
Abstract
Histone deacetylase (HDAC) inhibitors are emerging as a novel class of potentially therapeutic agents for treating acute injuries of the central nervous system (CNS). In this review, we summarize data regarding the effects of HDAC inhibitor administration in models of acute CNS injury and discuss issues warranting clinical trials. We have previously shown that the pan-HDAC inhibitor ITF2357, a compound shown to be safe and effective in humans, improves functional recovery and attenuates tissue damage when administered as late as 24 h after injury. Using a well-characterized, clinically relevant mouse model of closed head injury, we demonstrated that a single dose of ITF2357 administered 24 h after injury improves neurobehavioral recovery and reduces tissue damage. ITF2357-induced functional improvement was found to be sustained up to 14 d after trauma and was associated with augmented histone acetylation. Single postinjury administration of ITF2357 also attenuated injury-induced inflammatory responses, as indicated by reduced glial accumulation and activation as well as enhanced caspase-3 expression within microglia/macrophages after treatment. Because no specific therapeutic intervention is currently available for treating brain trauma patients, the ability to affect functional outcome by postinjury administration of HDAC inhibitors within a clinically feasible timeframe may be of great importance. Furthermore, a growing body of evidence indicates that HDAC inhibitors are beneficial for treating various forms of acute CNS injury including ischemic and hemorrhagic stroke. Because HDAC inhibitors are currently approved for other use, they represent a promising new avenue of treatment, and their use in the setting of CNS injury warrants clinical evaluation.
Similar content being viewed by others

Introduction
Acute central nervous system (CNS) injury is a major cause of prolonged morbidity and mortality in the adult population. Although this class of inflictions is comprised of two distinct pathologies, namely traumatic impact and stroke, it represents a common challenge in therapeutic development. More specifically, our current understanding of underlying pathological and protective mechanisms and their contribution to injury outcome is still greatly limited by the intrinsic complexity of the CNS and the difficulty to develop adequate models for investigating disease and evaluating potential therapies (1–4). Although substantial research efforts and many clinical trials have been conducted, no specific therapy for CNS trauma is currently clinically available, whereas tissue plasminogen activator (tPA) remains the only approved agent for treating ischemic stroke patients.
Despite varying initial causes of primary injury, CNS trauma as well as ischemic or hemorrhagic stroke share the common detriments of reduced blood flow and energy failure, resulting in cell death and tissue loss. Common mechanisms of cell damage include excitotoxicity, calcium overload, oxidative stress, acute inflammation and apoptosis (2,5). These similarities raise the notion that common therapeutic strategies may be useful in these settings. Indeed, strategies aimed at neutralizing mediators such as reactive oxygen species, inflammatory cytokines and pro-apoptotic factors have been considered for both stroke and trauma treatment (2,6). Enhancement of plasticity and repair mechanisms has also been the focus of extensive research in both contexts (7–9).
Interestingly, disruption of cellular acetylation homeostasis of histones and other proteins has recently been recognized as yet another common feature in neuropathological states (10). In particular, several studies have demonstrated that neurodegeneration is associated with a global decrease in histone acetylase transferase (HAT) activity, resulting in relative over-deacetylation (11,12). In light of this, histone deacetylase (HDAC) inhibitors were tested for therapeutic efficacy in various neurodegenerative diseases, yielding promising findings in models of Huntington disease, amyotrophic lateral sclerosis and experimental autoimmune encephalomyelitis (13–16). Given the involvement of neurodegeneration in the pathophysiology of acute CNS injury and the profound effect of histone acetylation status on gene expression, recent work has focused on evaluating the use of HDAC inhibitors in stroke and CNS trauma models. A growing body of evidence indicates beneficial effects of these agents in both stroke and trauma, yet substantial issues affecting drug development must be resolved before these exciting findings can be translated into clinically applicable therapeutics.
Cellular Protein Acetylation
Under normal conditions, stability of cellular acetylation homeostasis is preserved by maintenance of an appropriate balance between two discrete sets of enzymes that facilitate forward and backward modifications (17,18). These enzymes, HAT and HDAC, have been the focus of extensive research owing to the key role of histones in cellular function and disease (19,20). The activity of HATs and HDACs governs the level of histone acetylation. Generally, enhanced acetylation induces chromatin remodeling to a loosely packed configuration that enables subsequent gene transcription, whereas increased deacetylation fosters chromatin condensation and reduced gene expression. It should be noted, however, that nonhistone proteins linked to microtubule stability, metabolism and aging have also been shown to serve as substrates for certain HDACs (21–24), highlighting the importance of acetylation as a posttranslational mode of regulation.
Five main subtypes of HDACs have been identified in humans thus far (25). Class I HDACs include HDACs 1, 2, 3 and 8, all of which harbor Zn2+-dependent deacetylase activity. Class II HDACs can be further subdivided into class IIa and class IIb isoforms, which, similar to HDAC class I enzymes, also require Zn2+ for optimal activity. Class IIa HDAC enzymes (HDACs 4, 5, 7 and 9) display tissue-specific expression patterns and have been suggested to interact with various other proteins via an extended N-terminal domain (22,26–28). The HDAC IIb class includes HDAC 6 and HDAC 10. Interestingly, HDAC 6 contains two independent catalytic domains and deacetylases α-tubulin within the cytoplasm (22). The exact functions of HDAC 10 remain unknown. Class III HDACs are also known as sirtuins. These enzymes display structural and functional divergence from other HDACs and require NAD+ for their enzymatic activity (29). Finally, class IV consists of a single member, HDAC 11, which shares certain characteristics with class I and class II HDACs, but has been suggested to facilitate different physiological roles (30).
Disruption of Acetylation Homeostasis During Neurodegeneration
Maintenance of an adequate balance between the amount and activity of HATs and HDACs is pivotal for neuronal survival under normal conditions (12,31). As the HAT/HDAC balance profoundly affects chromatin arrangement and gene expression, any alteration disrupting this tightly regulated mechanism is likely to result in inadequate gene transcription and harbor detrimental effects on cell fate. Indeed, neurodegenerative states, involving significant cell death and loss of function, have been associated with disrupted acetylation homeostasis (18). Particularly, several studies have established a global decrease in HAT activity during neurodegeneration, inevitably resulting in relative over-deacetylation (11,12). Importantly, rather than a quantitative surge, the tilt in acetylation homeostasis does not involve an increase in HDAC quantity or activity; it is due to the loss of balance stemming from reduced HAT activity.
The role of transcriptional dysregulation in Huntington disease was initially suggested (32), prompting studies that demonstrated beneficial effects of HDAC inhibitor treatment in rodent models of this disease (14,16,33). Subsequently, the efficacy of HDAC inhibitors was shown in other models of neurodegenerative pathologies including amyotrophic lateral sclerosis, Alzheimer disease, spinal muscular atrophy and experimental autoimmune encephalomyelitis (13,15,25,34–36). It is interesting to note that CNS injury such as stroke or trauma also shares some common pathologies with the early stages of Alzheimer disease such as neuroinflammation, accumulation of extracellular β-amyloid (Aβ) and hyperphosphorylated Tau protein. Moreover, traumatic brain injury (TBI) is thought to be an environmental risk factor for Alzheimer disease (37) and was found to accelerate Aβ deposition in humans (38). It is therefore not surprising that HDAC inhibitors are being investigating for a wide array of neurodegenerative disorders. The wide-ranging neuroprotective potential indicated by this work, taken together with the fact that several HDAC inhibitors were already used clinically for other indications including epilepsy, sickle cell anemia and T-cell lymphoma (39–41), and the vital need for developing new therapeutics for acute CNS injury, fueled research efforts aimed at evaluating the use of HDAC inhibitors for treating stroke and CNS trauma.
HDAC Inhibitor Treatment for Acute CNS Insults
As mentioned, beneficial effects brought about by HDAC inhibitor treatment were demonstrated in a model of experimental autoimmune encephalomyelis (13). Interestingly, these effects were shown to include a reduction in inflammatory responses, neuronal death and axon loss, alongside an enhancement in antioxidant and antiexcitotoxic capacity. Given the involvement of both inflammation and reactive oxygen species production in injury pathophysiology (2,5), it would be conceivable that HDAC inhibitors may also convey substantial benefits when tested in models of acute CNS insults. Several recent studies have used experimental stroke models for evaluating the effects of HDAC inhibitors on injury outcome. Beneficial effects induced by various agents harboring HDAC inhibitor activity have been established in a range of focal ischemia models as well as in a model of intracerebral hemorrhage (ICH). Because this line of evidence has instigated a wide research interest that has been recently reviewed by others (42), we shall focus on highlighting key findings that are associated with common denominators of stroke and trauma and will proceed to review all available data regarding the effects of HDAC inhibitor treatment in the setting of traumatic CNS injury, a newly emerging topic with potentially significant clinical implications.
HDAC Inhibitors in Ischemic Stroke and ICH
Protective effects induced by various HDAC inhibitors have been shown in experimental models of focal CNS ischemia. Studies focused on evaluating the effects brought about by HDAC inhibitors belonging to two chemical groups, namely the small carboxylates sodium butyrate (SB), valproic acid (VPA) and sodium 4-phenylbutyrate (4-PBA) and hydroxamate-containing HDAC inhibitors trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA).
VPA, clinically used as an anticonvulsant and mood-stabilizing drug, was tested in transient and permanent middle cerebral artery occlusion (MCAO) rat models. Postinjury administration of this HDAC inhibitor led to a reduction in both lesion volume as well as ischemia-induced neurological deficits (43,44). Because improvement in functional outcome is undoubtedly a major end-point measure for evaluating the efficacy of any treatment given to stroke patients, the functional benefit of VPA demonstrated under experimental conditions represents an encouraging observation and an essential link when one considers the translation of experimental data to the clinic. Similarly, SB and 4-PBA have been shown to induce amelioration of tissue damage and improvement of functional outcome after experimental permanent MCAO and in a mouse model of hypoxia-ischemia injury (43,45,46). The ability of postinsult treatment with SB to stimulate neurogenesis in the ischemic brain was also demonstrated recently (47).
Hydroxamate-containing HDAC inhibitors TSA and SAHA were shown to convey a significant decrease in lesion volume after MCAO (43,48–50). A reduction of about 30% in the volume of infracted tissue was demonstrated 24 h after insult, and this result was augmented even further when lesion volume was measured at later time points (48). A similar effect of TSA was shown after permanent middle cerebral artery occlusion, indicating the therapeutic efficacy of the compound in this more severe ischemic injury model (43). A key finding of the latter report was the ability of postinjury administration of TSA to reduce ischemia-induced neurological deficits as well as lesion volume. However, it should be considered that in the majority of studies conducted thus far, HDAC inhibitor administration was initiated before (or immediately after) the onset of MCAO, measures that are not feasible when treating stroke-inflicted individuals. Nevertheless, the protective effects of the tested HDAC inhibitors were well established and have incited research efforts aimed at elucidating mechanisms that may underlie these benefits.
The notion that the protective role of VPA, SB, TSA and SAHA after ischemic CNS injury may be associated with their inhibitory activity is strengthened by observations indicating maintenance of adequate histone H3 acetylation levels upon treatment with these agents, as opposed to the robust decrease seen after ischemic insult in nontreated animals (43,44,49). The protective effect against ischemia-hypoxia injury that is brought about by 4-PBA was not specifically attributed to its HDAC-inhibiting activity (46). It should be considered in this context that all HDAC inhibitors tested in stroke models so far are known to exert substantial inhibitory activity on class I HDAC (51,52). Hence, it is conceivable that inhibition of histone deacetylation contributes to the overall protective effects obtained by treatment with these agents after ischemic CNS injury, at least in part. This notion is further supported by the ability of VPA, a compound displaying selectivity toward class I HDAC enzymes, to convey protection (43,44,52).
As mentioned, the ability of HDAC enzymes to deacetylate nonhistone targets has been reported (21–24). Consequently, administration of HDAC inhibitors would induce a reduction in the deacetylation of nonhistone entities known to be targeted by HDACs, including transcription factors. Because several transcription factors regulating the expression of pro-survival molecules are deacetylated by HDACs, HDAC inhibitor treatment may lead to increased transcription of neuroprotective factors due to transcription factor activation (18). In any case, reduced deacetylation of histones and transcription factors is expected to lead to increased expression of target gene products via enhanced transcription. Indeed, the protective effects obtained by HDAC inhibitor treatment in experimental models of ischemic stroke have been associated with increased expression of neuroprotective factors including heat shock protein 70 (HSP70), B-cell lymphoma 2 (Bcl-2), phosphorylated Akt (pAkt), gelsolin and p21cip/waf1 (43–45,49,50). However, the precise mechanism by which these factors are upregulated warrants further investigation. Interestingly, HDAC inhibitor administration was found to concomitantly reduce ischemia-induced upregulation of caspase-3 and p53 (43,44). This would suggest the involvement of multiple mechanisms in HDAC inhibitor-induced neuroprotection and may indicate that the effects of these agents on specific transcription factors may be time- or cell type-dependent (18,53).
In their study, Kim et al. (43) suggested that neuroprotection from permanent MCAO obtained by postinsult administration of HDAC inhibitors is likely to involve suppression of ischemia-induced inflammation, since they report on attenuated microglial activation and decreased accumulation of cyclooxygenase-2 after treatment. These findings coincide with the recognized antiinflammatory properties of these compounds (54) and may contribute to overall functional improvement, given the detrimental implications of early inflammatory responses on brain injury outcome. However, as inflammation is now known to convey a time- and context-dependent dual role in the secondary cascade of molecular events after CNS injury, with both deleterious and beneficial implications on functional outcome, caution should be exercised when interpreting such findings. In particular, the limit of the timeframe during which suppression of inflammatory responses would be desirable should be considered when determining the appropriate dosing regimen for post-CNS injury HDAC inhibitor treatment (55,56).
ICH-induced inflammation has been shown to represent a key factor leading to secondary brain damage, suggesting that antiinflammatory approaches may be used to treat hemorrhagic stroke (57). Despite this understanding, data regarding the possible use of HDAC inhibitors for this indication remain scarce, with only one published report directly addressing the issue. In their series of experiments, Sinn et al. (58) used a rat model of ICH to examine the effects of postinsult VPA treatment. A dose of 300 mg/kg VPA was administered intraperitoneally twice a day after ICH induction. This was found to result in augmented functional recovery up to 4 wks after injury, alongside reduced hematoma expansion and perihematomal cell death (58). Similar to findings reported in ischemic stroke models, higher levels of acetylated histone H3 were observed after treatment compared with a control group. Additionally, the functional improvement was accompanied by increased expression of several effectors including the phosphorylated forms of extracellular signal-activated kinase (ERK) and its downstream target, cyclic adenosine monophosphate response element-binding (CREB), the neuroprotective factors HSP70 and pAkt and the antiapoptotic mediators Bcl-2 and Bcl-2 extra long (Bcl-xl). Concurrently, the expression of the pro-apoptotic factor Bcl-2-associated x protein (Bax) and caspase activities were reduced, suggesting attenuation of apoptotic cell death signals. The investigators further report on reduced mRNA levels of FAS ligand, interleukin-6, matrix metalloproteinase 9, chemokine (c-c motif) ligand (CCL)-4, CCL2 and tPA, yet the role of these changes in overall improvement remains to be further established. VPA also demonstrated considerable antiinflammatory effects, highlighted by attenuated inflammatory cell infiltration. Taken together, the results obtained using the ICH model indicate a protective role for VPA in hemorrhagic cerebral injury that is most likely mediated by several mechanisms that work in concert to yield the overall benefit. It is likely that these mechanisms include enhancement of neuroprotective factors, possibly via increased transcription, as well as an antiinflammatory effect. Altogether, the body of evidence regarding ischemic and hemorrhagic stroke supports the potential use of HDAC inhibitors in treating both forms of injury despite their divergent etiologies. Hence, HDAC inhibitor therapy may represent a common class of therapeutics with applications in both indications. Following the same line of thought, the pathophysiological similarities between stroke and TBI suggest that HDAC inhibitors may prove protective when administered in the setting of traumatic impact.
HDAC Inhibitors and Traumatic CNS Injury
CNS trauma and TBI in particular are a major health care problem worldwide. Approximately 1.5 million new TBI cases occur annually in the United States alone, with mortality rates ranging between 35–40% in severe patients (59). Despite the incidence of these injuries and their substantial socioeconomic implications, no specific pharmacological intervention is currently available for clinical use. Specifically, the efforts to develop new therapies for treating affected patients have repeatedly failed in translation from experimental lab data to the clinical setting. In light of this, identification of new therapeutic classes that may be applicable in TBI is of great importance. The accumulating data regarding the protective effects of HDAC inhibitors in stroke models has led to a growing interest in their potential usefulness in TBI. Although this issue has only been brought forth during the past couple of years, the available data indicate significant improvement in injury outcome and encourages further consideration of HDAC inhibitor therapy for treating traumatic CNS injuries.
It was initially reported that the HDAC inhibitor 4-dimethylamino-N-[5-(2-mercaptoacetylamino) pentyl] benzamide (DMA-PB), when given immediately after lateral fluid percussion, attenuates the postinjury decrease in acetylated histone H3 levels within hippocampal tissue (60). This observation was accompanied by decreased density of phagocytic microglia within the same anatomical area and a slight decrease in neurodegeneration that did not reach statistical significance. These findings suggested that HDAC inhibitors such as DMA-PB may inhibit TBI-induced inflammation. However, the interpretation of these results was greatly hindered by several shortcomings of the initial report. First, the overall functional value of HDAC therapy was not shown. Second, as the ability to effectively intervene in posttrauma pathological processes is limited by a narrow timeframe for efficacious drug administration and belated arrival of injured individuals to the hospital (61), the ability to administer HDAC inhibitors within a clinically feasible timeframe after TBI must be addressed in the scope of evaluating their potential clinical value. Finally, it should be advantageous to conduct additional study in another experimental model that is of high relevance to the human pathological setting.
Enhanced Functional Recovery and Attenuated Tissue Damage After Postinjury HDAC Inhibitor Treatment
We conducted a series of experiments using the HDAC inhibitor ITF2357 (53). These experiments examined the functional and molecular consequences of ITF2357 treatment in a well-established mouse model of closed head injury (CHI). ITF2357 was chosen as the candidate HDAC inhibitor in light of its potent pan-HDAC-inhibiting activity and favorable safety profile in humans (52,62). Mice subjected to CHI were treated with ITF2357 1 or 24 h after injury and displayed significant greater long-term recovery of neurological function compared with vehicle-treated mice. Moreover, ITF2357 significantly reduced the number of degenerating cells (evaluated by Fluoro Jade staining) on day 3 as well as day 21 after injury compared with control. Moreover, lesion volume (evaluated by 2,3,5-triphenyltetrazolium chloride [TTC]) was 22% smaller than in control (P ≤ 0.01) (53). Our results indicated that ITF2357 is beneficial in the setting of CHI, demonstrating for the first time the ability of postinjury HDAC inhibitor treatment to enhance functional, neurobehavioral outcome after brain trauma. Of particular importance was the sustainable benefit observed after a single administration of ITF2357 given 24 h after injury. As opposed to immediate postinsult administration, the interpretation of which is greatly limited by lack of clinical applicability, our data may be relevant to the treatment of TBI patients, since many arrive late to seek medical attention. It is noteworthy that a single dose of ITF2357 given at 24 h after injury was sufficient to induce improved functional recovery for over 1 week. Further study of even later time of administration or repeated postinjury ITF2357 dosing is warranted.
Similar to findings reported in ischemic stroke models (43,49), ITF2357 was found to reduce lesion volume after CHI. This result would indicate that postinjury administration of the compound not only facilitates improved functional recovery, but also reduces underlying tissue damage, a notion further corroborated by its ability to reduce the extent of neurodegeneration (53). The neuroprotective properties of ITF2357 were associated with its ability to restore adequate histone acetylation levels. Particularly, HDAC inhibitor treatment attenuated the robust decrease in the levels of acetylated histone H3 observed in nontreated injured animals. This effect confirmed the availability and expected activity of ITF2357, and, as the attenuated decrease of histone H3 preceded the reduction in lesion volume and improvement in functional performance, may well have contributed to these benefits. Interestingly, HDAC inhibitor administration also attenuated the decrease in the levels of HSP70 after injury but only slightly affected pAkt expression. It should be noted in this context that the effects of HDAC inhibitors on both HSP70 and pAkt appear to be dose and agent dependent. Faraco et al. (49) demonstrated that the extent of the effect of SAHA on HSP70 varies with different administered doses. Additionally, these investigators did not observe any effect of SAHA on pAkt, whereas a significant induction was reported using SB or TSA (43). As HSP70 and pAkt levels were evaluated at later times after CHI, the lack of any change in pAkt levels may reflect the presence of prominent apoptotic processes in the injured tissue within this timeframe (53,63,64). Given the well-established neuroprotective nature of HSP70, a role for maintenance of higher levels of this factor in HDAC-induced neuroprotection from brain trauma is plausible. Furthermore, the similarity in the findings obtained in stroke and trauma models may indicate the involvement of common protective mechanisms in neuroprotection, including increased expression of HSP70. The precise mechanism by which HSP70 is upregulated in vivo remains unknown; yet, in vitro experiments suggest the involvement of the phosphatidylinositol 3-kinase/Akt pathway and Sp1 acetylation in its induction (65).
HDAC inhibitors were shown to convey antiinflammatory effects in both stroke and trauma models—namely, a reduction in postinsult cyclooxygenase-2 levels and attenuated accumulation of phagocytic microglia (43,60). It was also shown that 4-PBA and SB inhibit inflammatory markers including nitric oxide, inducible nitric oxide synthase, interleukin-6 and tumor necrosis factor-α in vitro (46,66). After CHI, we demonstrated a reduction in microglia/macrophage accumulation within the injured tissue upon ITF2357 treatment (53). Importantly, microglia/macrophages at the area of trauma were apoptotic, suggesting accelerated clearance of these cells from the injured tissue. At the same time, the number of astrocytes and their level of activation were also reduced, indicating a global reduction in the amount of inflammatory cells present after injury. It remains to be determined whether reduced post-CHI gliosis after HDAC inhibitor treatment is the consequence of enhanced clearance of activated microglia/macrophages via apoptosis and whether it may involve additional mechanisms such as chemokine inhibition, as shown in models of inflammatory bowel disease and ICH (58,67,68).
Certainly, the induction of tumor cell death by HDAC inhibitors is the basis for their clinical use in human cancer patients (69,70). As a matter of fact, acetylation of p53 has been proposed as a mechanism for tumor cell death induced by TSA, SAHA and ITF2357 (71). After CHI, we observed an increase in total p53 levels within the trauma area after ITF2357 treatment. Given the presence of many glial cells within the tissue after injury, the involvement of p53 acetylation in microglial/macrophage apoptosis warrants examination and may well contribute to the antiinflammatory and functional effects of ITF2357.
Recently, the ability of VPA to provide neuroprotection from TBI and improve cognitive function was experimentally demonstrated. A set of studies using a rat brain trauma model was conducted in an effort to determine whether postinjury administration of valproate can decrease blood-brain barrier permeability, reduce neural damage and improve neurobehavioral outcome (72). A dose of 400 mg/kg valproate administered intraperitoneally increased histone acetylation and reduced the activity of glycogen synthase kinase 3 in the hippocampus. When 400 mg/kg valproate was administered 30 min after injury, it improved blood-brain barrier permeability. The same dose was also effective in reducing cortical contusion and hippocampal dendritic damage and, most importantly, improved motor function and spatial memory. The observed enhancement in memory function may be associated with previous observations by the same group in which it was demonstrated that SB administration is capable of improving learning and memory in brain-injured mice when given concurrently with behavioral testing (73). It should also be considered that HDAC inhibitors have previously been shown to augment memory and synaptic plasticity and promote neuronal outgrowth (74–76). This occurrence was suggested to be mediated, at least in part, via CREB-binding protein-dependent p53 acetylation (74).
VPA-induced neuroprotection was also demonstrated after optic nerve crush (77). Specifically, increased survival of retinal ganglion cells was observed after subcutaneous VPA administration given twice daily at 300 mg/kg. A similar effect was obtained with a single intra-vitreal injection of VPA immediately after injury. In addition, axonal regeneration in culture was enhanced in VPA-treated retinal ganglion cells compared with controls. Molecular changes included a decrease in caspase-3 activity, induction of CREB and phosphorylated extracellular signal-activated kinase (pERK) but no change in histone acetylation. However, the VPA doses used in this study are rather high and may even be toxic. Indeed, in a recent study, Matalon et al. (78) compared the ability of VPA and ITF2357 to induce the expression of HIV-1 in a dose-response study that included the plasma concentrations of each HDAC inhibitor achieved in humans. The effects of VPA were achieved in concentrations that are often toxic to humans (in the mmol/L range), whereas ITF2357 yielded a more pronounced effect at lower 10−5 concentrations. Thus, ITF2357 appears to be both a more promising and a safer candidate for clinical use as a neuroprotectant.
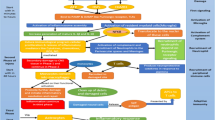
Taken together, the studies conducted using CNS trauma models establish a neuroprotective role for HDAC inhibitor treatment after traumatic impact. The reported neurobehavioral beneficial effects have been associated with changes in various molecular pathways and suggest the involvement of several mechanisms in overall protection, as summarized in Figure 1. The urgent requirement for identifying and testing new pharmacological entities for clinical use dictates the need for focused and efficient evaluation of the usefulness of HDAC-inhibiting compounds in the context of acute CNS injury. The main aspects warranting investigation are the unraveling of mechanisms that underlie HDAC inhibitor-mediated neuroprotection and, most importantly, resolution of issues concerning the safety and efficacy of this treatment.

Functional and molecular effects of HDAC inhibitor treatment in stroke and CNS trauma. Examples of common cell damage mechanisms shared by traumatic CNS impact and stroke include excitotoxicity, calcium overload, oxidative stress, acute inflammation and apoptosis. The discovery that relative over-deacetylation is a hallmark of neurodegenerative disorders led to the hypothesis that its correction will improve injury outcome. HDAC inhibitor administration was shown to improve neurobehavioral performance, decrease lesion volume, reduce BBB permeability and attenuate neurodegeneration. This step has been suggested to be mediated by multiple mechanisms leading to the restoration of acetylation homeostasis, most likely involving augmented acetylation of histones as well as nonhistone targets, as described in the text. Functional improvement was associated with a multitude of cellular and molecular changes affecting factors known to convey neuroprotective or antiapoptotic properties and with antiinflammatory effects including attenuated gliosis as well as reduced levels of inflammatory mediators. Ac-p53, acetylated p53; COX-2, cyclooxygenase-2; GSK-3, glycogen synthase kinase 3; iNOS, inducible nitric oxide synthase; MMP9, matrix metalloproteinase 9; TF, transcription factor; TNFα, tumor necrosis factor-α.
Considerations in Pharmacological Development
Studies conducted using various HDAC-inhibiting compounds reveal their effectiveness in promoting neuroprotection and improving postinjury outcome. However, the majority of these studies used agents that display pan-HADC-in-hibiting activity or, at the very best, partial class selectivity. In particular, the small carboxylate inhibitors used for evaluating the effects of HDAC inhibition in stroke models harbor HDAC class I selectivity in the case of VPA and are selective for both class I and class IIa HDACs in the case of SB and 4-PBA. Similarly, SAHA and TSA target both class I and class II HDACs. SAHA also targets class IV enzymes (51,52). The effects of HDAC inhibitors in CNS trauma have only been investigated using VPA, ITF2357, SB and MDA-PB, all of which are not selective toward a particular HDAC isoform (52,79). Hence, it remains unclear whether overall functional benefits result from multiple mechanisms activated concurrently or alternatively, whether functional improvement may be achieved by inhibiting individual HDAC isoforms. The ability to induce functional improvement by administering VPA indicates that increased histone acetylation contributes to neuroprotection. This result can be suggested since class I HDACs primarily target histones as substrates and are inhibited by VPA. However, because class II HDACs are capable of deacetylating nonhistone targets including transcription factors, their inhibition may well contribute to any increase in gene transcription occurring upon administration of a class I-class II combined inhibitor (SB, 4-PBA, SAHA and TSA). Furthermore, because each class of enzymes consists of several isoforms, it remains unclear whether a specific isoform can be implicated in facilitating a particular effect. It should also be noted that investigating the involvement of other classes of HDACs including HDAC IIb, III and IV in neuroprotection would require the design of inhibitors selective for these substrates. Although these issues are mechanistic in nature, they carry significant practical implications. Increasing the specificity of HDAC inhibitors is not only desirable for better defining underlying molecular mechanisms of action but, even more importantly, will reduce nonspecific side effects. Along these lines, the development of isoform-selective HDAC inhibitors has been pursued (80–84). The usefulness of selective HDAC inhibition in shedding new light on the role carried out by specific HDACs in the context of acute CNS injury was demonstrated in a recent study by Rivieccio et al. (85). In their report, these investigators show that selective HDAC6 inhibition protects against oxidative stress-induced neurodegeneration and promotes neurite growth in cortical neurons and dorsal root ganglion neurons (85). Another study indicated that mercaptoacetamide-based HDAC inhibitors may provide protection that is not accompanied by the toxicity displayed by hydroxamate-based compounds at high concentrations (79). This would indicate that selectively inhibiting particular HDACs may still convey protection without interfering with the activity of other isoforms. However, one should bear in mind that narrowing the range of inhibitory activity may also compromise the desired beneficial effect. This concern highlights the fragile balance between the need to identify therapeutics that achieve sustainable overall functional improvement and, at the same time, to minimize undesired side effects. Altogether, it would appear that the benefits of applying pan-HDAC inhibitors for acute CNS treatment currently outweigh their disadvantages in terms of selectivity. This result is suggested given the narrow therapeutic repertoire for treating acute CNS injury-affected patients and the already approved clinical use of HDAC inhibitors such as valproate and ITF2357 for a variety of indications and in ongoing clinical trials (39,40,62).
In addition to the issues outlined here, drugs administered for treating CNS disorders must also be able to efficiently penetrate the barriers separating this system from the periphery. Indeed, the ability of HDAC inhibitors to penetrate the blood-brain barrier (BBB) and the blood-retina barrier is well established by their reported neuroprotective effects (43,49,53). Yet, it should be noted that the degree of barrier penetration may well vary with time after injury. Because BBB permeability is modified by various mediators present after either stroke or trauma, including reactive oxygen species, inflammatory cytokines and other factors affecting endothelial cells (86–88), the effects introduced by a given dose of any drug should be established within a standardized timeframe, and the dynamic changes in BBB integrity must be considered when administering the drug systemically. Particular caution in determining the appropriate intervention schedule for HDAC inhibitor treatment is called for in light of its pronounced antiinflammatory effect. This step is warranted given that postinjury inflammatory responses within the CNS are known to convey both detrimental and beneficial roles in a time- and context-dependent manner (56,89–91). Hence, careful selection of the time at which an antiinflammatory agent is given after acute CNS injury is pivotal for assuring that it exerts a beneficial effect while not interfering with recovery processes. In regard to the potential practicality of applying HDAC inhibitors for treating acute CNS injury, findings demonstrating functional benefits obtained after administration as late as 24 h after injury and with repeated dosing are promising and encourage further pursuit of this new avenue of therapy (50,53).
Future Prospects
HDAC inhibitors represent a promising new class of therapeutics for acute CNS injury. As several of these compounds are already used clinically and reveal a favorable safety profile in humans, widening the range of indications for which they are used to include stroke and CNS trauma may provide an efficient and timely means of expanding the lacking range of therapeutic agents available for treating affected patients. Because optimization of the druglike properties and dosing regimen of candidate HDAC inhibitors will surely be required for successfully translating experimental data to the clinic, future research efforts and the development of novel HDAC inhibitors should focus on addressing these issues as well as unraveling particular mechanisms contributing to functional benefit.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Gallen CC. (2004) Strategic challenges in neurotherapeutic pharmaceutical development. NeuroRx. 1:165–80.
Leker RR, Shohami E, Constantini S. (2002) Experimental models of head trauma. Acta Neurochir. Suppl. 83:49–54.
Lipinski C, Hopkins A. (2004) Navigating chemical space for biology and medicine. Nature. 432:855–61.
Morganti-Kossmann MC, Yan E, Bye N. (2010) Animal models of traumatic brain injury: is there an optimal model to reproduce human brain injury in the laboratory? Injury. 41 Suppl 1:S10–3.
Bramlett HM, Dietrich WD. (2004) Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J. Cereb. Blood Flow Metab. 24:133–50.
Candelario-Jalil E. (2009) Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr. Opin. Investig. Drugs 10:644–54.
Benowitz LI, Carmichael ST. (2010) Promoting axonal rewiring to improve outcome after stroke. Neurobiol. Dis. 37:259–66.
De FP, Fellus J, Polito MZ, Thompson JW, Moser RS, DeLuca J. (2009) The new neuroscience frontier: promoting neuroplasticity and brain repair in traumatic brain injury. Clin. Neuropsychol. 23:1391–9.
Pape TL, Rosenow J, Lewis G. (2006) Transcranial magnetic stimulation: a possible treatment for TBI. J. Head Trauma Rehabil. 21:437–51.
Hahnen E, Hauke J, Trankle C, Eyupoglu IY, Wirth B, Blumcke I. (2008) Histone deacetylase inhibitors: possible implications for neurodegenerative disorders. Expert Opin. Investig. Drugs. 17:169–84.
Jin K, Mao XO, Simon RP, Greenberg DA. (2001) Cyclic AMP response element binding protein (CREB) and CREB binding protein (CBP) in global cerebral ischemia. J. Mol. Neurosci. 16:49–56.
Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL. (2003) Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 22:6537–49.
Camelo S, et al. (2005) Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 164:10–21.
Gardian G, et al. (2005) Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 280:556–63.
Ryu H, et al. (2005) Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 93:1087–98.
Thomas EA, et al. (2008) The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 105:15564–9.
Jenuwein T, Allis CD. (2001) Translating the histone code. Science. 293:1074–80.
Saha RN, Pahan K. (2006) HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 13:539–50.
Kouzarides T. (2007) Chromatin modifications and their function. Cell. 128:693–705.
Kruhlak MJ, et al. (2001) Regulation of global acetylation in mitosis through loss of histone acetyltransferases and deacetylases from chromatin. J. Biol. Chem. 276:38307–19.
Glozak MA, Sengupta N, Zhang X, Seto E. (2005) Acetylation and deacetylation of non-histone proteins. Gene. 363:15–23.
Hubbert C, et al. (2002) HDAC6 is a microtubule-associated deacetylase. Nature. 417:455–8.
Michan S, Sinclair D. (2007) Sirtuins in mammals: insights into their biological function. Biochem. J. 404:1–13.
Zhang Y, et al. (2003) HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 22:1168–79.
Kazantsev AG, Thompson LM. (2008) Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 7:854–68.
Hildmann C, Riester D, Schwienhorst A. (2007) Histone deacetylases: an important class of cellular regulators with a variety of functions. Appl. Microbiol. Biotechnol. 75:487–97.
Wen YD, et al. (2000) The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. U. S. A. 97:7202–7.
Yang WM, Tsai SC, Wen YD, Fejer G, Seto E. (2002) Functional domains of histone deacetylase-3. J. Biol. Chem. 277:9447–54.
Blander G, Guarente L. (2004) The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 73:417–35.
Gao L, Cueto MA, Asselbergs F, Atadja P. (2002) Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 277:25748–55.
Boutillier AL, Trinh E, Loeffler JP. (2003) Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J. Neurochem. 84:814–28.
Steffan JS, et al. (2000) The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. U. S. A. 97:6763–8.
Hockly E, et al. (2003) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A. 100:2041–6.
Avila AM, et al. (2007) Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 117:659–71.
Kilgore M, et al. (2010) Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 35:870–80.
Langley B, Gensert JM, Beal MF, Ratan RR. (2005) Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr. Drug Targets CNS Neurol. Disord. 4:41–50.
Lye TC, Shores EA. (2000) Traumatic brain injury as a risk factor for Alzheimer’s disease: a review. Neuropsychol. Rev. 10:115–29.
Roberts GW, et al. (1994) Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 57:419–25.
Atweh GF, et al. (1999) Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood. 93:1790–7.
Gerstner T, Bell N, Konig S. (2008) Oral valproic acid for epilepsy: long-term experience in therapy and side effects. Expert Opin. Pharmacother. 9:285–92.
Khan O, La Thangue NB. (2008) Drug insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat. Clin. Pract. Oncol. 5:714–26.
Langley B, Brochier C, Rivieccio MA. (2009) Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke. 40:2899–905.
Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. (2007) Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J. Pharmacol. Exp. Ther. 321:892–901.
Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. (2004) Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J. Neurochem. 89:1358–67.
Langley B, et al. (2008) Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21 (waf1/cip1) in cell cycle-independent neuroprotection. J. Neurosci. 28:163–76.
Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y. (2004) Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol. Pharmacol. 66:899–908.
Kim HJ, Leeds P, Chuang DM. (2009) The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J. Neurochem. 110:1226–40.
Endres M, et al. (2000) DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci. 20:3175–81.
Faraco G, et al. (2006) Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol. Pharmacol. 70:1876–84.
Yildirim F, et al. (2008) Inhibition of histone deacetylation protects wildtype but not gelsolindeficient mice from ischemic brain injury. Exp. Neurol. 210:531–42.
Bolden JE, Peart MJ, Johnstone RW. (2006) Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 5:769–84.
Khan N, et al. (2008) Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 409:581–9.
Shein NA, et al. (2009) Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 23:4266–75.
Dinarello CA. (2010) Anti-inflammatory agents: present and future. Cell. 140:935–50.
Emsley HC, Tyrrell PJ. (2002) Inflammation and infection in clinical stroke. J. Cereb. Blood Flow Metab. 22:1399–419.
Shohami E, Ginis I, Hallenbeck JM. (1999) Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 10:119–30.
Aronowski J, Hall CE. (2005) New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol. Res. 27:268–79.
Sinn DI, et al. (2007) Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol. Dis. 26:464–72.
Beauchamp K, Mutlak H, Smith WR, Shohami E, Stahel PF. (2008) Pharmacology of traumatic brain injury: where is the “golden bullet”? Mol. Med. 14:731–40.
Zhang B, et al. (2008) HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 1226:181–91.
Moppett IK. (2007) Traumatic brain injury: assessment, resuscitation and early management. Br. J. Anaesth. 99:18–31.
Vojinovic J, et al. (2011) Safety and efficacy of an oral histone deacetylase inhibitor in systemiconset juvenile idiopathic arthritis. Arthritis Rheum. 63:1452–8.
Grosjean MB, et al. (2007) Immunohistochemical characterization of Fas (CD95) and Fas Ligand (FasL/CD95L) expression in the injured brain: relationship with neuronal cell death and inflammatory mediators. Histol. Histopathol. 22:235–50.
Yatsiv I, et al. (2005) Erythropoietin is neuroprotective, improves functional recovery, and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. FASEB J. 19:1701–3.
Marinova Z, et al. (2009) Valproic acid induces functional heat-shock protein 70 via class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J. Neurochem. 111:976–87.
Huuskonen J, Suuronen T, Nuutinen T, Kyrylenko S, Salminen A. (2004) Regulation of microglial inflammatory response by sodium butyrate and short-chain fatty acids. Br. J. Pharmacol. 141:874–80.
Glauben R, et al. (2006) Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J. Immunol. 176:5015–22.
Glauben R, et al. (2008) Histone deacetylases: novel targets for prevention of colitis-associated cancer in mice. Gut. 57:613–22.
Armeanu S, et al. (2005) Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and ITF2357. J. Hepatol. 42:210–17.
Barbetti V, et al. (2008) Selective anti-leukaemic activity of low-dose histone deacetylase inhibitor ITF2357 on AML1/ETO-positive cells. Oncogene. 27:1767–78.
Carlisi D, et al. (2008) Histone deacetylase inhibitors induce in human hepatoma HepG2 cells acetylation of p53 and histones in correlation with apoptotic effects. Int. J. Oncol. 32:177–84.
Dash PK, et al. (2010) Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS One. 5:e11383.
Dash PK, Orsi SA, Moore AN. (2009) Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience. 163:1–8.
Gaub P, et al. (2010) HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 17:1392–1408.
Levenson JM, Sweatt JD. (2005) Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 6:108–18.
Vecsey CG, et al. (2007) Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J. Neurosci. 27:6128–40.
Biermann J, et al. (2010) Valproic acid-mediated neuroprotection and regeneration in injured retinal ganglion cells. Invest. Ophthalmol. Vis. Sci. 51:526–34.
Matalon S, et al. (2010) The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J. Acquir. Immune Defic. Syndr. 54:1–9.
Kozikowski AP, et al. (2007) Functional differences in epigenetic modulators: superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J. Med. Chem. 50:3054–61.
Balasubramanian S, Verner E, Buggy JJ. (2009) Isoform-specific histone deacetylase inhibitors: the next step? Cancer Lett. 280:211–21.
Bertrand P. (2010) Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 45:2095–116.
Bieliauskas AV, Pflum MK. (2008) Isoformselective histone deacetylase inhibitors. Chem. Soc. Rev. 37:1402–13.
Butler KV, Kozikowski AP. (2008) Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr. Pharm. Des. 14:505–28.
Thomas EA. (2009) Focal nature of neurological disorders necessitates isotype-selective histone deacetylase (HDAC) inhibitors. Mol. Neurobiol. 40:33–45.
Rivieccio MA, et al. (2009) HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. U. S. A. 106:19599–604.
Sandoval KE, Witt KA. (2008) Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 32:200–19.
Shapira Y, Setton D, Artru AA, Shohami E. (1993) Blood-brain barrier permeability, cerebral edema, and neurologic function after closed head injury in rats. Anesth. Analg. 77:141–8.
Witt KA, Mark KS, Sandoval KE, Davis TP. (2008) Reoxygenation stress on blood-brain barrier paracellular permeability and edema in the rat. Microvasc. Res. 75:91–6.
Morganti-Kossman MC, et al. (1997) Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol. Psychiatry. 2:133–6.
Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. (2002) Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr. Opin. Crit. Care 8:101–5.
Scherbel U, et al. (1999) Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc. Natl. Acad. Sci. U. S. A. 96:8721–6.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shein, N.A., Shohami, E. Histone Deacetylase Inhibitors as Therapeutic Agents for Acute Central Nervous System Injuries. Mol Med 17, 448–456 (2011). https://doi.org/10.2119/molmed.2011.00038
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00038