
Abstract
Histone deacetylases inhibitors (HDACi) include a growing number of drugs that share the ability to inhibit the enzymatic activity of some or all the HDACs. Experimental and preclinical evidence indicates that these epigenetic drugs not only can be effective in the treatment of malignancies, inflammatory diseases and degenerative disorders, but also in the treatment of genetic diseases, such as muscular dystrophies. The ability of HDACi to counter the progression of muscular dystrophies points to HDACs as a crucial link between specific genetic mutations and downstream determinants of disease progression. It also suggests the contribution of epigenetic events to the pathogenesis of muscular dystrophies. Here we describe the experimental evidence supporting the key role of HDACs in the control of the transcriptional networks underlying the potential of dystrophic muscles either to activate compensatory regeneration or to undergo fibroadipogenic degeneration. Studies performed in mouse models of Duchenne muscular dystrophy (DMD) indicate that dystrophin deficiency leads to deregulated HDAC activity, which perturbs downstream networks and can be restored directly, by HDAC blockade, or indirectly, by reexpression of dystrophin. This evidence supports the current view that HDACi are emerging candidate drugs for pharmacological interventions in muscular dystrophies, and reveals unexpected common beneficial outcomes of pharmacological treatment or gene therapy.
Similar content being viewed by others


Muscular Dystrophies—From Gene Mutation to Pathogenesis
Muscular dystrophies (MDs) include a heterogeneous group of genetic diseases invariably leading to muscle degeneration and impaired function. Mutation of nearly 30 genes gives rise to various forms of muscular dystrophy, which differ in age of onset, severity, and muscle groups affected (1).
The most common MD is the Duchenne muscular dystrophy (DMD), a severe recessive X-linked disease which affects one in 3,500 males, characterized by rapid progression of muscle degeneration, eventually leading to loss of ambulation and death within the second decade of life. This disorder is caused by mutations in the dystrophin gene that result in the complete absence of, or very infrequently, in the expression of, a truncated, nonfunctional protein. The dystrophin gene is the largest in the human genome and its coded protein is an important structural component of skeletal myofibers (2). Dystrophin interacts with a group of peripheral membrane and transmembrane proteins through the C-terminal domain, to form the dystrophin-associated protein complex (DAPC), which provides the molecular link between the cytoskeleton and the extracellular matrix of skeletal myofibers (3). As such, DAPC supports sarcolemmal integrity during muscle contraction. Essential components of the DAPC are the dystrophin-glycoprotein complex (DGC), which includes dystrophin, dystroglycans (DG) and sarcoglycans (SCG), and other associated proteins, such as a-dystrobrevin, syntrophins, neuronal nitric oxide synthase (nNOS), growth factor receptor-bound protein-2 (Grb2), caveolin-3 and sarcospan. In addition, enzymes (LARGE) and proteins with putative enzymatic activity (for example, fukutin-related protein [FKRP]) interact with DGC and catalyze DG glycosylation. Mutations of individual members of the DAPC cause distinct forms of muscular dystrophies, emphasizing the importance of these genes (4,5). Faulty muscle structure caused by the absence of extracellular or intracellular structural proteins results in cell membrane instability, initiating a cascade of deleterious events, such as uncontrolled calcium influx, apoptosis and necrosis, inflammation and replacement of muscle with fibrotic tissue and fat (1,6).
Muscle degeneration and regeneration are two key events underlying the pathogenesis of several muscular diseases, and in particular muscular dystrophies. The adult skeletal muscle possesses a remarkable regenerative potential that supports the repair of injured myofibers (7). Despite that, severe muscle loss is the invariable outcome of muscular dystrophies, leading to subverted muscle structure, and formation of fibrotic scars and massive fatty infiltration—a process called fibroadipose degeneration. This detrimental outcome typically compromises muscle function, alters the tissue environment and probably limits the potential effectiveness of regenerative approaches.
Pharmacological strategies for the treatment of muscular dystrophies are normally designed to counter the disease progression by targeting events downstream of the genetic mutation, such as inflammation, fibrosis, fat deposition and calcium homeostasis, or by promoting endogenous regeneration, or by upregulating compensatory proteins (8,9). Because of the hurdles that still prevent the application to dystrophic patients of gene- and cell-mediated therapies, pharmacological strategies provide a unique, immediate and suitable resource for the treatment of the current generation of dystrophic patients.
Compensatory Regeneration Potential of Dystrophic Muscles
Clinical and experimental evidence indicates that compensatory regeneration tends to counterbalance degeneration of dystrophic muscles at early stages of the disease (10). Regeneration of diseased or injured muscles occurs at expense of an heterogeneous population of resident muscle stem cells—the satellite cells—and possibly other cells endowed with an inducible myogenic potential (10,11). How and whether these muscle progenitor cells (MPC) cooperate to promote myonuclear turnover in physiological conditions, and to regenerate injured or diseased muscles, is still not understood fully. However, at advanced stages of muscular dystrophies the functional exhaustion of satellite cells compromises the beneficial effect of compensatory regeneration, leading to disease exacerbation and complicating the success of therapeutic interventions. A gradual decline in the regenerative properties of muscle typically is observed during aging also, likely as consequence of changes in the environmental cues that direct satellite cell-mediated tissue maintenance and repair (12). A recent work from Blau et al. (13) has established the first link between replicative senescence of MPC and disease progression in the mouse model of DMD (mdx mice), showing that mdx mice with shortened telomeres in muscle cells develop anticipated and more severe signs of disease. This evidence suggests that the intrinsic failure of muscle stem cells, to support long-term compensatory regeneration of dystrophin-deficient myofibers, might contribute to DMD pathogenesis and disease progression, possibly owing to the limited proliferation potential imposed by telomere shortening. An interesting perspective indicated by this work is that interventions that rejuvenate skeletal muscles (14) and/or bypass the replicative senescence (15) could be used to extend the regeneration potential of dystrophic muscles.
Muscle satellite cells have long been regarded as the sole source of myonuclei in muscle repair, however, the recent identification of different pluripotent cell types in muscles and in other adult tissues has challenged the widely held view that tissue-specific stem cells are predetermined to a specific tissue lineage. Indeed, adult stem cells isolated from various tissues appear to differentiate in vitro and in vivo into multiple lineages, depending on environmental cues or specific culture conditions. Progenitor cells isolated from bone marrow (BM) (16,17,18), from adult muscles (17,19), from embryonic and adult vasculature (20) and from various mesenchymal-derived tissues (21) can differentiate into the myogenic lineage. The actual contribution of these alternative muscle progenitors to myofiber turnover and repair is unclear (18,22); however, these alternative sources of MPC could be exploited to support therapeutic strategies to regenerate diseased or aged muscles.
Muscle-derived pluripotent cells also can contribute to muscle regeneration indirectly, by cell-to-cell interactions or by releasing cues within the regenerative environment. Regeneration cues typically instruct MPC to repair injured muscles correctly. The identification of muscle-derived interstitial cells that can adopt multiple lineages and contribute, either directly or indirectly, to muscle regeneration (23,24,25) is beginning to shed light on the functional interactions between the different cell types that participate in muscle regeneration. Muscle interstitial cells probably belong to a heterogeneous population endowed with limited pluripotency that display overlapping cell surface markers and similar biological properties. Interestingly, the finding that resident muscle interstitial cells retain the ability to turn into fibroadipocytes in response to signals released by degenerating muscles provided breakthrough information on the pathogenesis of neuromuscular disorders (24,25). Since ectopic fat formation and scar tissue are detrimental outcomes of many degenerative muscular disorders, these cells are interesting candidates as cellular determinants of disease progression. An important biological property of one of these cell populations—also called fibroadipocyte progenitors (FAPs)—relates to their reciprocal interactions with myofibers and satellite cells. In resting muscles, the interaction with intact myofibers prevents their conversion into fibroadipocytes (25); however, muscle injury stimulates these cells to produce paracrine factors that promote satellite cell-mediated regeneration (24). By contrast, in degenerating muscles, such as dystrophic muscles at advanced stages of disease, these cells turn into fibroadypocytes, which mediate fat deposition and fibrosis, thereby disrupting the environment conducive for muscle regeneration (24,25). This evidence indicates that differences in the signals derived from regenerative or degenerative muscles determine fate and function of these cells, and this might ultimately influence muscle ability to repair or undergo fibroadipogenic degeneration (26). Knowing the mechanism by which regeneration cues are converted into the epigenetic information that regulates the cell decision to adopt either the myogenic or the fibroadipogenic lineage will fill an important gap of knowledge and will provide the rationale for selective interventions toward shifting the balance between muscle regeneration and fat deposition for the treatment of muscular diseases.
Intracellular Conversion of External Cues into the Epigenetic Control of Gene Expression—DAPC Signaling to HDAC-Regulated Networks
How are regeneration cues converted into chromatin modifications that regulate gene expression in MPC and other components of the muscle regenerative environment? A number of studies have shown that extracellular signal-activated kinases can target chromatin-modifying complexes directly and regulate their function and distribution in satellite cells (27,28,29), thereby linking regeneration cues with changes of gene expression in muscle progenitors. Likewise, membrane-associated complexes, such as DAPC, have been suggested to modulate intracellular signaling to the nucleus. Previous studies indicated that DAPC regulates intramuscular generation of nitric oxide (NO), by documenting NO deficiency in dystrophic muscles (30). Independent reports have shown that NO-mediated S-nitrosylation of HDAC2 influences gene expression in different cell types by altering the acetylation status of histones at specific loci (31,32). The molecular connection between DAPC and chromatin has been described in muscle cells, by showing that NO signaling regulates HDAC2 activity by S-nytrosylation, which inhibits HDAC2-mediated gene repression (32). In muscles of mdx mice, the absence of dystrophin at the sarcolemma delocalizes and downregulates NO synthase (nNOS), leading to deficient S-nitrosylation of HDAC2 and constitutive inhibition of HDAC2-target genes. It is likely that DAPC-regulated NO signaling to HDAC2 mediates transcriptional adaptation to mechanical cues, such as those derived from myofiber contraction. Thus, the interruption of DAPC-NO signaling to HDAC2 in dystrophic muscles might compromise myofiber adaptation to contraction and contribute to muscle degeneration. One key gene controlled by HDAC2 in muscle cells is follistatin (33), the endogenous antagonist of the most potent inhibitor of skeletal myogenesis—myostatin (34). Although the precise contribution of the follistatin/myostatin (and other TGF-β members) network to the pathogenesis of muscular dystrophies is unclear currently (35), it is likely that the physical and functional interactions between these proteins regulate the size of adult skeletal muscles, in response to different stimuli, including mechanical cues. Thus, it is possible that HDAC-mediated control of follistatin transcription is important to maintain proper muscle size and structure postcontraction, and that an unbalanced follistatin/myostatin activity, which is caused by deregulated HDAC2 function in dystrophic muscles (32), contributes to DMD pathogenesis. Indeed, direct inhibition of myostatin (36,37) or delivery of follistatin (38,39) exerted similar beneficial effects in mdx mice—the mouse model of DMD—and are currently exploited for clinical trials in DMD (40,41). Likewise, direct inhibition of HDAC2 by HDACi, or inactivation by either nitric oxide donors or by reconstitution of the dystrophin-NO signaling (32) leads to derepression of follistatin, which mediates the ability of HDACi and NO signaling to stimulate myogenesis in vitro (42,43) and counters muscle degeneration in mdx mice (33,44)—see also next paragraph. Taken together, these findings identify in the HDAC-regulated myostatin/follistatin axis an important common target for therapeutic interventions that can be translated immediately into clinical practice.
A large amount of work illustrated the mechanism by which HDACs regulate muscle specific gene transcription in satellite cells, by controlling the activity of myogenic bHLH proteins (MyoD, Myf5, myogenin and MRF4) and MEF2 family factors (MEF2A, MEF2B, MEF2C and MEF2D) (45). These transcriptional activators cooperate to activate the expression of muscle genes (46) and non-coding RNAs involved in the activation of the myogenic program (47). Histone acetyltransferases (HATs) and histone deacetylases (HDACs) function antagonistically to regulate muscle gene expression, via control of histone acetylation—a modification that promotes gene expression—by direct interactions with myogenic bHLH proteins and MEF2 family factors. In undifferentiated myoblasts, association of MyoD and MEF2 factors mediates HDAC recruitment to the chromatin of target genes and prevents hyperacetylation and unscheduled activation of muscle gene expression (48). Interestingly, while class I HDACs (HDAC1 and HDAC2) show constitutive nuclear localization and preferentially associate with MyoD (49,50), class II HDAC members, HDAC4 and 5, shuttle between the nucleus and the cytoplasm and are dedicated repressors of MEF2-dependent transcription (51,52). Upon differentiation, displacement of HDACs from the chromatin of target genes correlates with the hyperacetylation at muscle loci and activation of muscle gene transcription genes (27). Moreover, acetylation of MyoD (and possibly other muscle bHLH proteins) and MEF2 factors contribute to stimulate transcription of target genes (53,54,55). Thus, the balance between acetylation and deacetylation is a critical determinant of muscle gene transcription and is emerging as a crucial target of interventions toward manipulating the regenerative potential of muscle stem cells.
Recent evidence demonstrates a reciprocal control between HDACs and muscle-specific microRNA (miRNAs). miRNAs are emerging as important regulators of muscle development, homeostasis and regeneration, and several studies reported on altered levels of miRNAs in muscular dystrophies and other muscular disorders (56,57). A number of muscle-specific and nonspecific miRNAs are induced during myogenesis, and many genes controlling myoblast proliferation and differentiation are the targets of miRNAs (58). An interplay between HDACs and miRNAs was described originally by Chen et al., who showed that miRNA-1 (miR-1) and miRNA-133 (miR-133) are clustered on the same chromosomal loci and transcribed together in a tissue-specific manner during development (59). Importantly, these two muscle-specific miRNAs (also called “myomiR”) have distinct roles in modulating skeletal muscle proliferation and differentiation. miRNA1 is induced during myogenic differentiation and downregulates HDAC4, leading to derepression of MEF2-activated gene transcription. By contrast, miR-133 enhances myoblast proliferation by repressing serum response factor (SRF) (59). Following this initial observation, a number of additional myomiR has been described (60). One important aspect of these miRNAs consists of their control by most of the same epigenetic regulators that typically control the expression of muscle genes. For instance, myomiR generally are regulated by MyoD- and MEF2-responsive promoters (47,61,62,63), which typically are regulated by HDACs. Thus, by extension, HDACi are predicted to promote the expression of myomiR in myoblasts.
The relationship between DAPC signaling to HDAC2 and the expression of miRNAs involved in the pathogenesis of DMD has been elucidated recently by Bozzoni’s group, which documented a reduced expression of miR1 and miR29 in muscles of dystrophic mdx mice, in coincidence with the absence of dystrophin and the consequent reduction of NO-mediated nytrosylation of HDAC2. The same authors demonstrated by chromatin immunoprecipitation that the expression of these myomiR is regulated by HDAC2. Indeed, restoring the NO signaling to HDAC2 in dystrophic mice, by exon skipping, leads to derepression of specific miRNAs (64). This evidence indicates a deregulated epigenetic control of miRNAs in dystrophic muscles, as a consequence of the primary genetic defect—mutations affecting dystrophin expression—as also suggested by previous works (65). Importantly, DAPC signaling to HDAC2-regulated miRNAs appears to have an important role in the pathogenesis of muscular dystrophies. In fact, reduced levels of miR-1 correlates with an upregulation of one of its target genes, the glucose-6-phosphate dehydroge-nase (G6PD), which is involved in the generation of antioxidant molecules that protect muscles against oxidative damage. Moreover, reduced levels of miR29 coincide with an augmented expression of two targets, such as elastin and collagen, which are implicated in muscle fibrosis. Collectively, these data demonstrate that deregulated DAPC/NO signaling to HDAC2 in dystrophic muscles is an important epigenetic contributor to DMD pathogenesis, as it controls a network consisting of miR and target genes involved in two key events of disease progression—fibrosis and oxidative stress. Given the predominant role of class I HDACs (which preferentially target MyoD) (50,49) in the control of miR-regulated networks, it is predicted that selective inhibition of class I HDACs can have beneficial effects on dystrophic muscles comparable to those observed with global HDACi (see paragraph below). Moreover, a distinct network of potential relevance in muscular dystrophy is composed by miRNA and class II HDACs and consists of miR-1-mediated suppression of HDAC4. In regenerating skeletal muscle, miR-1 expression is induced by mTOR signaling to MyoD, and miR-1-mediated suppression of histone deacetylase 4 (HDAC4) results in production of follistatin and subsequent myocyte fusion (66). While it has been shown that follistatin promoter is regulated selectively by class I HDACs (33), it is possible that HDAC4 controls follistatin expression indirectly or through MEF2 binding sites identified in the distal elements of follistatin promoter (PL Puri’s lab, unpublished data).
Thus, HDAC-regulated expression of genes and miRNAs involved in muscle regeneration defines a network downstream of DAPC that supervises the epigenetic control of gene expression in normal and dystrophic muscles, and therefore contributes to compensatory regeneration of dystrophic muscles or muscle degeneration during disease progression. Importantly, this network is susceptible to pharmacological manipulation by HDACi or by interventions that recover the integrity of DAPC. Therefore, pharmacological interventions that target HDAC (that is, HDACi) have the potential to direct this network toward compensatory regeneration, instead of fibroadipogenic degeneration.
Epigenetic Pharmacology in Muscular Dystrophies—Molecular Determinants of Cell and Gene Selectivity of HDACi
The beneficial potential of HDACi in the treatment of muscular dystrophies has been exploited in two distinct mouse disease models—the mdx mice, which are deficient for dystrophin expression and simulate DMD, and the α-sarcoglycan null mice, a model of limb-girdle muscular dystrophy type 2D (LGMD2D) (33). Although these mice show a milder “disease phenotype,” when compared with dystrophic patients, they recapitulate most of the salient pathogenetic events and reproduce signs and features of disease progression. As such, they provide the most amenable and approachable disease model for exploratory and preclinical evaluation of experimental interventions in muscular dystrophies. In both models, long-term exposure to distinct HDACi—trichostatin A (TSA), valproic acid (VPA) and phenylbutyrate—effectively countered disease progression (33). These compounds are general inhibitors of HDACs (and are therefore called pan-HDACi) that promote formation of skeletal myotubes with an increased size in vitro (42,43). When administered to dystrophic mice, these HDACi exerted similar beneficial effects. In particular, in mdx mice, HDACi increased the cross-sectional area (CSA) of myofibers, decreased the cellular (inflammatory) infiltrate and prevented the formation of fibrotic scars, which contribute to counter the muscle loss and the functional decline that are observed typically in mdx mice (33). Interestingly, the extent by which HDACi ameliorates the mdx phenotype varies significantly among these compounds, with TSA being the most effective drugs at defined concentrations (TSA 0.6 mg/kg, delivered by daily intra-peritoneal injection) (33). The differences among these drugs are probably ascribed to their different pharmacological profile and bioavailability. Remarkably, these effects were achieved in the absence of dystrophin restoration or utrophin upregulation, indicating that HDACi target events downstream of DAPC. Indeed, similar beneficial effects were observed in mdx mice upon delivery of NO (67), which mediates DAPC signaling to HDAC2 (32). This is consistent with a global deficiency of NO observed in dystrophin-deficient muscles (30,65). A common mediator of the beneficial effects exerted by HDACi and NO is follistatin (see paragraph above), which mediates the increased CSA observed in myofibers of mdx mice exposed to either NO donors or HDACi (43,44,33). While a relationship between increased CSA and protection of muscle integrity has been proposed (68), it should be noted that follistatin can target other pathogenic events of muscular dystrophies, such as fibrosis, independently (69).
Current studies are seeking to define the relative ability to counter DMD progression with a number of different HDACi, which have been used in clinical practice for some time (VPA and phenyl-butyrate) or have been approved recently for the treatment of cancer and other diseases. Among them, we have tested the suberoylanilide hydroxamic acid (SAHA) and ITF2357 (givinostat) in the treatment of mdx mice MS275 (33). These compounds all were effective in ameliorating the dystrophic phenotype of mdx mice (unpublished data). A dose-finding study was performed by Colussi et al. with SAHA, using escalating doses ranging from 0.3 to 100 mg/kg/day delivered to mdx mice for 3 months (70). This study identified an efficacy in recovering functional and histological parameters within a window of doses between 0.6 and 5 mg of SAHA, with evident reduction of the beneficial effects with doses lower than 0.6 mg and higher than 5 mg. While the reason is still unclear for the dose-dependent response of mdx mice to SAHA, this evidence indicates that dose-finding studies should be extended to all HDACi used for the experimental treatment of muscular dystrophies. The same study reported on an interesting correlation between mdx exposure to SAHA (5 mg/kg/day) and restoration of the profile of specific plasma proteins that were found to be expressed differentially in mdx mice versus their normal counterpart (70). An interesting insight into the specific role of individual HDACs in the pathogenesis of muscular dystrophy is suggested by the comparable efficacy of MS275, which selectively inhibits class I HDACs (33), and pan HDACi. This suggests that inhibition of class I HDACs is sufficient to exert most of the beneficial effects observed in HDACi-treated mdx mice, once again emphasizing the key contribution to mdx pathogenesis by class I HDACs. Consistently, class II HDAC specific inhibitors have shown an opposite effect in vitro, inhibiting rather than promoting skeletal myogenesis (71).
One of the hurdles for the translation into clinical trials of experimental drugs endowed with therapeutic potential in animal models of muscular dystrophy is the absence of information on critical pharmacological parameters in children—an issue that often complicates the use of these drugs in DMD boys. A notable exception is represented by ITF2357, which is being tested currently in a phase I safety study in children affected by Systemic Onset Juvenile Arthritis (SOJIA) (72). Preclinical studies show the effectiveness of ITF2357 in preventing disease progression in mdx mice after prolonged exposure (PL Puri’s lab, unpublished data). These studies are predicated to provide the key information for the translation of ITF2357 into clinical trials for DMD.
Studies performed in animal models have revealed a somehow surprising selective effect of HDACi that is in apparent conflict with the expected widespread activity of these drugs, which cause global inhibition of ubiquitous HDACs and the consequent hyperacetylation in all organs and tissues. Clinical trials with HDACi confirmed a relative selectivity of action of HDACi, which within certain doses cause more minor side effects than those expected (73). HDACi appear to target specific cell types, which are enriched in organs and tissues subjected to high nuclear turnover—that is, resident stem cells of rapidly self-renewing organs and tissues (74). This might explain the selective activity of HDACi toward tumor cells and the most prominent side effects observed in treated organisms that derive from the HDACi activity on rapidly self-renewing organs, such as bone marrow, hair follicle, skin and intestine (75). This evidence leads to the speculation that HDACi might target specific cellular populations, such as cancer cells or other cell types activated in response to physiological or pathological conditions, selectively—for example, compensatory regeneration in degenerative disorders, such as muscular dystrophies. Remarkable features shared by these cell types are their relative stemness or multipotency and tendency to self-renew and proliferate. Consistently, embryonic stem cells are particularly sensitive to HDACi (76,77), and HDACi are especially active during embryo development (78). Collectively, these observations point to pluripotent cells (both embryonic stem cells and adult, resident stem cells) as preferential target of HDACi, and suggest that stem cell-specific chromatin signature could provide the “permissive ground” for changes in gene expression, upon exposure to HDACi.
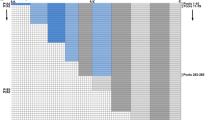
A number of studies have begun to address this issue. We previously have shown a stage-dependent effect of HDACi in muscle cells in vitro and in vivo (42,43). These studies demonstrated that the ability of HDACi to promote muscle gene expression and to implement skeletal myogenesis is restricted to proliferating muscle precursors, such as cultures of undifferentiated myoblasts during in vitro myogenesis, and muscle progenitors during somitogenesis and regeneration of injured muscles in adult life. These findings prompted us to hypothesize that tissue progenitors and terminally differentiated progeny respond differently to HDACi. To test this hypothesis, we have examined the changes in gene expression induced by TSA in undifferentiated human primary myoblasts versus terminally differentiated human myotubes, by microarray analysis. Figure 1A shows that TSA perturbs the gene expression profile in myoblasts significantly, but not in myotubes (the complete list of upregulated and down-regulated genes has been deposited in NCBI’s Gene Expression Omnibus [79] and is accessible through GEO series accession number GSE27073 [https://doi.org/www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE27073]).

(A) Gene expression analysis by microarray in human skeletal myoblasts (undifferentiated mononucleated cells, cultured in growth medium—15% FBS) and human skeletal myotubes (preformed myotubes, cultured in differentiation medium—2% horse serum) exposed to the HDACi TSA (50 nmol/L) for 24 h. contr.: control. (B) Representation of categories of genes induced by TSA in human skeletal myoblasts. (C) List of common genes found upregulated upon TSA treatment in both human skeletal myoblasts (exposed to TSA for 24 h in growth medium) and in mouse C2C12 myoblasts (exposed to TSA for 24 h in growth medium and then incubated in differentiation medium, without TSA). (D) Distribution of epigenetic marks (H3K4me3 and H3K27me3) in TSA-induced genes in human skeletal myoblasts.
The large majority of the genes upregulated by TSA in myoblasts were involved in the myogenic differentiation program (Figure 1B) and showed a significant overlap with the genes induced in C2C12 mouse myoblasts exposed to TSA and subsequently incubated in differentiation medium (43) (Figure 1C). Thus, in myoblasts, HDACi anticipate the expression of genes that are normally induced during differentiation. Interestingly, genome-wide location analysis showed that the genes induced by TSA in myoblasts showed specific epigenetic signatures, consisting of either coexistence of comparable levels of H3K4 trimethylation (H3K4me3) and H3-K27me3 (42% of induced genes) or predominant enrichment for H3K4 me3 (57% of induced genes) (Figure 1D). In contrast, virtually none (1%) of induced genes showed a selective enrichment of the repressive mark H3-K27me3. Thus, the absence of bivalence or preexisting activator marks (that is, H3K4me3) precludes gene activation by HDACi, indicating the importance of functional interactions between HDACs and the chromatin modifying complexes responsible for preexisting histone modifications—for example, polycomb and thritorax complexes. A bivalent chromatin structure is an epigenetic signature that identifies genes poised for transcription that typically are enriched in embryonic stem cells or other pluripotent cell types (80,81). During cellular differentiation, “chromatin bivalency” typically is resolved into chromatin conformations either permissive or repressive for gene transcription (82–84). Resolution of this “bivalency,” together with other epigenetic modifications (that is, DNA methylation), is likely to confer resistance to HDACi in myotubes and perhaps other terminally differentiated tissues. In contrast, pluripotent tissue progenitors or adult stem cells, which are enriched in “bivalent genes,” are more susceptible to changes in gene expression when exposed to HDACi, which presumably contribute to resolve the bivalence by inducing hyperacetylation (see schematic illustration in Figure 2). Of note, we have observed both upregulated and downregulated genes in myoblasts exposed to TSA. We do not know if downregulated genes are affected directly or indirectly by HDACi.

Schematic illustration of the epigenetic status of HDACi-responsive genes in muscle progenitors exposed to differentiation cues.
Future genome-wide studies of the epigenetic profile of muscle stem cells isolated from normal and dystrophic skeletal muscles exposed to HDACi will shed light on the chromatin landscape that predicts responsiveness to HDACi.
Recent work is providing further support to the existence of epigenetic determinants of HDACi selectivity in regulating gene expression (85,86). An emerging mechanism relates to the relationship between histone acetyltransferases (HATs) and HDACs that appears more dynamic than expected and involves other histonemodifying enzymes, such as histone lysine methyltransferases (HKMTs). Through large-scale strategies, Wang et al. provided evidence that both HATs and HDACs are recruited to transcribed regions of active genes by phosphorylated Pol II (86). These studies indicate that the function of the majority of HDACs in the genome is to remove acetyl groups at active genes during the process of transcriptional initiation and elongation, thus resetting the chromatin structure for a next round of transcription. For instance, genes that are primed by H3K4me3 are poised for activation and subjected to a dynamic cycle of acetylation and deacetylation by transient HAT/HDAC binding. Conversely, silent genes deprived of any H3K4 methylation show no HDACs binding and are not susceptible to acetylation upon treatment with HDACi. Thus, H3K4me3 appears to be a prerequisite for histone hyperacetylation and transcription of those genes that are activated by HDACi (86). This work provides new fundamental insight on the mechanisms by which HDACi affect gene expression, and explains why genes uniquely enriched in H3K27me3 are insensitive to HDACi (see Figure 1). In an independent study, Karantzali and colleagues demonstrated that HDACi preferentially target genes enriched for H3K4me3 and H3-K27me3 in embryonic stem (ES) cells, further indicating the relationship between “chromatin bivalency,” stem cells and response to HDACi (85).
On the basis of the evidence described above, we are tempted to speculate that adult stem cells, which are activated in high-turnover organs and tissues or upon injury and diseases (that is, during compensatory regeneration), or transformed (cancer) stem cells are the preferential target of HDACi. It will be interesting to explore the potential synergism between pharmacological interventions that promote “chromatin bivalence” and HDACi. The activation of tissue-associated stem cells is imparted by extrinsic signals (that is, regeneration cues), which are converted into chromatin modification by intracellular signaling pathways (27). Thus, one interesting perspective is to modulate the signaling to the chromatin in specific cell types pharmacologically to confer responsiveness to HDACi. We have recently shown that p38 signaling, which is elicited in muscle stem cells by regeneration cues, such as TNFα, directs polycomb repressive complex 2 (PRC2)-mediated repression of Pax7, via H3K27me3 (28,87). Pharmacological inhibition of this signaling in activated satellite cells, by anti-TNF antibodies or EzH2 inhibitors, might be used to induce “chromatin bivalence” of key genes and “sensitize” to HDACi cells that would otherwise be resistant.
Conclusions: Toward Clinical Trials with HDACi in Muscular Dystrophies
The available data indicate the HDACi are strong candidates for the pharmacological treatment of DMD. Unlike with steroids, which currently are used as palliative treatment in muscular dystrophies, the molecular rationale and the epigenetic basis of the beneficial effect of HDACi are being revealed. In particular, HDAC-regulated networks downstream of dystrophin appear to be key targets of HDACi and might mediate their ability to promote regeneration at the expense of degenerative events. Future studies should identify the cell types that mediate HDACi effects in dystrophic muscles, and the epigenetic signatures predictive of response to HDACi. This information might be instrumental to predict patient responsiveness to HDACi and to identify markers of response to treatment. An interesting perspective suggested by the identification of DAPC signaling to HDACs, as a target of pharmacological interventions, is the potential extension of HDACi to the treatment of different types of muscular dystrophies.
The current availability of HDACi in clinical practice (about 100 clinical trials) indicates the opportunity for an immediate translation of these drugs into pharmacological treatments of DMD in human patients. Current efforts should be dedicated to the search of the optimal compounds and conditions for HDACi-based clinical trials.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Dalkilic I, Kunkel LM. (2003) Muscular dystrophies: genes to pathogenesis. Curr. Opin. Genet. Dev. 13:231–8.
Hoffman EP, Brown RH Jr, Kunkel LM. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 51:919–28.
Ervasti JM. (2007) Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta. 1772:108–17.
Mendell JR, Boué DR, Martin PT. (2006) The congenital muscular dystrophies: recent advances and molecular insights. Pediatr. Dev. Pathol. 9427–43.
Davies KE, Nowak KJ. (2006) Molecular mechanisms of muscular dystrophies: old and new players. Nat. Rev. Mol. Cell. Biol. 7:762–73.
Batchelor CL, Winder SJ. (2006) Sparks, signals and shock absorbers: how dystrophin loss causes muscular dystrophy. Trends Cell. Biol. 16:198–205.
Kuang S, Rudnicki MA. (2008) The emerging biology of satellite cells and their therapeutic potential. Trends Mol. Med. 14:82–91.
Engvall E, Wewer UM. (2003) The new frontier in muscular dystrophy research: booster genes. FASEB J. 17:1579–84.
Mozzetta C, Minetti G, Puri PL. (2009) Regenerative pharmacology in the treatment of genetic diseases: the paradigm of muscular dystrophy. Int. J. Biochem. Cell Biol. 41:701–10.
Shi X, Garry DJ. (2006) Muscle stem cells in development, regeneration, and disease. Genes. Dev. 20:1692–708.
Péault B, et al. (2007) Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol. Ther. 15:867–77.
Gopinath SD, Rando TA. (2008) Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell. 7:590–8.
Sacco A, et al. (2010) Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell. 143:1059–71.
Conboy IM, et al. (2005) Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 433:760–4.
Jaskelioff M, et al. (2011) Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 469:102–6.
Ferrari G, et al. (1998) Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 279:1528–30.
Gussoni E, et al. (1999) Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature. 401:390–4.
LaBarge MA, Blau HM. (2002) Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell. 111:589–601.
Asakura A, Seale P, Girgis-Gabardo A, Rudnicki MA. (2002) Myogenic specification of side population cells in skeletal muscle. J. Cell Biol. 159:123–34.
Minasi MG, et al. (2002) The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development. 129:2773–83.
Young HE, et al. (2001) Human reserve pluripotent mesenchymal stem cells are present in the connective tissues of skeletal muscle and dermis derived from fetal, adult, and geriatric donors. Anat. Rec. 264:51–62.
Sherwood RI, et al. (2004) Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 119:543–54.
Mitchell KJ, et al. (2010) Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat. Cell Biol. 12:257–66.
Joe AW, et al. (2010) Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12:153–63.
Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. (2010) Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 12:143–52.
Rodeheffer MS. (2010) Tipping the scale: muscle versus fat. Nat. Cell Biol. 12:102–4.
Guasconi V, Puri PL. (2009) Chromatin: the interface between extrinsic cues and the epigenetic regulation of muscle regeneration. Trends Cell Biol. 19:286–94.
Palacios D, et al. (2010) TNF/p38±/polycomb signaling to Pa×7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 7:455–629.
Acharyya S, et al. (2010) TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy. PLoS One. 5:e12479.
Wehling M, Spencer MJ, Tidball JG. (2001) A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 155:123–31.
Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. (2008) S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 455:411–5.
Colussi C, et al. (2009) HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. U. S. A. 106:1679.
Minetti GC, et al. (2006) Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 12:1147–50.
Lee SJ. (2004) Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 20:61–86.
Tsuchida K. (2008) Targeting myostatin for therapies against muscle-wasting disorders. Curr. Opin. Drug Discov. Devel. 11:487–94.
Bogdanovich S, Perkins KJ, Krag TO, Whittemore LA, Khurana TS. (2005) Myostatin propeptidemediated amelioration of dystrophic pathophysiology. FASEB J. 19:543–9.
Bogdanovich S, et al. (2002) Functional improvement of dystrophic muscle by myostatin blockade. Nature. 420:418–21.
Nakatani M, et al. (2008) Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 22:477–87.
Haidet AM, et al. (2008) Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc. Natl. Acad. Sci. U. S. A.105:4318–22.
Wagner KR, Lechtzin N, Judge DP. (2007) Current treatment of adult Duchenne muscular dystrophy. Biochim. Biophys. Acta. 1772:229–37.
Rodino-Klapac LR, et al. (2009) Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve. 39:283–96.
Iezzi S, Cossu G, Nervi C, Sartorelli V, Puri PL. (2002) Stage-specific modulation of skeletal myogenesis by inhibitors of nuclear deacetylases. Proc. Natl. Acad. Sci. U. S. A. 99:7757–62.
Iezzi S, et al. (2004) Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev. Cell. 6:673–84.
Pisconti A, et al. (2006) Follistatin induction by nitric oxide through cyclic GMP: a tightly regulated signaling pathway that controls myoblast fusion. J. Cell Biol. 172:233–44.
Puri PL, Sartorelli V. (2000) Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J. Cell Physiol. 185:155–73.
Palacios D, Puri PL. (2006) The epigenetic network regulating muscle development and regeneration. J. Cell Physiol. 207:1–11.
Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. (2006) Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. U. S. A. 103:8721–6.
McKinsey TA, Zhang CL, Olson EN. (2001) Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 11:497–504.
Puri PL, et al. (2001) Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell. 8:885–97.
Mal A, Sturniolo M, Schiltz RL, Ghosh MK, Harter ML. (2001) A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. EMBO J. 20:1739–53.
McKinsey TA, Zhang CL, Olson EN. (2000) Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. U. S. A. 97:14400–5.
Lu J, McKinsey TA, Zhang CL, Olson EN. (2000) Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell. 6:233–44.
Sartorelli V, et al. (1999) Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol. Cell. 4:725–34.
Ma K, Chan JK, Zhu G, Wu Z. (2005) Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol. Cell. Biol. 25:3575–82.
Angelelli C, et al. (2008) Differentiation-dependent lysine 4 acetylation enhances MEF2C binding to DNA in skeletal muscle cells. Nucleic Acids Res. 36:915–28.
Greco S, et al. (2009) Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 23:3335–46.
Eisenberg I, et al. (2007) Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. U. S. A. 104:17016–21.
Williams AH, Liu N, van Rooij E, Olson EN. (2009) MicroRNA control of muscle development and disease. Curr. Opin. Cell Biol. 21:461–9.
Chen JF, et al. (2006) The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 38:228–33.
Eisenberg I, Alexander MS, Kunkel LM. (2009) miRNAS in normal and diseased skeletal muscle. J. Cell. Mol. Med. 13:2–11.
Rosenberg MI, Georges SA, Asawachaicharn A, Analau E, Tapscott SJ. (2006) MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J. Cell Biol. 175:77–85.
Liu N, et al. (2007) An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. U. S. A. 104:20844–9.
Mallappa C, et al. (2010) Myogenic microRNA expression requires ATP-dependent chromatin remodeling enzyme function. Mol. Cell. Biol. 30:3176–86.
Cacchiarelli D, et al. (2010) MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 12:341–51.
Colussi C, et al. (2009) Nitric oxide deficiency determines global chromatin changes in Duchenne muscular dystrophy. FASEB J. 23:2131–41.
Sun Y, et al. (2010) Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J. Cell Biol. 28:189:1157–69.
Brunelli S, et al. (2007) Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc. Natl. Acad. Sci. U. S. A. 104:264–9.
Zammit PS, Partridge TA. (2002) Sizing up muscular dystrophy. Nat. Med. 8:1355–6.
Aoki F, Kojima I. (2007) Therapeutic potential of follistatin to promote tissue regeneration and prevent tissue fibrosis. Endocr. J. 54:849–54.
Colussi C, et al. (2010). Proteomic profile of differentially expressed plasma proteins from dystrophic mice and following suberoylainide hydroxamic acid treatment. Proteomics Clin. Appl. 4:71–83.
Nebbioso A, et al. (2009) Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep. 10:776–82.
Vojinovic J, Damjanov N. (2011) HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol. Med. 17:397–403.
Elaut G, Rogiers V, Vanhaecke T. (2007) The pharmaceutical potential of histone deacetylase inhibitors. Curr. Pharm. Des. 13:2584–620.
Barker N, Bartfeld S, Clevers H. (2010) Tissue-resident adult stem cell populations of rapidly self-renewing organs. Cell Stem Cell. 7:656–70.
Duvic M, Vu J. (2007) Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert. Opin. Investig. Drugs. 16:1111–20.
Ware CB, et al. (2009). Histone deacetylase inhibition elicits an evolutionarily conserved self-renewal program in embryonic stem cells. Cell Stem Cell. 4: 359–69.
Liang J, et al. (2008) Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat. Cell Biol. 10:731–9.
Menegola E, Di Renzo F, Broccia ML, Giavini E. (2006) Inhibition of histone deacetylase as a new mechanism of teratogenesis. Birth Defects Res. C Embryo Today. 78:345–53.
Edgar R, Domrachev M, Lash AE. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30:207–10.
Bernstein BE, et al. (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 125:315–26.
Azuara V, et al. (2006) Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 8:532–8.
Lee MG, et al. (2006) Functional interplay between histone demethylase and deacetylase enzymes. Mol. Cell. Biol. 26:6395–402.
Boyer LA, et al. (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 441:349–53.
Mohn F, et al. (2008) Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 30:755–66.
Karantzali E, et al. (2008) Histone deacetylase inhibition accelerates the early events of stem cell differentiation: transcriptomic and epigenetic analysis. Genome Biol. 9:R65.
Wang Z, et al. (2009) Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 138:1019–31.
Mozzetta C, et al. (2011) Selective control of Pax7 expression by TNF-activated p38±/polycomb repressive complex 2 (PRC2) signaling during muscle satellite cell differentiation. Cell Cycle. 10:191–8.
Acknowledgments
PL Puri is an Associate Telethon Scientist of the Dulbecco Telethon Institute (DTI) and Associate Investigator of San-ford Children’s Health Research Center. C Mozzetta is a recipient of AFM post-doc fellowship. The authors thank all members of PL Puri’s lab for productive discussion and comments during manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Consalvi, S., Saccone, V., Giordani, L. et al. Histone Deacetylase Inhibitors in the Treatment of Muscular Dystrophies: Epigenetic Drugs for Genetic Diseases. Mol Med 17, 457–465 (2011). https://doi.org/10.2119/molmed.2011.00049
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00049