lupus has included the use of cyclophosphamide and corticosteroids, both of which improve renal function but have other deleterious side effects (6). Although most patients respond to this treatment, it is estimated that 35% suffer at least one episode of renal relapse and 5% to 20% develop end-stage renal disease (7). Furthermore, treatment with cyclophosphamide and corticosteroids is non-specific and inhibits normal cellular function.
Extensive research has focused on identifying genes and inflammatory mediators involved in disease. Over the last decade, great strides have been made in understanding the complex pathogenesis underlying LN, and our increased understanding of molecular immunology has led the way for the development of new immunomodulatory therapies (8–10). The use of specific immunomodulators, including monoclonal antibody therapy, has shown promising results, although these agents are still somewhat nonspecific and target groups of cells (6,11). To target lupus more effectively, it is imperative that we uncover the underlying mechanisms that trigger disease activity.
Epigenetics and Lupus
While environmental as well as genetic factors are associated with SLE, there is no specific biomarker to indicate disease pathogenesis, despite decades of extensive work in the understanding of the etiopathogenesis of SLE. Additionally, wide-ranging analysis of both genetics and environmental pathogens has yet to reveal a common mechanism for the initiation of disease. The term “epigenetics” describes heritable alterations of phenotypic traits that are not based on changes in DNA sequence (12). Epigenetics may explain many aspects of the transmission of genetic information ranging from the silencing of the whole chromosome to specific gene activation, as well as nutritional and environmental effects on gene expression patterns. One important epigenetic regulator of gene expression is the modification of histone proteins. Histones are alkaline proteins that package and order the DNA into structural chromatin units. Core histone proteins are classified as H2A, H2B, H3 and H4. Two of each of the core histones assemble to form one octameric nucleosome core around which the DNA wraps. A linker histone (H1) binds the nucleosome, thus locking the DNA into place. The discovery of histone modification has initiated a flurry of investigations into how histone proteins may regulate gene expression (13).
Substantial evidence exists that histone proteins may play a critical role in the regulation of a wide variety of normal cellular functions and pathologies (14). For genes to be transcribed, DNA must uncoil from around the histone proteins. This is accomplished with the assistance of histone acetylases (HAT), which acetylate the lysine residues in core histones leading to transcriptionally active chromatin. Conversely, HDAC removes the acetyl groups from the lysine residues, condensing the chromatin and silencing gene expression. HDAC inhibitors (HDACi) block this action and can result in hyperacetylation of histones, thereby affecting gene expression. Owing to their conserved catalytic domain, HDACs have been actively targeted as a therapeutic target. The reversible nature of these epigenetic maintainer enzymes provides a plausible treatment for skewing the differentiation of a cell type to a different phenotype and may provide viable treatment options to ameliorate disease (15,16). Indeed, the vast majorities of studies with pan HDACi in cell culture systems and in vivo disease models of cancers have resulted in FDA approval of these classes of drugs for the treatment of cancer (17). The present review will discuss the current knowledge of histone proteins’ modification for the treatment of SLE as well as the molecular mechanisms by which HDACi may modulate disease.
HDAC Inhibitors
In the human genome, there have been approximately 2,000 genes that have been identified as transcription factors. To date, only 18 genes have been identified as HDAC and these are subdivided into four classes based on structural and functional similarities (18). Class I HDACs (HDAC1, -2, -3, -6, and -8) are expressed in a wide range of tissues and found exclusively in the nucleus (19,20,21). In contrast, class II HDACs (HDAC4, -5, -7, and -9) have a more limited cellular expression, including immune cells, and are found both in the nucleus and the cytoplasm (21–25). Class III HDACs, known as sirtuins or SIRTs (silent mating type information regulation 2 homolog), are thought to play important roles in the regulation of metabolism and apoptosis via deacetylation of cytosolic and mitochondrial enzymes. Sirt1–Sirt7 are homologues of yeast Sir2 and form a structurally distinct class of nicotinamide adenine dinucleotide (NAD)-dependent enzymes and have been reviewed previously (26,27). HDAC1, -2 and -3 are ubiquitously expressed in the various immune tissues (28) and their expression in human peripheral blood mononuclear cells (PBMCs) is increased by polyclonal activators (phorbol ester and α-CD3 antibodies) and HDACi, but not by lipopolysaccharide (29). Currently, several HDACi, including hydroxyl-butyrate, suberoylanilide hydroxamic acid (SAHA), Trochostain A (TSA), MS275, PXD 101, FR901228, ITF2357 and others designed to inhibit specific classes of HDAC proteins, are in various stages of development (30–32).
Genetics, Epigenetics and Lupus
In regard to SLE, studies between monozygotic and dizygotic twins have provided insight into the role of genetic components and epigenetics on identifying specific susceptible genes. In SLE, several alleles have been identified as risk factors including major histocompatibility complex region, IFN-signaling molecules, transduction signaling genes, components of the complement cascade and others (33–36). Many of the SLE susceptibility genes also are associated with pathways regulating other autoimmune disorders, including rheumatoid arthritis (3,16,37). Direct comparison of identical twins provides an excellent model for identifying epigenetic factors that may play a role in disease pathogenesis. Studies in monozygotic twins have revealed differences with DNA methylation and histone acetylation, and may explain the existence of genome-wide epigenetic differences that result in altered phenotypes (38–40). Javierre et al. recently published data concerning DNA methylation status collected from identical twins discordant for SLE and two other related systemic autoimmune diseases. They found only SLE samples exhibited significant changes in DNA methylation status at both the global and sequence-specific levels in comparison with their healthy discordant twins and compared with unrelated matched normal controls (39). Their studies support the hypothesis for epigenetic alterations playing a critical role in SLE pathogenesis.
Drug-Induced SLE and HDAC
Nearly 30 years ago, a study by Lin-twin et al. showed that decreased acetylation status resulted in a higher incidence of autoantibodies (41). Indeed, drug-induced lupus has been reported by patients taking pharmacologic agents such as procanamide (a pharmaceutical antiar-rhythmic agent used for the medical treatment of cardiac arrhythmias) if the patients showed decreased acetylation (42,43). Conversely, reports by Portanova et al. found that acetylation did not induce a lupus phenotype or alter antibody production (44). In larger prospective studies, Mongey et al. (45) reported patients receiving procanamide showed a persistently higher frequency of developing autoantibodies, although the majority of the patients did not develop drug-related lupus. Furthermore their studies suggested that although acetylation status correlated with IgG antibodies to the H2A–H2B dimer complex, this was not a risk factor for the development of procanamide-related lupus.
Lupus Models For HDAC Inhibition
Studies of human lupus have been aided by the availability of animal models that in many ways resemble the human disease, including autoantibody production and glomerulonephritis. Additionally, congenic strains have allowed the study into the genetics of SLE at both polygenic and at the single-gene levels. There are several different strains of mice that develop various aspects of disease similar to human lupus. Two such strains are the MRL/MPJ-Faslpr (MRL/lpr) and the NZB/NZW F1 mouse strain. The common immunological and clinical manifestations of SLE in both of these strains include hyperactive B and T cells, high titers of several autoantibodies directed against nuclear antigens, a defect in the clearance of immune complexes, and immune complex-mediated glomerulonephritis that eventually leads to renal failure and death (46–49). MRL/MPJ mice with a mutation in the lpr gene alters the Fas receptor and prevents autoreactive T cells from undergoing deletion in the thymus (50). Subsequently these animals develop systemic autoimmunity, massive lymphadenopathy associated with proliferation of aberrant T cells, arthritis and immune complex glomerulonephritis similar to human lupus and die at around 24 weeks from renal disease. The composite genome distribution of autoantibodies produced by these mice are similar in spectrum to those seen in human lupus including anti-double-stranded DNA antibodies and anti-Sm antibodies (51,52). New Zealand Black x New Zealand White (NZB/W) F1 female mice develop autoimmunity characterized by high levels of antinuclear antibodies, hemolytic anemia, proteinuria and progressive immune complex glomerulonephritis and have served as a model for autoimmune disease since the early 1960s (51).
In the MRL/lpr murine model of lupus, CD4+ T cells play an essential role in disease pathogenesis in the kidney and facilitate polyclonal B-cell activation, autoantibody production, immune complex formation and direct infiltration by production of inflammatory mediators in the kidney (53–55). The effector T cells including Th1, Th2 helper cells and follicular helper T (Tfh) cells are largely responsible for B-cell activation, class switching and induction of long-lived plasma and memory cells which are thought to play a major role in the pathogenesis of lupus (56,57). Conversely, regulatory T (Treg) cells are key mediators of peripheral tolerance by suppressing T effector cells and are known to be decreased either in number or function in lupus (58–62). Therefore, it may be argued that aberrant differentiation of multipotential naive CD4+ T cells into distinct lineages including Th1, Th2, Tfh and Treg cells are responsible for the pathogenesis of lupus.
T Cells and HDAC Inhibitors
An epigenetic switch occurs when a stable cell type changes to another stable cell type without changes in DNA sequences (63,64). It has been proposed recently that three categories of signals (epigenerator, epigenetic initiator and epigenetic maintainer) are required in the establishment of a stable cell (65). Epigenerator signals generated by self-antigen or environmental factors may induce naïve CD4+ T cells to differentiate into specific effector cell types. These cellular processes depend on modulation of lineage specific master transcription factors (epigenetic initiators) for T effector and Treg cells (i.e. Foxp3 for Tr, for example, BCL6 for Tfh, T bet for Th1 and GATA-3 for Th2) (62). Subsequently, the epigenetic maintainers including chromatin modifying enzymes (that is, histone acetyltransferase, deacetylases, methyltransferase, demethylase, DNA methylation enzymes and microRNAs) establish a chromatin landscape by changing DNA methylation, histone modification, histone variants and nucleosome positioning, resulting in terminally differentiated cell types (65). Among different epigenetic maintainers, histone acetylation is one of the most widespread modifiers of histones and serves as a key regulator of chromatin structure and gene transcription, and thus may play a key factor in the differentiation of CD4+ T cells (66).
Using two HDACi (TSA and SAHA), we have demonstrated that these agents decrease kidney disease in both the MRL/lpr and NZB/NZW F1 mice lupus mouse models (67–69). In MRL/lpr mice, we demonstrated that TSA and SAHA decreased inflammatory cytokine (IL-12, IFN-γ, IL-6 and IL-10) production in isolated splenocytes while increasing accumulation of acetylated histones H3 and H4 in total cellular chromatin. Although we did not find decreased production of IL-1β in splenocytes in our studies treated with HDACi, Susick et al. showed TSA decreased IL-1β-induced metabolic dysfunction in pancreatic cells (70). Inhibition of IL-1β could have therapeutic effects in SLE as deletion of IL-1β in lupus mice has been shown to ameliorate disease (71). We demonstrated that HDACi decreased inflammatory mediator production in renal mesangial cells both in vitro and in vivo with reduced disease as demonstrated by decreased inflammatory mediator production, proteinuria and glomerulonephritis. The decrease in disease was in part by upregulation of Foxp3+CD4+CD25+ (Treg) cells (69). This may be important in the pathogenesis of SLE, as human studies have demonstrated the numbers of Treg cells in SLE patients was related inversely to both the serum level of anti-dsDNA and disease activity (72,73).
Several studies have supported a role for HDAC inhibition in inflammatory diseases (74,75) and have focused on Treg cells as they have been reported to be crucial in the mediation of inflammatory bowel disease (IBD) (24). It was reported that HDAC inhibition in vivo increased Foxp3 gene expression, as well as the production and suppressive function of Treg cells. Of particular interest is the role of HDAC9 on Treg cell function. Studies have suggested that HDAC9 proved particularly important in regulating Foxp3-dependent suppression (24,76). Optimal Treg cell function required acetylation of several lysines in the forkhead domain of Foxp3, and Foxp3 acetylation enhanced binding of Foxp3 to the IL-2 promoter and suppressed endogenous IL-2 production (24). Moreover, recent studies have demonstrated that overexpression of HDAC9 in Treg cells decreased their suppressive capacity (72). We currently are examining the role of HDAC9 in lupus. We found the expression levels of HDAC9 were decreased in MRL/lpr mice compared with MRL/MJP mice (unpublished data). This increase in HDAC9 in Treg cells may explain the decrease in suppressive activity of Treg cells in MRL/lpr mice consistent with previously published studies (77).
It is established that the effector T-cell lineage shows great plasticity. While Th17 cells play a crucial role in the immunologic response to microbial infection, they also are induced in SLE (78,79). When TSA was added to cultures of human T cells in vitro, profound negative effects on the emergence of IL-17-producing cells from Treg cells were observed, implying that Treg cell differentiation into IL-17-producing cells depends on histone/protein deacetylase activity (80). In this regard, the addition of HDACi to lupus mice might skew the T-cell population to that of a Treg-cell population. Both SAHA and TSA inhibited the production of IL-12 and IL-23. Furthermore, the HDACi selectively blocked the production of Th1-attracting chemokines CXCL9, CXCL10 and CXCL11 (80). Therefore, it can be concluded the reduction of Th1- and Th17-inducing cytokines as well as Th1-attracting chemokines may represent relevant mechanisms through which HDACi exert their immunomodulatory effects.
In our investigations to understand further the interplay between various histone modifications including acetylation, methylation and lupus disease, we performed differential expression histone modification analysis in splenocytes from the MRL/lpr mouse model of lupus. Using stable isotope labeling in combination with mass spectrometry, we found global site-specific hypermethylation (except H3 K4 methylation) and hypoacetylation in histone H3 and H4 in MRL/lpr mice compared with control MRL/MPJ mice (66). Moreover, histone modifications such as H3 K18 methylation, H4 K31 methylation and H4 K31 acetylation were expressed differentially in MRL/lpr mice compared with controls. We also observed in vivo administration of the HDACi TSA corrected the site-specific hypoacetylation states on H3 and H4 in MRL/lpr mice with improvement of disease phenotype showing that histone modifications can therefore be reset with histone deacetylase inhibition in vivo (66).
Within the Th subpopulation, there also is an imbalance between Th1 and Th2 cytokine production in SLE (81–83). Activated Th2 cells overproduce IL-6, IL-12, IL-10 and tumor necrosis-α, whereas Th1 cells under produce IL-2, IFN-γ and transforming growth factor-β (TGF-β) (84α86). Moreover, activated Th cells overexpress cell-surface CD40 ligand (CD154) that persists over an extended time interval compared with activated normal T cells (87). The ongoing interaction between CD154 on T cells and CD40 on B cells, in the presence of high levels of IL-6 and IL-10 and in the absence of effective regulatory CD8+ T cytotoxic/suppressor cells, persistently activates B cells, resulting in high-output immunoglobulin production. This altered homeostasis ultimately leads to polyclonal hypergammaglobulinemia. Recent studies have shown the HDACi ITF2357 inhibits IL-6 receptor signaling (88) and inflammation in colitis (89) and traumatic brain injury (90) by decreasing inflammatory mediator production triggered by glial cell activation (91). IL-6 receptor inhibition currently is being pursued in clinical trials as a means to ameliorate SLE (92), possibly through the inhibition of plasmacytoid dendritic cells (93). Reddy and coworkers recently have shown the HDACi SAHA and ITF2357 directly decrease dendritic cell function through increased expression of the dendritic cell inhibitor indoleamine 2,3-dioxygenase (94). This would suggest that, in addition to dampening receptor responsiveness to inflammatory molecules, HDACi may upregulate immuno-suppressive genes.
Several other reports have shown TSA to exert a dampening effect on T cell activation with stimulation. Nambiar and coworkers showed TSA reduced the expression of the T cell receptor zeta chain gene. Additionally, TSA upregulated the expression of its homologous gene Fc (epsilon) receptor I γ chain (95). These effects were associated with a decreased intracytoplasmic-free calcium response and altered tyrosine phosphorylation patterns of cytosolic proteins. Along with these effects, the expression of the IL-2 gene was suppressed. The effects of TSA on human T cells were predominantly immunosuppressive and reminiscent of the signaling aberrations that have been described in patients with systemic lupus erythematosus (95). Further evidence supporting a role for HDAC inhibition of T-cell activation was demonstrated in the graft versus host disease (GVHD) model. This model has been used to elucidate complications that may arise from allergenic bone marrow transplantation which is widely viewed as a T cellmediated response. After bone marrow transplantation, T cells present in the graft, either as contaminants or intentionally introduced into the host, attack the tissues of the transplant recipient after perceiving host tissues as antigenically foreign. Recent strategies to prevent GVHD have included the ablation of T cells by an anti-CD3 conditioning regime for the recipient. However, anti-CD3 administration often has been associated with cytokine storm syndrome (96). Li et al. reported that SAHA reduced the cytokine storm associated with anti-CD3 preconditioning (97). Furthermore, they demonstrated in NZB/W lupus mice that anti-CD3 and SAHA preconditioning not only prevented GVHD disease when given cells from nonautoimmune donor cells, but also reversed glomerulonephritis in the NZB/W diseased mice. Their studies showed an effective dosing regimen of SAHA of δ 200 µg/g. In our studies, we have shown SAHA administration of 50 µg/g decreased lupus disease progression in NZB/W animals while adverse effects with prolonged administration of SAHA greater than 50 µg/g were noted (67). Taken together, these results indicate that conditioning with anti-CD3 and HDACi represent a radiation-free treatment regime for the prevention of GVHD which may have clinical applications for treating cancers and rheumatic disorders. T-cell differentiation and activation may be modulated by HDAC inhibitors as they may act at various stages of naïve T-cell development (Figure 1).

Reported mechanisms by which histone deacetylase inhibitors modulate naïve T-cell differentiation to that of a T-regulatory cell phenotype.
B Cells and HDAC Inhibition
B cells have been believed to play a critical role in SLE, including the loss of B-cell tolerance and hyperresponsiveness of B cells to immune stimulation in lupus-prone strains. Autoreactive B cells have been recognized for over 50 years as a central component of disease. Using B-cell deficient progeny of SLE-prone MRL/lpr mice bred to mice with the Jh (B-cell deficient) mutation, Schmoclik et al. reported no signs of autoimmune kidney destruction or vasculitis. In contrast, wild type littermates with intact B cells had severe nephritis and vasculitis as well as autoantibodies (49,98). Similar findings have been reported in NZB/W F1 animals (99). Although in our studies we found little effect of TSA on splenic B-cell profiles in MRL/lpr mice, Lu et al. reported TSA strongly inhibited germline and post-switch immunoglobulin transcripts in primary splenic B cells from MRL/lpr mice (100). Furthermore, they observed by chromatin immunoprecipitation assays that HDAC1 was recruited to the 3′-IgH enhancer. Overexpression of HDAC1 increased the activity of IgH enhancers, especially 3′-IgH enhancers. These findings implicate HDAC in IgH gene transcription via activation of 3′-IgH enhancers.
Autoantibodies are a hallmark of lupus nephritis; studies by van Bavel et al. reported that elevated autoantibody production was observed in acetylated apoptotic cells compared with non-acetylated apoptotic cells (101). This would suggest acetylation may increase autoantibody production in lupus; we found in our studies that lupus mice treated with TSA did not show a decreased production of autoantibodies (69). It may be somewhat surprising that autoantibody production was not decreased with HDAC inhibition. It has been established that, although B cells are required for lupus nephritis (49,102), autoantibody generation is not necessarily indicative of active disease (103).
One significant advance made in identifying lupus susceptibility genes in the last decade is the discovery of the role of IFN-α in lupus. In 2003, Baechler et al. reported an increase in IFN-α inducible genes in the peripheral blood from patients with severe lupus (104). This has been coined the “interferon signature” and several later studies have verified these results and further clarified the role of IFN-α-inducible genes in SLE. Several IFN-regulated genes have been shown to be induced in patients with SLE (21,35,105). Plasmacytoid dendritic cells represent a major cell type that is responsible for IFN-α production. TSA has been shown to block IFN-α production by plasmacytoid dendritic cells cultured in vitro in the presence of serum obtained from SLE patients (93).
Transcription Factors and HDAC Inhibition
Class III group of HDACs, collectively called sirtuins (SIRTs), have been implicated in chromatin silencing, cell survival and aging (106). Unlike the class I and II HDACs, the deacetylase activity of SIRTs is dependent on the NAD-to-NADH ratio of the cell and is considered a sensor of redox signaling (107). During oxidative stress, cells alter their NAD-to-NADH ratio to increase the NAD, which elevates the deacetylase activity of SIRTs. Conversely, NADH and nicotinamide act as inhibitors of this family. In a recent study, Hu et al. showed SIRT-1 expression was decreased with SIRT-1 siRNA injection in MRL/lpr lupus mice (108). Additionally, transcription of P300, PCAF and HDAC7 were decreased as compared with noninjected animals. Moreover, serum anti-dsDNA antibody levels, renal IgG deposition and renal pathological scores, particularly tubulointerstitial scores, decreased significantly, showing SIRT1 overexpression and decreased histone acetylation is implicated in lupus in MRL/lpr mice. In human lupus, reports also suggest a role for SIRT1 in disease (109). Lupus patients with active disease showed a global histone H3/H4 hypoacetylation in the CD4+ T cells compared with controls (109). The degree of histone H3 acetylation correlated negatively with increased disease activity in lupus patients as measured by the systemic lupus erythematosus disease activity index.
Although several studies have shown inhibition of HDAC being beneficial for autoimmune diseases, reports by Kuwatsuka et al. suggest decreased levels of antibodies against HDAC3 actually may be associated with the severity of autoimmune disease (110). They found in systemic sclerosis (SSc) patients, the production of autoantibodies against HDACs, IgG and IgM anti-HDAC3 were significantly lower than in normal controls. Furthermore, they reported the decreased levels of IgG anti-HDAC3 antibodies were specific to SSc, while IgG anti-HDAC3 antibody levels in SLE were similar and slightly increased relative to normal controls. These data suggest an increase in anti-HDAC3 antibodies may be protective in SSc, while anti-HDAC3 autoantibodies may be pathogenic in SLE patients.
Future Directions for SLE
The genetic studies in human and murine lupus suggest that polymorphisms in different genes with different combinations contribute to the lupus pathogenesis (111). Several gene specific knockout studies in mice have shown different ways to elicit the lupus phenotype, perhaps by the creation of specific pathways and networks of aberrant gene expression (112). Positive or negative feedback loops involving transcriptional regulatory proteins are the fundamental principle of epigenetic inheritance of gene expression in a cell-specific context (113). The change in the epigenetic landscape generated by a specific class or subclass of HDACi may dampen the positive inflammatory feedback loop initiated to induce production of cytokines and chemokines, which favors infiltration of inflammatory cells to target organs and facilitates autoantibody deposition in kidney tissue in lupus. To this end, recent studies showed the HDACi ITF2357 selectively inhibited function of the mutated JAK2 (V617F) gene associated with polycythemia vera (114). Furthermore, ITF2357 has shown efficacy in treating juvenile idiopathic arthritis (115).
Several studies using microarray analyses have demonstrated that 1% to 3% of genes are either upregulated or downregulated with lupus pathogenesis (116,117). Depending upon the cell type and context, HDACi also affect 1% to 5% genes (66). It may be argued that HDACi represent a better approach than targeted therapy because it may affect aberrant expression in specific genes to alter the global inflammatory landscape. It has been reported that pan-HDACi may elicit side effects similar to lupus symptoms such as fatigue, low platelet count, thromboembolism and others. It is highly likely that the development of iso-form-specific HDACi to target specific abnormal genes may dampen the hyper-responsive lupus phenotype leading to better efficacy with fewer side effects. Indeed, selective therapeutics targeting HDAC may reverse the expression of several genes in the spleen and kidney that show altered expression in lupus. Delineating the mechanisms by which specific HDACi treat lupus remains in its early stages of investigation.
Epigenerator signals, including environmental cues or niches (such as viral, bacterial infection, self antigen, etc.) induce intracellular signals that activate or decrease epigenetic initiators (transcription factors, non-coding RNAs, etc.). Once induced, an epigenetic maintainer (histone acetylases and deacety-lases) coordinates the overall cell response. Once the stable chromatin landscape change occurs as a result of aberrant expression of chromatin-modifying enzymes in a particular cell type, it will differentiate the cell terminally without the requirement of other additional initiating signals. The persistence of chromatin modifying enzymes requires the cooperation between both the initiator and maintainer signals for terminal differentiation of effector cells. This may be where epigenetic modulators such as HDACi have the efficacy to alter the cell phenotype.
HDACi have been considered for the treatment of systemic lupus erythematosus based upon in vitro cell culture and in vivo studies in murine models of lupus, but clinical efficacy remains to be established. Although there is a great need for new lupus therapies, progress has been slow. Recently, Belimumab was the first therapeutic approved by the FDA for the treatment of SLE although its efficacy in treating patients with active renal disease was not studied. Furthermore, African Americans with SLE did not significantly respond to treatment. Lupus is a complex disease without a large enough patient base to attract the attention of most pharmaceutical companies. This, coupled with the expenses associated with lupus trials and the recent unsuccessful study outcome for other lupus therapeutics, illustrates the need for new strategies for targeting lupus. Modifying specific histone proteins using HDACi may prove to be a successful therapeutic approach.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Shapira Y, Agmon-Levin N, Shoenfeld Y. (2010) Defining and analyzing geoepidemiology and human autoimmunity. J.Autoimmun. 34:J168–77.
Hahn BH. (2003) Systemic lupus erythematosus and accelerated atherosclerosis. N. Engl. J. Med. 349:2379–80.
Centola M, et al. (2007) Gene expression profiles of systemic lupus erythematosus and rheumatoid arthritis. Expert Rev. Clin. Immunol. 3:797–806.
Bruce IN. (2005) Atherogenesis and autoimmune disease: the model of lupus. Lupus. 14:687–690.
Valesini G, Conti F. (2011) The persistent challenge of lupus nephritis. Clin. Rev. Allergy Immunol. 40:135–7.
Daikh DI, Wofsy D. (1998) On the horizon: clinical trials of new immunosuppressive strategies for autoimmune diseases. Transplant. Proc. 30:4027–8.
Borchers AT, Naguwa SM, Shoenfeld Y, Gershwin ME. (2010) The geoepidemiology of systemic lupus erythematosus. Autoimmun. Rev. 9:A277–87.
Aran AA, Putterman C. (2008) Treatment of lupus nephritis: facing the era of immunotherapy. Panminerva Med. 50:235–45.
Giles I, Putterman C. (2008) Autoantibodies and other biomarkers — pathological consequences (1). Lupus. 17:241–246.
La Cava A. Targeting B cells with biologics in systemic lupus erythematosus. Expert Opin. Biol. Ther. 10:1555–61.
Burmester GR, Horneff G, Emmrich F. (1992) Management of early inflammatory arthritis. Intervention with immunomodulatory agents: monoclonal antibody therapy. Baillieres Clin. Rheumatol. 6:415–34.
Choi JK. (2010) Systems biology and epigenetic gene regulation. IET Syst. Biol. 4:289.
Selvi BR, Krishna DV, Ostwal YB, Kundu TK. (2010) Small molecule modulators of histone acetylation and methylation: a disease perspective. Biochim. Biophys. Acta. 1799:810–28.
Perl A. (2010) Pathogenic mechanisms in systemic lupus erythematosus. Autoimmunity. 43:1–6.
Willyard C. (2010) The saving switch. Nat. Med. 16:18–21.
Karberg S. (2009) Switching on epigenetic therapy. Cell. 139:1029–31.
Marks PA. (2007) Discovery and development of SAHA as an anticancer agent. Oncogene. 26:1351–6.
McGee-Lawrence ME, Westendorf JJ. (2011) Histone deacetylases in skeletal development and bone mass maintenance. Gene. 474:1–11.
Montgomery RL, et al. (2007) Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes. Dev. 21:1790–802.
Montgomery RL, et al. (2008) Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Invest. 118:3588–97.
Haberland M, Mokalled MH, Montgomery RL, Olson EN. (2009) Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes. Dev. 23:1625–30.
Zhang K, et al. (2002) Histone acetylation and deacetylation: identification of acetylation and methylation sites of HeLa histone H4 by mass spectrometry. Mol. Cell. Proteomics. 1:500–8.
Chang S, et al. (2006) Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 126:321–34.
de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology. 138:583–94.
Zhou X, Marks PA, Rifkind RA, Richon VM. (2001) Cloning and characterization of a histone deacetylase, HDAC9. Proc. Natl. Acad. Sci. U. S. A. 98:10572–7.
Glauben R, Siegmund B. (2009) Molecular basis of histone deacetylase inhibitors as new drugs for the treatment of inflammatory diseases and cancer. Methods Mol. Biol. 512:365–76.
Blanchard F, Chipoy C. (2005) Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases? Drug Discov. Today. 10:197–204.
Guardiola AR, Yao TP. (2002) Molecular cloning and characterization of a novel histone deacetylase HDAC10. J. Biol. Chem. 277:3350–6.
Dangond F, Gullans SR. (1998) Differential expression of human histone deacetylase mRNAs in response to immune cell apoptosis induction by trichostatin A and butyrate. Biochem. Biophys. Res. Commun. 247:833–7.
Chavan AV, Somani RR. (2010) HDAC inhibitors — new generation of target specific treatment. Mini Rev. Med. Chem. 10:1263–76.
Beumer JH, Tawbi H. (2010) Role of histone deacetylases and their inhibitors in cancer biology and treatment. Curr. Clin. Pharmacol 5:196–208.
Rambaldi A, et al. (2010) A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br. J. Haematol. 150:446–55.
Balomenos D, Rumold R, Theofilopoulos AN. (1998) Interferon-gamma is required for lupuslike disease and lymphoaccumulation in MRL-lpr mice. J. Clin. Invest. 101:364–71.
Jevnikar AM, Grusby MJ, Glimcher LH. (1994) Prevention of nephritis in major histocompatibility complex class II-deficient MRL-lpr mice. J. Exp. Med. 179:1137–43.
Baechler EC, Gregersen PK, Behrens TW. (2004) The emerging role of interferon in human systemic lupus erythematosus. Curr. Opin. Immunol. 16:801–7.
Lemay S, Mao C, Singh AK. (1996) Cytokine gene expression in the MRL/lpr model of lupus nephritis. Kidney Int. 50:85–93.
Hemminki K, Li X, Sundquist J, Sundquist K. (2009) Familial associations of rheumatoid arthritis with autoimmune diseases and related conditions. Arthritis Rheum. 60:661–8.
Fong KY, Boey ML. (1998) The genetics of systemic lupus erythematosus. Ann. Acad. Med. Singapore. 27:42–6.
Javierre BM, et al. (2010) Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 20:170–9.
Ballestar E, Esteller M, Richardson BC. (2006) The epigenetic face of systemic lupus erythematosus. J. Immunol. 176:7143–7.
Litwin A, Adams LE, Zimmer H, Hess EV. (1981) Immunologic effects of hydralazine in hypertensive patients. Arthritis Rheum. 24:1074–8.
Uetrecht JP, Freeman RW, Woosley RL. (1981) The implications of procainamide metabolism to its induction of lupus. Arthritis Rheum. 24:994–1003.
Marsden JR, Mason GG, Coburn PR, Rawlins MD, Shuster S. (1985) Drug acetylation and expression of lupus erythematosus. Eur. J. Clin. Pharmacol. 28:387–90.
Portanova JP, Small CJ, Kohler PF. (1985) No demonstrable relationship between IgM and IgG antinuclear antibody levels and acetylator phenotype in patients with systemic lupus erythematosus. Arthritis Rheum. 28:995–8.
Mongey AB, Sim E, Risch A, Hess E. (1999) Acetylation status is associated with serological changes but not clinically significant disease in patients receiving procainamide. J. Rheumatol. 26:1721–6.
Andrews BS, et al. (1978) Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J. Exp. Med. 148:1198–215.
Bouzahzah F, Jung S, Craft J. (2003) CD4+ T cells from lupus-prone mice avoid antigen-specific tolerance induction in vivo. J. Immunol. 170:741–8.
Chan O, Madaio MP, Shlomchik MJ. (1997) The roles of B cells in MRL/lpr murine lupus. Ann. N. Y. Acad. Sci. 815:75–87.
Chan OT, Madaio MP, Shlomchik MJ. (1999) B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J. Immun. 163:3592–6.
Cohen PL, Eisenberg RA. (1991) Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–69.
Theofilopoulos AN, Dixon FJ. (1985) Murine models of systemic lupus erythematosus. Adv. Immunol. 37:269–390.
Theofilopoulos AN, Kono DH. (1998) Mechanisms and genetics of autoimmunity. Ann. N. Y. Acad. Sci. 841:225–35.
Fairhurst AM, et al. (2009) Type I interferons produced by resident renal cells may promote endorgan disease in autoantibody-mediated glomerulonephritis. J. Immunol. 183:6831–8.
Zeller GC, Hirahashi J, Schwarting A, Sharpe AH, Kelley VR. (2006) Inducible co-stimulator null MRL-Faslpr mice: uncoupling of autoantibodies and T cell responses in lupus. J. Am. Soc. Nephrol. 17:122–30.
Odegard JM, et al. (2009) ICOS controls effector function but not trafficking receptor expression of kidney-infiltrating effector T cells in murine lupus. J. Immunol. 182:4076–84.
Vinuesa CG, Sanz I, Cook MC. (2009) Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 9:845–57.
DiPlacido LD, Craft J. (2010) Emerging from the shadows: follicular helper T cells in autoimmunity. Arthritis Rheum. 62:6–8.
Simpson N, et al. (2010) Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 62:234–44.
Yan XJ, et al. (2009) Indole-3-carbinol improves survival in lupus-prone mice by inducing tandem B- and T-cell differentiation blockades. Clin. Immunol. 131:481–94.
Valencia X, Yarboro C, Illei G, Lipsky PE. (2007) Deficient CD4+CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J. Immunol. 178:2579–88.
Miyara M, et al. (2005) Global natural regulatory T cell depletion in active systemic lupus erythematosus. J. Immunol. 175:8392–400.
Gerli R, et al. (2009) Identification of regulatory T cells in systemic lupus erythematosus. Autoimmun. Rev. 8:426–30.
Iliopoulos D, Oikonomou P, Messinis I, Tsezou A. (2009) Correlation of promoter hypermethylation in hTERT, DAPK and MGMT genes with cervical oncogenesis progression. Oncol. Rep. 22:199–204.
Iliopoulos D, Satra M, Drakaki A, Poultsides GA, Tsezou A. (2009) Epigenetic regulation of hTERT promoter in hepatocellular carcinomas. Int. J. Oncol. 34:391–9.
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. (2009) An operational definition of epigenetics. Genes. Dev. 23:781–3.
Garcia BA, Busby SA, Shabanowitz J, Hunt DF, Mishra N. (2005) Resetting the epigenetic histone code in the MRL-lpr/lpr mouse model of lupus by histone deacetylase inhibition. J. Proteome Res. 4:2032–42.
Reilly CM, et al. (2004) Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J. Immunol. 173:4171–8.
Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS. (2003) Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Invest. 111:539–52.
Reilly CM, et al. (2008) The histone deacetylase inhibitor trichostatin A upregulates regulatory T cells and modulates autoimmunity in NZB/W F1 mice. J. Autoimmun. 31:123–30.
Susick L, Senanayake T, Veluthakal R, Woster PM, Kowluru A. (2009) A novel histone deacetylase inhibitor prevents IL-1beta induced metabolic dysfunction in pancreatic beta-cells. J. Cell Mol. Med. 13:1877–85.
Voronov E, et al. (2006) IL-1 beta-deficient mice are resistant to induction of experimental SLE. Eur. Cytokine Netw. 17:109–16.
Horwitz DA. (2008) Regulatory T cells in systemic lupus erythematosus: past, present and future. Arthritis Res. Ther. 10:227.
Bonelli M, von Dalwigk K, Savitskaya A, Smolen JS, Scheinecker C. (2008) Foxp3 expression in CD4+ T cells of patients with systemic lupus erythematosus (SLE): a comparative phenotypic analysis. Ann. Rheum. Dis. 67:664–71.
Shuttleworth SJ, Bailey SG, Townsend PA. (2010) Histone deacetylase inhibitors: new promise in the treatment of immune and inflammatory diseases. Curr. Drug Targets. 11:1430–8.
Glauben R, et al. (2006) Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J. Immunol. 176:5015–22.
Tao R, et al. (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 13:1299–307.
Tao R, et al. (2007) Histone deacetylase inhibitors and transplantation. Curr. Opin. Immunol. 19:589–95.
Mok MY, Wu HJ, Lo Y, Lau CS. The relation of interleukin 17 (IL-17) and IL-23 to Th1/Th2 cytokines and disease activity in systemic lupus erythematosus. J. Rheumatol. 37:2046–52.
Ma J, et al. (2010) The imbalance between regulatory and IL-17-secreting CD4+ T cells in lupus patients. Clin. Rheumatol. 29:1251–8.
Bosisio D, et al. (2008) Blocking TH17-polarizing cytokines by histone deacetylase inhibitors in vitro and in vivo. J. Leukoc. Biol. 84:1540–8.
Lit LC, et al. (2007) Elevated gene expression of Th1/Th2 associated transcription factors is correlated with disease activity in patients with systemic lupus erythematosus. J. Rheumatol. 34:89–96.
Sun D, et al. (2004) Regulation of immune function by calorie restriction and cyclophosphamide treatment in lupus-prone NZB/NZW F1 mice. Cell. Immunol. 228:54–65.
Crispin JC, et al. (2003) Immunoregulatory defects in patients with systemic lupus erythematosus in clinical remission. Lupus. 12:386–93.
Enghard P, Langnickel D, Riemekasten G. (2006) T cell cytokine imbalance towards production of IFN-gamma and IL-10 in NZB/W F1 lupus-prone mice is associated with autoantibody levels and nephritis. Scand. J. Rheumatol. 35:209–16.
Kikawada E, Lenda DM, Kelley VR. (2003) IL-12 deficiency in MRL-Fas(lpr) mice delays nephritis and intrarenal IFN-gamma expression, and diminishes systemic pathology. J. Immunol. 170:3915–25.
Raziuddin S, al-Janadi MA, al-Wabel AA. (1991) Soluble interleukin 2 receptor levels in serum and its relationship to T cell abnormality and clinical manifestations of the disease in patients with systemic lupus erythematosus. J. Rheumatol. 18:831–6.
Toubi E, Shoenfeld Y. (2004) The role of CD40-CD154 interactions in autoimmunity and the benefit of disrupting this pathway. Autoimmunity. 37:457–64.
Todoerti K, et al. (2010) Pleiotropic anti-myeloma activity of ITF2357: inhibition of interleukin-6 receptor signaling and repression of miR-19a and miR-19b. Haematologica. 95:260–9.
Glauben R, et al. (2008) Histone deacetylases: novel targets for prevention of colitis-associated cancer in mice. Gut. 57:613–22.
Shein NA, et al. (2009) Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 23:4266–75.
Faraco G, et al. (2009) Histone deacetylase (HDAC) inhibitors reduce the glial inflammatory response in vitro and in vivo. Neurobiol. Dis. 36:269–79.
Illei GG, et al. (2010) Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 62:542–52.
Salvi V, et al. (2010) Trichostatin A blocks type I interferon production by activated plasmacytoid dendritic cells. Immunobiology. 215:756–61.
Reddy P, et al. (2008) Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J. Clin. Invest. 118:2562–73.
Nambiar MP, Warke VG, Fisher CU, Tsokos GC. (2002) Effect of trichostatin A on human T cells resembles signaling abnormalities in T cells of patients with systemic lupus erythematosus: a new mechanism for TCR zeta chain deficiency and abnormal signaling. J. Cell. Biochem. 85:459–69.
Kircher B, Latzer K, Gastl G, Nachbaur D. (2003) Comparative in vitro study of the immunomodulatory activity of humanized and chimeric anti-CD25 monoclonal antibodies. Clin. Exp. Immunol. 134:426–30.
Li N, et al. (2008) HDAC inhibitor reduces cytokine storm and facilitates induction of chimerism that reverses lupus in anti-CD3 conditioning regimen. Proc. Natl. Acad. Sci. U. S. A. 105:4796–801.
Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. (1994) The role of B cells in lpr/lpr-induced autoimmunity. J. Exp. Med. 180:1295–306.
Ye YL, Suen JL, Chen YY, Chiang BL. (1998) Phenotypic and functional analysis of activated B cells of autoimmune NZB x NZW F1 mice. Scand. J. Immunol. 47:122–6.
Lu ZP, Ju ZL, Shi GY, Zhang JW, Sun J. (2005) Histone deacetylase inhibitor Trichostatin A reduces anti-DNA autoantibody production and represses IgH gene transcription. Biochem. Biophys. Res. Commun. 330:204–9.
van Bavel CC, Dieker JW, Tamboer WP, van der Vlag J, Berden JH. (2010) Lupus-derived monoclonal autoantibodies against apoptotic chromatin recognize acetylated conformational epitopes. Mol. Immunol. 48:248–56.
Ahuja A, et al. (2007) Depletion of B cells in murine lupus: efficacy and resistance. J. Immunol. 179:3351–61.
Haas KM, et al. (2010) Protective and pathogenic roles for B cells during systemic autoimmunity in NZB/W F1 mice. J. Immunol. 184:4789–800.
Baechler EC, et al. (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. U. S. A. 100:2610–5.
Bleesing JJ, et al. (2001) Immunophenotypic profiles in families with autoimmune lymphopro-liferative syndrome. Blood. 98:2466–73.
North BJ, Verdin E. (2004) Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 5:224.
Imai S, Armstrong CM, Kaeberlein M, Guarente L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 403:795–800.
Hu N, Long H, Zhao M, Yin H, Lu Q. (2009) Aberrant expression pattern of histone acetylation modifiers and mitigation of lupus by SIRT1-siRNA in MRL/lpr mice. Scand. J. Rheumatol. 38:464–71.
Hu N, et al. (2008) Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 35:804–10.
Kuwatsuka Y, et al. (2009) Decreased levels of autoantibody against histone deacetylase 3 in patients with systemic sclerosis. Autoimmunity. 42:120–5.
Crow MK. (2008) Collaboration, genetic associations, and lupus erythematosus. N. Engl. J. Med. 358:956–61.
Liu K, Mohan C. (2006) What do mouse models teach us about human SLE? Clin. Immunol. 119:123–30.
Sedighi M, Sengupta AM. (2007) Epigenetic chromatin silencing: bistability and front propagation. Phys. Biol. 4:246–55.
Guerini V, et al. (2008) The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F). Leukemia. 22:740–7.
Vojinovic J, Damjanov N. (2011) HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol. Med. 17:397–403.
Bauer JW, Bilgic H, Baechler EC. (2009) Geneexpression profiling in rheumatic disease: tools and therapeutic potential. Nat. Rev. Rheumatol. 5:257–65.
van Baarsen LG, Bos CL, van der Pouw Kraan TC, Verweij CL. (2009) Transcription profiling of rheumatic diseases. Arthritis Res. Ther. 11:207.
Thanou-Stavraki A, Sawalha AH. (2011) An update on belimumab for the treatment of lupus. Biologics. 5:33–43.
Acknowledgments
The authors would like to thank C Dinarello for his insightful comments with the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Reilly, C.M., Regna, N. & Mishra, N. HDAC Inhibition in Lupus Models. Mol Med 17, 417–425 (2011). https://doi.org/10.2119/molmed.2011.00055
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00055