
Abstract
A defining feature of multiple sclerosis (MS) is the occurrence of clinical relapses separated by periods of clinical stability. Better understanding of the events underlying clinical relapse might suggest new approaches to treatment. The objective of this study was to measure changes in the expression of RNA in the blood during relapse. We used microarrays to measure mRNA expression in paired samples from 14 MS patients during clinical relapse and while stable. Seventy-one transcripts changed expression at the P< 0.001 significance level. The most notable finding was decreased expression of transcripts with regulatory function, expressed primarily in non-T cells. These decreased transcripts included the interleukin-1 receptor antagonist, which had a corresponding decrease in the protein concentration in serum. Transcripts with increased expression were expressed primarily in T cells. Pathways analysis suggested involvement of the cytokine network, coagulation and complement cascades, IL-10 signaling and NF-κB signaling. We conclude that there are alterations of mRNA expression in both T cells and non-T cells during MS relapse.
Similar content being viewed by others

Introduction
Multiple sclerosis (MS) is an inflammatory and demyelinating disease of the central nervous system (CNS), which is thought to be immune-mediated (1). One of the characteristic features of MS is the fluctuating disease activity, with clinical relapses separated by remissions. The immunologic events leading to relapse are not well defined, and better understanding of these events might suggest new treatment strategies.
Events occurring inside the CNS are difficult to study, but relapses likely are triggered by events outside the CNS. Many relapses follow minor infections, such as viral upper respiratory infections (2,3). Treatments which interfere with the migration of lymphocytes into the CNS greatly reduce the number of relapses (4). Thus, changes in gene expression in peripheral blood leukocytes might clarify the mechanism of MS relapses. Previous studies using microarray technology to measure gene expression changes in MS relapse had interesting results, but are limited by multiple factors, including the available microarrays, the use of unpaired samples, small sample sizes, and possible alteration of gene expression during sample preparation (5–8). We have extended these findings using a larger population of subjects with paired relapse and stable samples from each, and comprehensive measurements of mRNA expression.
Materials and Methods
Study Subjects
Heparinized blood samples were obtained from 14 patients with relapsing-remitting MS during an acute exacerbation and while clinically stable. The relapse specimen was obtained within the first wk of a clinical relapse, and all relapse specimens were obtained before initiation of corticosteroid treatment. The patients were evaluated by a single experienced neurologist (JW Lindsey), and relapses were defined by the occurrence of new symptoms and physical findings (nine subjects) or the worsening of existing symptoms (five subjects) with a duration of at least 48 h in the absence of infection or other illness. The symptoms and deficits defining the relapse were typical for MS, and included hemiparesis, paraparesis, sensory loss in various distributions, unilateral ataxia, and myelopathy. The stable specimens were obtained at least 1 month before or 3 months after a clinical relapse. Six subjects had the stable sample collected first, and eight had the relapse sample collected first. The mean interval between the two samples was 307 d (range 63 to 672 d). Nine patients were female, and five were male. Six patients were on interferon at both times, two were on no disease-modifying treatment, four were on glatiramer acetate at both times, and two were untreated at time of relapse and on glatiramer when the stable specimen was collected. Although the patients were on different treatments, the paired design of the study should minimize the known effects of treatment on RNA expression. The inclusion of the two patients whose treatment changed did not alter the results significantly. Serum specimens were collected from 20 patients in relapse and while stable, including 13 of the 14 patients in the microarray experiment. Specimen collection was approved by the Committee for the Protection of Human Subjects at the University of Texas Health Science Center at Houston, and all subjects signed informed consent before blood collection.
T-Cell and Non-T-Cell Isolation
Peripheral blood mononuclear cells (PBMC) from patients were isolated with Ficoll density gradients, lysed in Trizol (Invitrogen, Carlsbad, CA, USA), and stored frozen at −80°C, and total RNA was extracted using combined Trizol and RNeasy spin columns (Qiagen, Valencia, CA, USA). For the comparison of T and non-T cells, two buffy coats were obtained from the Gulf Coast Regional Blood Center and PBMC were isolated with Ficoll. T cells were isolated with positive selection with mouse monoclonal anti-CD4 and CD8 antibodies (Southern Biotech, Birmingham, AL, USA) and Dynal antimouse IgG magnetic beads (Invitrogen). The positively selected cells were lysed with Trizol while still bound to the magnetic beads. The unbound cells also were lysed in Trizol as non-T cells. Serum was separated from cells and stored at −80°C.
RNA Expression
RNA expression was profiled on Illumina (San Diego, CA, USA) beadchip humanRef-8 v3.0 microarrays at the University of Texas-Houston Quantitative Genomics Core Laboratory. Two hundred ng of total RNA were amplified and purified using Illumina TotalPrep RNA Amplification Kit (Ambion, Austin, TX, USA) following kit instructions. Amplified cRNA was subsequently purified and concentration was measured by NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, DE, USA). An aliquot of 750 ng was loaded onto the microarray, hybridized at 58°C in an Illumina Hybridization Oven for 17 h, washed and incubated with streptavidin-Cy3 to detect the biotin-labeled cRNA. Arrays were dried and scanned with the Illumina BeadArray Reader. Each chip contains eight separate microarrays, and the samples were run on five separate chips in five different experiments with the stable and relapse specimen for each patient on the same chip. Data were extracted, background subtracted, and normalized by quantiles using Illumina BeadStudio 3.2, and then exported to a worksheet. These files for all experiments were combined in a single worksheet, and data for non-expressed transcripts were removed. The edited worksheet was then analyzed using BRB-ArrayTools 3.8.0 and Ingenuity (version 8.0). We identified genes that were differentially expressed between the two classes using the Class Comparison tool in BRB which utilizes a random-variance t test without correction for multiple testing but with calculation of the false discovery rate (9).
ELISA
Enzyme-linked immunosorbent assay (ELISA) for C1Q was performed as described using commercially available antibodies (Quidel, San Diego, CA, USA) (10), except the incubation with serum was decreased from overnight to 2 h. ELISA for interleukin-16 (IL-16) and interleukin-1 receptor antagonist (IL-1RA) were performed with commercial ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Web Deposition of Data
The microarray data have been deposited in the Gene Expression Omnibus, https://doi.org/www.ncbi.nlm.nih.gov/geo/, accession number GSE19224.
Results
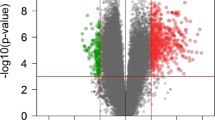
RNA expression of PBMC in relapse was compared with the corresponding stable sample for each of the 14 subjects using the paired class comparison tool in BRB. There were 14,421 expressed transcripts, and 71 of them changed expression in relapse at the P < 0.001 significance level, 46 increased and 25 decreased (Table 1). Since the P values are not corrected for multiple comparisons, we would expect that about 14 of these 71 transcripts are false positives. There were 530 transcripts which were significantly changed at the P < 0.01 significance level. The differences in expression between relapse and stable were typically modest. On initial inspection of the results, it appeared that most of the transcripts which decreased in relapse are expressed in antigen presenting cells. To further investigate this, we measured RNA expression in T cells and non-T cells from two normal controls and calculated the ratio of expression in non-T cells compared with T cells for each transcript. Nearly all the transcripts which decreased in relapse are expressed more highly in non-T cells (Figure 1A). We considered the possibility that this is due to a decrease in the number of non-T cells in the blood in relapse. But transcripts known to be expressed mainly in non-T cells such as CD11b and CD14 were not decreased significantly, and the 100 transcripts expressed most highly in non-T cells are not decreased consistently in relapse (Figure 1B). We verified our classification of transcripts as being expressed in non-T or T cells by comparison with the human data available on the Immunological Genome Project website (https://doi.org/www.imgen.org/). Of the 22 transcripts we classified as increased in non-T cells, 15 were clearly increased in non-T cells, 2 were equivocal, and 5 were not found in the database.

Changes in expression in T versus non-T cells. (A) The ratio of relapse versus stable and non-T versus T expression for the 71 transcripts with significance of P< 0.001. The ratio of relapse to stable is on the y axis on a linear scale, the ratio of non-T to T is on the xaxis on a logarithmic scale. (B) Ratio of relapse to stable versus non-T to T for the 100 transcripts with the highest bias to expression in non-T cells. There is no general decrease in transcripts expressed in non-T cells.
We investigated whether particular pathways were altered in relapse using both the pathways analysis tools in BRB and Ingenuity (Figure 2, Table 2). Several pathways were identified as having significant alterations. In general, in each pathway there were only a handful of significantly altered transcripts. For example, in the BioCarta cytokine network pathway, the changes significant at the P < 0.05 level were increased expression of mRNA for IL-16 and lymphotoxin-α and decreased expression of interferon-γ, IL-6 and IL-15. The prion pathway identified in both BioCarta and KEGG analysis has decreased expression of BCL2 and the prion protein as the only two changes significant at P < 0.05. We also analyzed the changes in expression by gene ontology. The most significant alterations were for the “immune system process” and “immune response” terms. The most significant changes included decreases in the interleukin 1 family transcript IL1F7 and decreases in multiple members of the leukocyte immunoglobulin-like receptor family.

Ingenuity pathways. Bar denotes statistical significance, squares indicate the fraction of transcripts in that pathway that are changed.
In unsupervised hierarchical clustering based on the 14,421 expressed transcripts, the samples clustered based on the experiment and subject (Figure 3). There were three large clusters, and two of the clusters were formed by the samples from a single experiment. For 11 of the 14 subjects, the nearest neighbor was the paired sample from that subject. There was no separation based on disease status (relapse versus stable), disease modifying treatment, or gender.

Hierarchical clustering. Dendrogram constructed using BRB ArrayTools with centered correlation and average linkage. The branch labels give the experiment number (1–5), the subject number (1–14), clinical status, treatment, and gender. The samples from the first two experiments formed large clusters. For 11 of the subjects, the nearest neighbor for the stable sample is the corresponding relapse sample from that subject. There was no tendency to cluster by clinical status, treatment, or gender. S, stable; R, relapse; I, interferon; G, glatiramer; N, no treatment; m, male; f, female.
We investigated whether the changes in mRNA expression correlated with changes in protein expression in serum for three of the transcripts. The decrease in IL-1 receptor antagonist mRNA was matched by a corresponding decrease in the amount of protein in the serum. In stable disease, the concentration of IL-1RA was 537 ± 414 pg/mL, with a mean decrease of 96 ± 178 pg/mL in relapse (P = 0.030, paired t test, n = 19, mean ± standard deviation). There was a modest correlation between the change in expression of protein on ELISA and change in mRNA on microarray (r = 0.39, n = 13). In contrast, the amount of IL-16 protein was not increased in relapse. The concentration of IL-16 in the stable samples was 197 ± 72 pg/mL, with a mean decrease of 4 ± 75 pg/mL in relapse (P not significant, n = 19). One of the most significantly decreased transcripts was the complement protein C1QB. C1QC also was decreased, but with a lower significance level. The C1Q protein is a complex of C1QA, C1QB and C1QC, and is synthesized in macrophages or dendritic cells and secreted into the serum (11). The amount of C1Q in stable disease was 78.7 ± 4.3 µg/mL with an increase of 0.5 ± 6.8 in relapse (not significant, n = 20).
Discussion
We have compared gene expression in relapse and remission in paired samples from 14 MS patients using microarrays which cover the known expressed mRNA in the human genome. We found significant changes in expression of several mRNA, which may clarify the nature of the disease process in MS. Several features of these results deserve comment.
First, the most salient finding is that there were changes in both T cells and non-T cells. The changes in non-T cells were primarily decreases in selected transcripts. Not all non-T transcripts were decreased, so this does not appear to be due to migration of these cells out of the blood in relapse, but rather to a change in the expressed genes. Where their function is known, many of the decreased transcripts have a regulatory or inhibitory role. CDKN1C and ZAK are both likely inhibitors of cell division, IL1RN inhibits the activity of interleukin-1, and PILRA has a tyrosine-based inhibitory motif and regulates the protein tyrosine phosphatase SHP-1, which is central in the control of cell signaling and may be altered in MS (12). The downregulation of mRNA for inhibitory proteins suggests that the non-T cells are becoming activated. Further study is needed to define the types of non-T cell involved.
In contrast, mRNA differentially expressed in T cells were increased predominantly in expression. The ones with the most significant changes in expression are involved in metabolic processes or are of unknown function. The most significant change in a cytokine mRNA is the increase in IL16, which is known to be chemotactic for cells bearing CD4. Other cytokine mRNA changes are decreased IFNG, IL6, and IL15.
Second, the changes in expression are modest in size, in the range of 70% to 130% of the baseline values. There are several potential explanations for this. It is probable that only a fraction of circulating PBMC are involved in the immunologic events causing a clinical relapse. Also, the disease process in MS is more continuously active than the clinical symptoms indicate, so many of our stable samples probably have similar changes. Finally, we used PBMC, which contain a variety of different cell types, so changes in mRNA expression in a single cell type may be obscured by expression of the same mRNA in other cell types.
Third, the changes in mRNA expression did not necessarily predict changes in protein concentration in serum. Multiple factors could contribute to this, including post-transcriptional regulation, rates of protein secretion into serum, the half-life of the protein in serum, and cell types other than PBMC secreting particular proteins. The microarray results are better interpreted as an indicator of the internal state of the PBMC rather than the external milieu in the serum.
There have been several previous studies of expression changes in relapse, and we compared transcripts reported to be altered in those studies with our set of 530 transcripts altered at a significance level of 0.01. The concordance between our results and previous studies is limited. This is not unexpected given differences in methods. The majority of previous studies compared small numbers of patients in relapse to a different group of stable patients. The large variability of gene expression between individuals seen in this study (Figure 3) suggests that any changes due to relapse are likely to be obscured. Only two studies used paired samples, and both of those studies tested only T cells positively selected by anti-CD3 antibody. This approach discards the information from non-T cells, which proved to be interesting in our study. It also might cause changes in mRNA expression in the T cells due to crosslinking of CD3, with unknown effects on the results. In this work, we isolated the PBMC and stabilized RNA as quickly as possible to minimize ex vivo changes in mRNA expression.
The most directly comparable study is that of Satoh et al. (8). This study included only six subjects with paired samples, used a relatively small microarray with 1,258 selected transcripts, and positively selected T cells with anti-CD3 antibody. They identified 43 differentially expressed genes, none of which were altered in this study. Achiron et al. have reported two studies, which compared unpaired samples, some from patients in relapse and others from different patients in remission (5,13). We replicated their findings that BCL2 is decreased and BNIP3 is increased. More recent work from the same group reports that expression of a small set of mRNA can be used to predict relapse (14), but the transcripts they used as predictors were not altered in our patients. Arthur et al. also reported a comparison of unpaired relapse and remission patients (6), but comparison with their results is difficult since they compared relapse to controls and remission to controls rather than directly comparing relapse and remission. They found decreased CDKN1C in both relapse and remission, and an increase in OGT in relapse, but not remission. Malmestrom et al. compared expression in 10 subjects with paired relapse and remission samples (7), but they used anti-CD3 antibody to positively select T cells, and their reported results cover only a limited range of transcripts and proteins in CD8 cells with no overlap with our findings. Finally, we found no overlap with the findings of Brynedal et al., who compared expression from unpaired blood samples from 10 stable patients and 14 patients in relapse (15). The poor agreement of our study and previous work is likely due to differences in microarrays, differences in the cell types used as the source of mRNA, the use of paired rather than unpaired samples, the limited results reported from some studies, and small sample sizes leading to false positive results.
The concordance of our pathway analysis with previous studies is likewise limited. We found that the NF-κB pathway was changed in agreement with Satoh et al. (8). We found changes in some transcripts involved in apoptosis as discussed above, but we did not find significant alterations in apoptotic pathways as reported by Achiron et al. and Arthur et al. (6,13).
We found better concordance with studies comparing MS patients and controls (Table 3). It is reasonable to expect that some of the mRNA changes that distinguish MS patients from controls might also distinguish MS relapses from remissions. Indeed, we found concordant changes in many transcripts which had previously been reported to be changed in MS (16–19). Of particular interest is the large number of concordant findings with the study of Corvol et al. (17). This is somewhat unexpected, since they studied gene expression in negatively selected CD4 T cells in patients with clinically isolated syndrome. This suggests that transcripts altered early in disease also change in relapse. Since their CD4 cells were 95% pure, and they still measured changes in transcripts expressed primarily in non-T cells, this raises the question of whether they are finding changes in the residual non-T cells or if these transcripts also are altered in CD4 cells. In addition to gene expression studies, we compared our results to the findings of genome-wide association studies in MS (20,21). Three of the loci identified in recent studies, IL7R, PTGER4 and TYK2, had changes in expression during relapse.
The three proteins we studied all have potential relevance to MS. C1Q has immune regulatory properties and decreases in C1Q are associated with autoimmunity (11). Although we could not demonstrate a change in serum C1Q in our 19 patients, others have measured a decrease in C1Q and other complement components in a larger group of MS patients in relapse (22). IL-16 has been implicated in autoimmune diseases (23), including MS and animal models of MS (24,25), and IL-16 is downregulated during interferon treatment of MS (26). IL-1RA has antiinflammatory activity, and decreases in IL-1RA would be expected to cause inflammation. The role of IL-1RA genotype as a risk factor for MS is controversial (27), but IL-1RA protein is increased during interferon or glatiramer treatment (28,29). The only previous study measuring IL-1RA protein in relapse found it was increased rather than decreased (30), but they compared IL-1RA levels in eight relapsing patients to a different group of stable patients.
One consideration in interpreting these results is that we tested patients only at two time points—the onset of clinical symptoms and while clinically stable. The hypothesized immune activation in the periphery should occur before inflammation in the CNS. By the time symptoms appear, the relevant changes in the peripheral blood may have declined or even resolved. One prospective study with blood samples done every 2 to 4 weeks found that stimulated IFN-γ production in vitro peaked 1 to 2 weeks before clinical symptoms, and continued to decline after the onset of symptoms (31). This is consistent with our observation of decreased IFNG mRNA at the time of onset of symptoms. Some of the changes measured in blood at the onset of symptoms may reflect the resolution of recent immune activation.
In conclusion, the observed changes in gene expression in PBMC with MS relapse suggest an activation of non-T cells and changes in function in T cells. Further investigation of the events occurring with MS relapse should be pursued. The optimum study would include more subjects, perform serial sampling of patients at frequent time intervals, use MRI rather than clinical relapse as the measure of MS disease activity, include a white cell differential to document the proportion of monocytes and lymphocytes and study more homogeneous populations of leukocytes.
Disclosure
We declare that the authors have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. (2000) Multiple sclerosis. N. Engl. J. Med. 343:938–52.
Sibley WA, Bamford CR, Clark K. (1985) Clinical viral infections and multiple sclerosis. Lancet. 1:1313–5.
Buljevac D, et al. (2002) Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain. 125:952–60.
Polman CH, et al. (2006) A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 354:899–910.
Achiron A, Gurevich M, Friedman N, Kaminski N, Mandel M. (2004) Blood transcriptional signatures of multiple sclerosis: unique gene expression of disease activity. Ann. Neurol. 55:410–7.
Arthur AT, et al. (2008) Genes implicated in multiple sclerosis pathogenesis from consilience of genotyping and expression profiles in relapse and remission. BMC Med. Genet. 9:17.
Malmestrom C, et al. (2008) Relapses in multiple sclerosis are associated with increased CD8(+) T-cell mediated cytotoxicity in CSF. J. Neuroimmunol 196:159–65.
Satoh J, Misawa T, Tabunoki H, Yamamura T. (2008) Molecular network analysis of T-cell transcriptome suggests aberrant regulation of gene expression by NF-kappaB as a biomarker for relapse of multiple sclerosis. Dis. Markers. 25:27–35.
Wright GW, Simon RM. (2003) A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 19:2448–55.
Dillon SP, D’Souza A, Kurien BT, Scofield RH. (2009) Systemic lupus erythematosus and C1q: A quantitative ELISA for determining C1q levels in serum. Biotechnol. J. 4:1210–4.
Lu JH, et al. (2008) The classical and regulatory functions of C1q in immunity and autoimmunity. Cell. Mol. Immunol. 5:9–21.
Christophi GP, et al. (2009) Macrophages of multiple sclerosis patients display deficient SHP-1 expression and enhanced inflammatory phenotype. Lab. Invest. 89:742–59.
Achiron A, Feldman A, Mandel M, Gurevich M. (2007) Impaired expression of peripheral blood apoptotic-related gene transcripts in acute multiple sclerosis relapse. Ann. N. Y. Acad. Sci. 1107:155–67.
Gurevich M, Tuller T, Rubinstein U, Or-Bach R, Achiron A. (2009) Prediction of acute multiple sclerosis relapses by transcription levels of peripheral blood cells. BMC Med. Genomics. 2:46.
Brynedal B, et al. (2009) Gene expression profiling in multiple sclerosis: A disease of the central nervous system, but with relapses triggered in the periphery? Neurobiol. Dis. 37:613–21.
Ramanathan M, et al. (2001) In vivo gene expression revealed by cDNA arrays: the pattern in relapsing-remitting multiple sclerosis patients compared with normal subjects. J. Neuroimmunol. 116:213–9.
Corvol JC, et al. (2008) Abrogation of T cell quiescence characterizes patients at high risk for multiple sclerosis after the initial neurological event. Proc. Natl. Acad. Sci. U. S. A. 105:11839–44.
Bomprezzi R, et al. (2003) Gene expression profile in multiple sclerosis patients and healthy controls: identifying pathways relevant to disease. Hum. Mol. Genet. 12:2191–9.
Iglesias AH, et al. (2004) Microarray detection of E2F pathway activation and other targets in multiple sclerosis peripheral blood mononuclear cells. J. Neuroimmunol. 150:163–77.
De Jager PL, et al. (2009) Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 41:776–82.
(2009) Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet. 41:824–8.
Cojocaru M, Serbanescu A, Cojocaru IM. (1993) Changes of serum complement and of circulating immune complexes in patients with multiple sclerosis. Rom. J. Intern. Med. 31:131–7.
Glass WG, Sarisky RT, Vecchio AM. (2006) Not-so-sweet sixteen: the role of IL-16 in infectious and immune-mediated inflammatory diseases. J. Interferon Cytokine Res. 26:511–20.
Skundric DS, Cai J, Cruikshank WW, Gveric D. (2006) Production of IL-16 correlates with CD4+Th1 inflammation and phosphorylation of axonal cytoskeleton in multiple sclerosis lesions. J. Neuroinflammation. 3:13.
Skundric DS, Zhou W, Cruikshank WW, Dai R. (2005) Increased levels of bioactive IL-16 correlate with disease activity during relapsing experimental autoimmune encephalomyelitis (EAE). J. Autoimmun. 25:206–14.
Annibali V, et al. (2007) Gene expression profiles reveal homeostatic dynamics during interferonbeta therapy in multiple sclerosis. Autoimmunity. 40:16–22.
Feakes R, et al. (2000) Interleukin 1 receptor antagonist (IL-1ra) in multiple sclerosis. J. Neuroimmunol. 105:96–101.
Comabella M, et al. (2008) Induction of serum soluble tumor necrosis factor receptor II (sTNF-RII) and interleukin-1 receptor antagonist (IL-1ra) by interferon beta-1b in patients with progressive multiple sclerosis. J. Neurol. 255:1136–41.
Burger D, et al. (2009) Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1beta in human monocytes and multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A. 106:4355–9.
Nicoletti F, et al. (1996) Circulating serum levels of IL-1ra in patients with relapsing remitting multiple sclerosis are normal during remission phases but significantly increased either during exacerbations or in response to IFN-beta treatment. Cytokine. 8:395–400.
Dettke M, Scheidt P, Prange H, Kirchner H. (1997) Correlation between interferon production and clinical disease activity in patients with multiple sclerosis. J. Clin. Immunol. 17:293–300.
Acknowledgments
We thank Landon Hatfield, Uffaf Khan, and the University of Texas-Houston Microarray Core Facility for technical assistance. This work was funded in part by the Clayton Foundation for Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lindsey, J.W., Agarwal, S.K. & Tan, F.K. Gene Expression Changes in Multiple Sclerosis Relapse Suggest Activation of T and Non-T Cells. Mol Med 17, 95–102 (2011). https://doi.org/10.2119/molmed.2010.00071
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2010.00071