
Abstract
Gender dimorphisms exist in the pathogenesis of a variety of cardiovascular, cardiopulmonary, neurodegenerative, and endocrine disorders. Estrogens exert immense influence on myocardial remodeling following ischemic insult, partially through paracrine growth hormone production by bone marrow mesenchymal stem cells (MSCs) and endothelial progenitor cells. Estrogens also facilitate the mobilization of endothelial progenitor cells to the ischemic myocardium and enhance neovascularization at the ischemic border zone. Moreover, estrogens limit pathological myocardial remodeling through the inhibitory effects on the proliferation of the cardiac fibroblasts. Androgens also may stimulate endothelial progenitor cell migration from the bone marrow, yet the larger role of androgens in disease pathogenesis is not well characterized. The beneficial effects of sex steroids include alteration of lipid metabolism in preadipocytes, modulation of bone metabolism and skeletal maturation, and prevention of osteoporosis through their effects on osteogenic precursors. In an example of sex steroid-specific effects, neural stem cells exhibit enhanced proliferation in response to estrogens, whereas androgens mediate inhibitory effects on their proliferation. Although stem cells can offer significant therapeutic benefits in various cardiovascular, neurodegenerative, endocrine disorders, and disorders of bone metabolism, a greater understanding of sex hormones on diverse stem cell populations is required to improve their ultimate clinical efficacy. In this review, we focus on the effects of estrogen and testosterone on various stem and progenitor cell types, and their relevant intracellular mechanisms.
Similar content being viewed by others

Introduction
Gender differences, represented in large measure by the differential effects of sex-specific hormones, exist in a variety of cardiovascular,(1–4) cardiopulmonary, (5,6) neurodegenerative, (7–9) endocrine (10) and metabolic bone diseases such as osteoporosis (11–13). Indeed, the role of sex steroids in cardiovascular disease has been studied extensively (14–27). Clinically, female patients show relative cardiac protection from acute infarctions and better outcome following myocardial infarction compared with males (21). Such gender dimorphisms may be due to the beneficial effects of estrogens or to the absence of the deleterious effects of androgens (28,29). Differences in estrogen receptor (ER) signaling also may play a significant role in outcome following cardiovascular diseases (21,30–32). In addition, gender differences in proinflam-matory signaling and immune responses have been described (33,34). Elegant work by Chaudry (4) and other investigators demonstrate that alteration of immune function by sex steroids can lead to therapeutic interventions and improved outcomes. A better understanding of sex hormone regulation from a cell biology perspective will be critical therefore in improving patient outcomes.
Stem cell transplantation has revolutionized the treatment of hematological disorders such as myelodysplastic syndrome and acute myeloid leukemia (35,36). In recent years, stem cell therapy has been used to improve postmyocardial infarction, ventricular repair, and remodeling mechanisms (37). In this context, stem cell therapy may be associated with better functional recovery of the infarcted ventricles in treated patients (38–40).
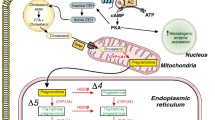
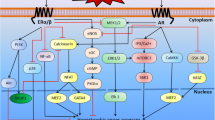
Recent studies reported the presence of estrogen and testosterone receptors on stem cells (41–43), suggesting that estrogen and testosterone may modify the function of those cells (18,19,44,45) (Figures 1 and 2). 17β-estradiol enhances the proliferation and migration of endothelial progenitor cells (EPCs) to the injured vessels, or ischemic myocardial tissues, which, through the process of homing, help in repair and regeneration to compensate for the lost tissue. It also has been postulated that vascular endothelial growth factor (VEGF) might be responsible for EPC migration in response to 17β-estradiol (44). On the contrary, the role of androgens on stem cells is controversial, but most studies suggest that androgens have inhibitory effects on stem cell functions, and anti-androgens could negate these effects (41). Thus, modification of the function of stem cells through estrogenic or anti-androgenic stimulus may help formulate “super stem cells” with better therapeutic efficacy. To achieve this goal, the role of sex hormones on stem cell function must be elucidated. It is the purpose of this review to summarize the current knowledge of the effects of estrogens and androgens on various stem cell populations.

Effects of estrogen on various stem cells and progenitor cells. ESC (embryonic stem cell); EPC (endothelial progenitor cell); MSC (mesenchymal stem cell); HSC (hematopoietic stem cell); CF (cardiac fibroblast); BMP (bone matrix protein); RUNX2/ CBFA1 (runt-related transcription factor 2/core-binding factor alpha).

Effects of androgens on various stem cells and progenitor cells. ESC (embryonic stem cell); EPC (endothelial progenitor cell); HSC (hematopoietic stem cell); NSC (neural stem cell); OPC (osteogenic progenitor cell).
Embryonic Stem Cells
The expression of estrogen receptors ER-α and ER-β in embryoid bodies takes place as early as d 2, suggesting the influence of estrogen on the differentiation and proliferation of human embryonic stem cells (ESCs) and embryoid bodies (43). The effects of estrogens on ESC proliferation have been demonstrated by Han et al. 17β-estradiol induces an increase of ([3] H) thymidine incorporation by murine ESCs and enhances DNA synthesis, which is in turn inhibited by anti-estrogen tamoxifen. Estrogens upregulate the expression of ERa and ERβ protein levels and increase mRNA expression of the proto-oncogenes c-fos, c-jun, and c-myc. In addition, 17β-estradiol activates the MAPK cascade as well as cyclin-dependent kinases, with associated increases in cyclins D1 and E, which are important intermediaries in cell cycle progression (42).
Although it is known that estrogens enhance the proliferation of embryonic stem cells, the role of androgens is not well understood. The presence of androgen receptors (AR) in ESCs has been detected as early as 4.5 d in mice and 5 d in humans, and it also was observed that the concentration of AR increases during differentiation of ESCs in a stage-dependent manner (46). Testosterone or dihydrotestosterone treatment is not associated with any significant change in androgen receptor mRNA expression level. But, in a contrast that suggests a testosterone depletion effect, nilutamide, a nonsteroidal antiandrogen, causes proliferation of ESCs through an increase of Akt protein expression and decreased cell-cycle inhibitor p27 (Kip1) expression (41). These findings suggest a definite role of both sex steroids in the differentiation and proliferation of embryonic stem cells.
Mesenchymal Stem Cells
MSCs are novel therapeutic agents for organ protection, and estrogens may enhance the protective function of MSCs by increasing or decreasing cytokine and growth factor production in these cells (47). Our previous study proved that gender differences exist in activated MSC function. In particular, lipopolysaccharide- (LPS) and hypoxia-induced VEGF production was significantly greater in female MSCs compared with male MSCs. Female MSCs express significantly less pro-inflammatory cytokines, TNF-α and IL-6, compared with male MSCs in response to acute LPS and hypoxia, suggesting their ability to limit inflammatory reactions (18). In males, TNFR1 regulates VEGF, TNF, and IL-6 production, whereas TNFR expression status does not affect cytokine and growth hormone production in females (19). We also observed gender differences in stem cell-mediated protection in a Langen-dorff preparation. Rat hearts were subjected to 25 min of warm global ischemia followed by 40 min of reperfusion and were assigned randomly to one of three groups: (a) vehicle treated; (b) male MSC treated; and (c) female MSC treated. Female MSC-treated hearts exhibited significantly improved contractility and compliance as compared with hearts treated with male MSC or vehicle (48).
Regarding the role of estrogens in osteogenic differentiation of MSCs, there is evidence that 17β-estradiol supports growth and differentiation mostly through the ERa receptor (49). This receptor bias may be attributed to interindividual variability and gender differences of osteoblast responses of MSCs to estrogen manifested by ERα polymorphism. In terms of male sex hormone effects, testosterone decreases the specific alkaline phosphatase activity in male MSCs but does not affect calcium deposition in either sex (50). Bone marrow MSCs, when exposed to osteogenic differentiation medium supplemented with 17β-estradiol, increase the expression of bone morphogenetic protein (BMP) and osteocalcin, and significantly increase the deposition of calcium (51,52). 17β-estradiol also stimulates the expression of osteogenic genes for ALP, collagen I, and TGF-β1 by MSCs (12). These observations suggest the bulk of the heavy lifting in bone metabolism/physiology is handled by the female sex steroids.
Clues to the molecular mechanisms underlying the role of sex hormones in MSC differentiation may be found in the ERK pathway. Resveratrol, a phytoestrogen found in red wine, stimulates the expression of osteoblastic markers such as RUNX2/CBFA1, osterix, and osteocalcin in human bone marrow mesenchymal stem cell cultures. This effect is associated with a rapid activation of ERK1 and ERK2, and also can be inhibited by the ERK inhibitor PD98059. Resveratrol enhances osteoblastic maturation and calcium deposition into the extracellular matrix (53) and its effects on osteoblastic differentiation are mediated through NO/cGMP pathway (54). Sex hormone involvement in MSC function also includes leptin- and vitamin D-linked increases in aromatase activity during osteogenesis, leading to osteogenic differentiation of MSCs (55) and significant E2 linked increases in the lipid stores of differentiated adipocytes (52).
Hematopoietic Stem Cells
There are gender differences in hematopoietic progenitor cell concentrations in cord blood samples collected from infants. Specifically, male infants had significantly higher median CD34+ cell concentrations than female infants (31.8/ µL compared with 30.2/ µL, respectively; P = 0.03). This relative increase is reflected in adult males, who, when compared with age-matched females, have an increased number of colony-forming cells, erythroblastic colonies, and granulocyte-macrophage colonies in their peripheral blood (56). This signifies that gender may affect the hematopoietic potential of cord blood transplants (57).
The effect of sex steroids on B lymphopoiesis is the subject of extensive, continuing study. Sex steroids suppress B lymphocyte production in murine bone marrow. Pre B-lymphocytes produce the heavy chain of IgM (µ chain) (58), under the influence of the IgM constant region gene (cµ) (59), which can act as a marker of pre-B cells. V(D)J recombination confers the ability of the immune system to respond to a vast number of foreign antigens, which occurs particularly in immature lymphocytes and is mediated by the recombination activating gene products Rag1 and Rag2 (60,61). 17β-estradiol treatment reduces cµ pre-B cells, associated with a decrease in Ig gene rearrangements and rag1 transcripts (62). It also has been demonstrated that ERα is predominantly responsible for mediating 17β-estradiol induced changes in B-cell precursors (63,64). These findings suggest that 17β-estradiol exerts negative influence on the production of B-lineage cells by modifying the differentiation, proliferation, and survival of early B-cell precursors.
A novel system for expansion of hematopoietic stem cells utilizes “selective amplifier genes” that encode fusion proteins (granulocyte colony-stimulating factor [G-CSF] mutant receptor and delta G-CSF mutant receptor) between the granulocyte colony-stimulating factor receptor (G-CSF-R) and the hormone-binding domain of estrogen receptor (ER). ERs were replaced with a mutant receptor (TmR), which specifically binds to 4-hydroxytamoxifen (Tm). Interleukin-3 (IL-3)-dependent Ba/F3 (mouse peripheral blood pro-B) cells and hematopoietic progenitor cells transduced with the fusion proteins showed IL-3-independent growth in response to Tm, whereas, the cells were insensitive to estrogen at concentrations up to10(−7) M to 10(−6) M. Murine bone marrow cells transduced with G-CSF-TmR and delta G-CSF-TmR formed colonies in methyl-cellulose medium in response to Tm, but no colonies appeared with 10(−7) M estrogen or without cytokines. These results suggest that the influences of endogenous estrogen can be ablated by using the G-CSF-TmR/Tm or delta G-CSF-TmR/ Tm system to expand hematopoietic stem cells with potential therapeutic application (65).
Androgens exert an inhibitory effect on B lymphopoiesis (66,67), but enhance erythropoietic differentiation (68) and thrombocytopoiesis (69). Cultures of human erythropoietic precursor cells collected from children’s normal marrow in the presence of erythropoietin demonstrated a significant increase in the number of colony-forming units (CFU-E) and burst-forming units (BFU-E) of derived colonies in the presence of androgens (10(−8) M or 10(−7) M). These colonies also showed increased uroporphyrinogen I synthase activity, indicating increased heme synthesis (68). Androgens also can rescue mature erythroid colony-forming cells from apoptosis induced by serum and growth factor deprivation (70), thus increasing erythrocyte population. Interestingly, castration of normal male mice leads to splenic enlargement and expansion of the B cell population, which is mediated via androgen receptors present in both immature B cells and marrow stromal cells. These effects can be reversed with androgen replacement (66), further elucidating the role of the male hormone in lymphopoiesis.
Cardiac Fibroblasts
Gender may affect the healing of ischemic myocardium through changes in the function of cardiac fibroblasts (CFs). These progenitor cells play a significant role in the remodeling of ischemic myocardium, and the signal transduction pathways controlling the proliferation of CFs under hypoxia-induced stress reveal significant gender differences. These studies found females to be resistant to hypoxia-induced inhibition of DNA synthesis associated with decreased expression of NFκB and increased expressions of p53 and bcl-2 in comparison to males (71,72). Estrogens exert modulatory effects on cardiac fibroblast function. 17β-estradiol inhibits proliferation and collagen synthesis (3H-proline incorporation) in male and female CFs in a similar way, and facilitates beneficial cardiac remodeling following ischemia. Consequently, hormone replacement therapy using 17β-estradiol may exert protective effects on post menopausal women against cardiovascular events (73).
Cardiac fibroblasts express estrogen receptor protein, and stimulation of CFs with 17β-estradiol causes nuclear translocation of these proteins, indicating one effect of estrogen on gene regulation (74). In cardiac fibroblasts, 17β-estradiol plays an inhibitory role on renin-angiotensin system-induced gene expression, signal transduction, and ECM remodeling. Angiotensin II increases fibroblast proliferation and synthesis of collagen types I and III through the upregulation of expression of the angiotensin AT(1) receptor gene and β1 integrins. 17β-estradiol can prevent these increases in proliferation and AT(1) receptor mRNA levels and can attenuate the collagen synthesis in response to angiotensin II. 17β-estradiol inhibits AngII-stimulated expression of β1 integrins significantly and attenuates collagen gel contraction (75).
Estrogens can improve cardiac fibroblast-mediated remodeling of ischemic myocardium through both genomic and nongenomic mechanisms. 17β-estradiol exerts an inhibitory effect on the growth of cardiac fibroblasts through both ERα and ERβ (76). Underlying molecular mechanisms include increased mitogen-activated protein (MAP) kinase p42/44 activation and decreased p38 activation (77). In addition, 17β-estradiol increases the steady-state mRNA level of transforming growth factor-β3 and fibronectin in these cells (78). A recent study has demonstrated that the selective estrogen receptor agonists PPT (4,4′,4″-[4-propyl-([1]H)-pyrazole-1,3,5-triyl] tris-phenol) for ER-α and DPN (2,3-bis[4-hydroxyphenyl]-propionitrile) for ER-β, stimulate the large-conductance Ca++-activated K+ (BK[Ca]) channels in cultured human cardiac fibroblasts (HCFs). In whole-cell configuration, depolarizing pulses evoked large outward currents (Ik) with an outward rectification, the amplitude of which was increased in the presence of DPN or PPT. Paxilline, a selective blocker of BK(Ca) channels, could reverse the DPN- or PPT-induced amplitude of Ik. However, no change in the transcriptional level of the BK(Ca)-channel a-subunit was observed by RT-PCR analysis in chronic treatment with these two compounds. These findings suggest that estrogen induces a rapid stimulatory effect on human cardiac fibroblasts via a nongenomic mechanism through the activation of BK(Ca)-channels (79). Building on this understanding of the molecular mechanisms in cardiac fibroblasts may enable us to modify the functions of these cells.
Endothelial Progenitor Cells
Blood contains endothelial progenitor cells (EPC), which can differentiate into endothelial cells and modulate healing of injured vessels. In one study on a healthy middle-aged population without known cardiovascular risk factors, it has been demonstrated that women exhibited a distinctly higher EPC colony-forming capacity (approximately 150%) and greater migratory activity (40%) compared with men (80). Moreover, human women with a higher plasma estrogen concentration showed a significantly higher level of circulating EPCs. Increase in the number of EPCs by 17β-estradiol is mediated by decreased rate of apoptosis through a caspase-8-dependent pathway (81). The effects of estrogen on EPCs are mediated via ERα receptor (82). However, another similar study failed to demonstrate significant gender-specific differences in the frequency of colony formation (83).
To explore sex hormone specificity in EPCs, ERα KO mice were treated with 17β -estradiol, which failed to induce migration, tube formation, adhesion, and estrogen-responsive element-dependent gene transcription activities. In bone marrow transplantation models, endogenous EPC migration and capillary density at the border zone of ischemic myocardium was reduced in 17β-estradiol treated ERα KO mice. Using a murine ischemic heart model, it also was shown that migration of EPCs into the ischemic border zone was impaired in ERα KO bone marrow transplant mice. Downregulation of VEGF also was noted in EPCs from ERa KO mice both in vivo and in vitro (44). It was postulated that 17β-estradiol mobilizes EPCs via endothelial nitric oxide synthase-mediated activation of matrix metalloproteinase-9 (84). Upregulation of MMP-9 results in the release of soluble Kit-ligand (sKitL), which facilitates the transfer of endothelial cells from the quiescent to proliferative pool (85).
Disease models in animals also have been employed to examine the effects of sex hormones on progenitor cell functionality. In spontaneously hypertensive rats, the number of differentiated and adherent EPCs derived from bone marrow was lower compared with age-matched normotensive rats. Treatment with 17β-estradiol significantly increases the number of EPCs. EPCs derived from hypertensive rats show low telomerase activity and early senescence. Estrogen treatment delays senescence and augments telomerase activity through PI3-K/ Akt pathway (86,87).
Regarding male sex hormone effects, recent clinical studies suggest that androgens increase the number of circulating EPCs through a possible effect on bone marrow. It has been revealed that hypogonadotrophic hypogonadal men have low circulating EPCs that increase significantly after androgen treatment (88). A direct effect of testosterone also was suggested by expression of androgen receptor (AR) mRNA and protein in human EPCs. Synthetic androgen, methyltrienolone (R1881), causes AR translocation in the nucleus, suggesting its activation increases proliferation, migration, and colony formation activity of these cells. Proliferation, migration, and colony formation activities of the EPCs could be abolished by pretreatment with flutamide (89). A greater understanding of these molecular mechanisms will yield insights into how gender differences affect the healing process in patients.
Preadipocytes
Regulation of lipid metabolism plays a key role in atherosclerosis and ischemic cardiac diseases. Estrogens control the expression of lipogenic genes such as leptin, perilipin, peroxisome proliferation activator receptor-delta, and lipoprotein lipase in adipocytes and estrogen supplementation helps to reduce adipose mass and adipocyte size and prevents development of obesity in postmenopausal state (90–92) by both genomic and nongenomic mechanisms (93). Genomic effects of estrogen are limited to the regulation of leptin and lipoprotein lipase expression whereas nongenomic effects are mediated via the s messenger systems, namely cAMP cascade and the phosphoinositide cascade. Activation of the cAMP cascade by estrogen is followed by activation of hormone-sensitive lipase leading to lipolysis in adipose tissues. Activation of protein kinase C through the PI3K cascade, controls the proliferation and differentiation of preadipocytes (93). Human preadipocytes (PAs) possess ERα protein and express ERa gene, but do not express ERβ receptors, indicating that effect of estrogen on adipogenesis is mediated through the ERα receptor (52,94,95). The effects of estrogens on ERa is site specific (95).
In humans, development of abdominal fat deposition is inversely proportionate to blood testosterone levels (96). Androgen receptors are found on preadipocytes (97,98) and the effects of testosterone on these cells also are site-specific. Castration is associated with increased proliferation and differentiation of epididymal and perirenal preadipocytes in male rats; whereas, peripubertal testosterone supplementation reduces inguinal and retroperitoneal fat depots of ovariectomized (OVX) rats. Testosterone decreases adipocyte proliferation without affecting adipocyte mean cell size or the size distribution profiles (99). Androgens act directly on fat cells by upregulating a 2-AR expression (100). Androgens also exert their modulatory effects on the transcription factor C/EBP α, which is a key regulator of the expression of adipogenic genes (101), providing molecular context for gender-based effects on adipocyte physiology.
Osteogenic Progenitors
Estrogen and testosterone play crucial roles in bone metabolism. Even in males, estrogen is critical for the pubertal growth spurt characterized by skeletal maturation, accrual of peak bone mass, and the maintenance of bone mass in the adult through its effects on remodeling and bone turnover (102).
In OVX rats, estrogen deficiency causes osteopenia and induces bone turnover. Endosteal bone formation in OVX rats is associated with an increased proliferation of both osteoblast precursor cells present in the marrow stroma and along the endosteal bone surface. The osteoclast surface (percent of the bone trabecular surface covered with osteoclasts) also increases in OVX rats following ovariectomy, suggesting that bone formation increases in correlation with bone resorption. Estradiol supplementation reverses both the increase in resorption and formation indices (103).
Interestingly, 17β-estradiol, through the ERα receptor, increases bone matrix protein (BMP)-2, and BMP-4 expression. It also attenuates the self renewal of early osteoblast progenitors (having limited self-renewal capacity) for both osteoblasts and osteoclasts. Both these effects slow down bone remodeling (104,105). Through TGF β, estrogen causes repression of T cell proliferation and differentiation, and inhibits INFγ production. This subsequently decreases TNF production and thus reduces osteo-clastic activity (106). Ovariectomy increases IL-12 and IL-18 secretion by macrophages which result in enhanced T cell activation and TNF production causing bone loss (107). IL-6 (108-110) and IL-7 also have been attributed to induced bone loss associated with estrogen deficiency (111), not only through increased osteoclastic activity but also by reducing bone deposition through downregulation of the osteoblast-specific transcription factor, core-binding factor a 1/Runx2 (112). In addition, 17β-estradiol exerts its inhibitory effect on osteoclasts through the regulation of VEGF production; ovariectomized mice were associated with increased VEGF production and increased osteoclastic activity (113).
Testosterone also has beneficial effects on bone metabolism in adult males mediated by androgen receptors. Different genomic and nongenomic pathways are believed to be involved in mediating the effects of testosterone on bone metabolism. The nongenomic effects are mediated via Akt activation (114) through stimulation of src kinase (115,116). MAP kinase signaling cascade also is activated with testosterone treatment resulting in increased expression of Raf-1 and ERK-2 (115). Insulin like growth factor-I (IGF-I), and insulin like growth factor binding proteins (IGFBP) also play a significant part. Androgen decreases insulin like growth factor binding protein IGFBP-4 which is inhibitory for osteoblasts, and increases IGFBP-2 and IGFBP-3 mRNA and protein levels, which have stimulatory effects on osteoblasts (117).
The genomic effect of testosterone is mediated by the increased osteoprotegerin (OPG) expression. Osteoprotegerin is a receptor activator of NF-κB ligand, which inhibits the differentiation of the osteoclast precursor into a mature osteoclast (118). However, the effect of testosterone on osteoprotegerin expression is controversial. Some authors have demonstrated that 5α-dihydrotestosterone (DHT) reduces OPG in a dose-dependent manner (119). In total, these observations underscore the importance of understanding the differential effects of sex hormones on bone metabolism and physiology.
Neural Stem Cells
Estrogens modulate neurogenesis during embryonic development. Estrogens induce the neuronal phenotype in embryonic stem cell culture and enhance proliferation of embryonic neural stem cells, increasing the ratio of neurons to glial cells (120,121). In combination with poly-L-ornithine/fibronectin, estrogens also have been shown to accelerate differentiation and maturation of neurons (121). Furthermore, estrogens also enhance differentiation and survival of dopaminergic neurons harvested from human neural stem cells, suggesting a possible role of estrogens in the transplantation of neural stem cells as a therapeutic approach for Parkinson’s disease (122). Like the previously discussed cell types, beneficial effects of estrogens on neurons are mediated through both genomic and nongenomic pathways (123,124).
Testosterone has a negative influence on neural stem cell proliferation. Nandrolone (19-Nortestosterone) reduces cell proliferation in neural stem cells stimulated with epidermal growth factor, which can be reversed by flutamide, a receptor antagonist. Nandrolone also decreases the BrdU labeling of neural stem cells in the dentate gyrus, indicating reduced cell proliferation in vivo (125). These observations serve to emphasize the differential role of gender specific hormones on neural cell ontogeny.
Conclusion
Sexual dimorphism clearly influences the function of various stem cell types throughout the body. A better understanding of the effects of estrogen and testosterone on these cells will allow investigators and clinicians to modulate the functions of these cells directly, with the ultimate goal of generating more potent stem cell applications for the treatment of human disease.
References
Kher A et al. (2005) Sex differences in the myocardial inflammatory response to acute injury. Shock. 23:1–10.
Deitch EA et al. (2007) Hormonally active women tolerate shock-trauma better than do men: a prospective study of over 4000 trauma patients. Ann. Surg. 246:447–53; discussion 453–5.
Choudhry MA et al. (2005) Gender differences in acute response to trauma-hemorrhage. Shock. 24 Suppl 1:101–6.
Choudhry MA, Bland KI, Chaudry IH. (2006) Gender and susceptibility to sepsis following trauma. Endocr. Metab. Immune Disord. Drug Targets. 6:127–35.
Lahm T et al. (2007) Endogenous estrogen attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction: the effects of sex and menstrual cycle. Am. J. Physiol. Endocrinol. Metab. 293:E865–71.
Deitch EA et al. (2008) Resistance of the female, as opposed to the male, intestine to I/R-mediated injury is associated with increased resistance to gut-induced distant organ injury. Shock. 29:78–83.
Schwendimann RN, Alekseeva N. (2007) Gender issues in multiple sclerosis. Int. Rev. Neurobiol. 79:377–92.
Shulman LM. (2007) Gender differences in Parkinson’s disease. Gend. Med. 4:8–18.
Lobo RA. (2007) Menopause and stroke and the effects of hormonal therapy. Climacteric. 10 Suppl 2:27–31.
Eugene D, Djemli A, Van Vliet G. (2005) Sexual dimorphism of thyroid function in newborns with congenital hypothyroidism. J. Clin. Endocrinol. Metab. 90:2696–700.
Feng W et al. (2007) Prevention of osteoporosis and hypogonadism by allogeneic ovarian transplantation in conjunction with intra-bone marrow-bone marrow transplantation. Transplantation. 84:1459–66.
Zhou S et al. (2001) Estrogen modulates estrogen receptor alpha and beta expression, osteogenic activity, and apoptosis in mesenchymal stem cells (MSCs) of osteoporotic mice. J. Cell. Biochem. Suppl. Suppl 36:144–55.
DiSilvio L, Jameson J, Gamie Z, Giannoudis PV, Tsiridis E. (2006) In vitro evaluation of the direct effect of estradiol on human osteoblasts (HOB) and human mesenchymal stem cells (h-MSCs). Injury. 37 Suppl 3:S33–42.
Vaccarino V, Krumholz HM, Berkman LF, Horwitz RI. (1995) Sex differences in mortality after myocardial infarction. Is there evidence for an increased risk for women? Circulation. 91: 1861–71.
Hodis HN, Mack WJ. (2002) Atherosclerosis imaging methods: assessing cardiovascular disease and evaluating the role of estrogen in the prevention of atherosclerosis. Am. J. Cardiol. 89:19E–27E; discussion 27E.
Paroo Z, Haist JV, Karmazyn M, Noble EG. (2002) Exercise improves postischemic cardiac function in males but not females: consequences of a novel sex-specific heat shock protein 70 response. Circ. Res. 90:911–7.
Herrington DM et al. (2000) Effects of estrogen replacement on the progression of coronaryartery atherosclerosis. N. Engl. J. Med. 343: 522–529.
Crisostomo PR et al. (2006) Sex dimorphisms in activated mesenchymal stem cell function. Shock. 26:571–4.
Crisostomo et al. (2007) Gender differences in injury induced mesenchymal stem cell apoptosis and VEGF, TNF, IL-6 expression: role of the 55 kDa TNF receptor (TNFR1). J. Mol. Cell. Cardiol. 42:142–9.
Pitcher JM et al. (2006) Endogenous estrogen mediates a higher threshold for endotoxin-induced myocardial protection in females. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290:R27–33.
Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. (2006) Estrogen receptor-alpha mediates acute myocardial protection in females. Am. J. Physiol. Heart Circ. Physiol. 290:H2204–9.
Baker L et al. (2003) The role of estrogen in cardiovascular disease. J. Surg. Res. 115:325–44.
Wang M et al. (2005) Role of endogenous testosterone in myocardial proinflammatory and proapoptotic signaling after acute ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 288:H221–6.
Nelson NT et al. (2006) Does endogenous testosterone mediate the lower preconditioning threshold in males? J. Surg. Res. 131:86–90.
Crisostomo PR, Wang M, Wairiuko GM, Morrell ED, Meldrum DR. (2006) Brief exposure to exogenous testosterone increases death signaling and adversely affects myocardial function after ischemia. Am. J. Physiol Regul Integr. Comp. Physiol. 290:R1168–74.
Nam UH et al. (2007) The effect of chronic exogenous androgen on myocardial function following acute ischemia-reperfusion in hosts with different baseline levels of sex steroids. J. Surg. Res. 142:113–8.
Meldrum DR. (2006) Estrogen increases protective proteins following trauma and hemorrhage. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290:R809–11.
Cavasin MA, Tao Z, Menon S, Yang XP. (2004) Gender differences in cardiac function during early remodeling after acute myocardial infarction in mice. Life Sci. 75:2181–92.
Cavasin MA, Tao ZY, Yu AL, Yang XP. (2006) Testosterone enhances early cardiac remodeling after myocardial infarction, causing rupture and degrading cardiac function. Am. J. Physiol. Heart Circ. Physiol. 290:H2043–50.
Pelzer T et al. (2005) The estrogen receptor-alpha agonist 16alpha-LE2 inhibits cardiac hypertrophy and improves hemodynamic function in estrogen-deficient spontaneously hypertensive rats. Cardiovasc. Res. 67:604–12.
Booth EA, Obeid NR, Lucchesi BR. (2005) Activation of estrogen receptor-alpha protects the in vivo rabbit heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 289: H2039–47.
Shearman AM et al. (2003) Association between estrogen receptor alpha gene variation and cardiovascular disease. JAMA. 290:2263–70.
McMurray RW, Ndebele K, Hardy KJ, Jenkins JK. (2001) 17-beta-estradiol suppresses IL-2 and IL-2 receptor. Cytokine. 14:324–33.
Crane-Godreau MA, Wira CR. (2005) Effects of estradiol on lipopolysaccharide and Pam3Cys stimulation of CCL20/macrophage inflammatory protein 3 alpha and tumor necrosis factor alpha production by uterine epithelial cells in culture. Infect. Immun. 73:4231–7.
de Witte T, Suciu S, Brand R, Muus P, Kroger N. (2007) Autologous stem cell transplantation in myelodysplastic syndromes. Semin. Hematol. 44:274–277.
Breems DA, Lowenberg B. (2007) Acute myeloid leukemia and the position of autologous stem cell transplantation. Semin. Hematol. 44:259–66.
Crisostomo PR, Meldrum DR. (2007) Stem cell delivery to the heart: clarifying methodology and mechanism. Crit. Care Med. 35:2654–6.
Mangi AA et al. (2003) Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat. Med. 9:1195–201.
Assmus B et al. (2002) Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction (TOPCARE-AMI). Circulation. 106:3009–17.
Erbs S et al. (2007) Restoration of microvascular function in the infarct-related artery by intracoronary transplantation of bone marrow progenitor cells in patients with acute myocardial infarction: the Doppler Substudy of the Reinfusion of Enriched Progenitor Cells and Infarct Remodeling in Acute Myocardial Infarction (REPAIR-AMI) trial. Circulation. 116:366–74.
Chang CY et al. (2006) Androgenic and antiandrogenic effects and expression of androgen receptor in mouse embryonic stem cells. Fertil. Steril. 85 Suppl 1:1195–203.
Han HJ, Heo JS, Lee YJ. (2006) Estradiol-17beta stimulates proliferation of mouse embryonic stem cells: involvement of MAPKs and CDKs as well as protooncogenes. Am. J. Physiol. Cell Physiol. 290:C1067–75.
Hong SH et al. (2004) Expression of estrogen receptor-alpha and -beta, glucocorticoid receptor, and progesterone receptor genes in human embryonic stem cells and embryoid bodies. Mol. Cells. 18:320–5.
Hamada H et al. (2006) Estrogen receptors alpha and beta mediate contribution of bone marrow-derived endothelial progenitor cells to functional recovery after myocardial infarction. Circulation. 114:2261–70.
Marin-Husstege M, Muggironi M, Raban D, Skoff RP, Casaccia-Bonnefil P. (2004) Oligodendrocyte progenitor proliferation and maturation is differentially regulated by male and female sex steroid hormones. Dev. Neurosci. 26:245–54.
Bremner WJ, Millar MR, Sharpe RM, Saunders PT. (1994) Immunohistochemical localization of androgen receptors in the rat testis: evidence for stage-dependent expression and regulation by androgens. Endocrinology. 135:1227–34.
Crisostomo PR et al. (2008) Human mesenchymal stem cells stimulated by TNF-alpha, LPS, or hypoxia produce growth factors by an NF kappa B-but not JNK-dependent mechanism. Am. J. Physiol. Cell. Physiol. 294:C675–82.
Crisostomo PR et al. (2007) In the adult mesenchymal stem cell population, source gender is a biologically relevant aspect of protective power. Surgery. 142:215–21.
Wang Q et al (2006) Temporal expression of estrogen receptor alpha in rat bone marrow mesenchymal stem cells. Biochem. Biophys. Res. Commun. 347:117–23.
Leskela HV et al. (2006) Estrogen receptor alpha genotype confers interindividual variability of response to estrogen and testosterone in mesenchymal-stem-cell-derived osteoblasts. Bone. 39:1026–34.
Fawell SE et al. (1990) Inhibition of estrogen receptor-DNA binding by the “pure” antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization. Proc. Natl. Acad. Sci. U. S. A. 87:6883–7.
Hong L, Colpan A, Peptan IA. (2006) Modulations of 17-beta estradiol on osteogenic and adipogenic differentiations of human mesenchymal stem cells. Tissue Eng. 12:2747–53.
Dai Z et al. (2007) Resveratrol enhances proliferation and osteoblastic differentiation in human mesenchymal stem cells via ER-dependent ERK1/2 activation. Phytomedicine. 14:806–14.
Song LH et al. (2006) Resveratrol prevents CsA inhibition of proliferation and osteoblastic differentiation of mouse bone marrow-derived mesenchymal stem cells through an ER/NO/cGMP pathway. Toxicol. In Vitro. 20:915–22.
Pino AM et al. (2006) Aromatase activity of human mesenchymal stem cells is stimulated by early differentiation, vitamin D and leptin. J. Endocrinol. 191:715–25.
Horner S, Pasternak G, Hehlmann R. (1997) A statistically significant sex difference in the number of colony-forming cells from human peripheral blood. Ann. Hematol. 74:259–63.
Aroviita P, Teramo K, Hiilesmaa V, Kekomaki R. (2005) Cord blood hematopoietic progenitor cell concentration and infant sex. Transfusion. 45: 613–21.
Kitamura D, Roes J, Kuhn R, Rajewsky K. (1991) A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 350:423–6.
Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. (2005) Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J. Exp. Med. 202:561–8.
Oettinger MA, Schatz DG, Gorka C, Baltimore D. (1990) RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 248:1517–23.
Mombaerts P et al. (1992) RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 68: 869–77.
Medina KL, Strasser A, Kincade PW. (2000) Estrogen influences the differentiation, proliferation, and survival of early B-lineage precursors. Blood. 95:2059–67.
Thurmond TS et al. (2000) Role of estrogen receptor alpha in hematopoietic stem cell development and B lymphocyte maturation in the male mouse. Endocrinology. 141:2309–18.
Smithson G, Couse JF, Lubahn DB, Korach KS, Kincade PW. (1998) The role of estrogen receptors and androgen receptors in sex steroid regulation of B lymphopoiesis. J. Immunol. 161:27–34.
Xu R et al. (1999) A selective amplifier gene for tamoxifen-inducible expansion of hematopoietic cells. J. Gene Med. 1:236–44.
Viselli SM, Reese KR, Fan J, Kovacs WJ, Olsen NJ. (1997) Androgens alter B cell development in normal male mice. Cell. Immunol. 182:99–104.
Erben RG, Eberle J, Stangassinger M. (2001) B lymphopoiesis is upregulated after orchiectomy and is correlated with estradiol but not testosterone serum levels in aged male rats. Horm. Metab. Res. 33:491–8.
Claustres M, Sultan C. (1986) Stimulatory effects of androgens on normal children’s bone marrow in culture: effects on BFU-E, CFU-E, and uropor-phyrinogen I synthase activity. Horm. Res. 23: 91–8.
Sullivan PS, Jackson CW, McDonald TP. (1995) Castration decreases thrombocytopoiesis and testosterone restores platelet production in castrated BALB/c mice: evidence that testosterone acts on a bipotential hematopoietic precursor cell. J. Lab. Clin. Med. 125:326–33.
Kim SW et al. (2005) Direct and indirect effects of androgens on survival of hematopoietic progenitor cells in vitro. J. Korean Med. Sci. 20:409–16.
Griffin M, Lee HW, Zhao L, Eghbali-Webb M. (2000) Gender-related differences in proliferative response of cardiac fibroblasts to hypoxia: effects of estrogen. Mol. Cell. Biochem. 215:21–30.
Zhao X, Eghbali-Webb M. (2002) Gender-related differences in basal and hypoxia-induced activation of signal transduction pathways controlling cell cycle progression and apoptosis, in cardiac fibroblasts. Endocrine. 18:137–45.
Dubey RK, Gillespie DG, Jackson EK, Keller PJ. (1998) 17Beta-estradiol, its metabolites, and progesterone inhibit cardiac fibroblast growth. Hypertension. 31:522–8.
Grohe C et al. (1997) Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. 416:107–12.
Zhou L, Shao Y, Huang Y, Yao T, Lu LM. (2007) 17beta-estradiol inhibits angiotensin II-induced collagen synthesis of cultured rat cardiac fibroblasts via modulating angiotensin II receptors. Eur. J. Pharmacol. 567:186–92.
Watanabe T et al. (2003) 17 beta-estradiol inhibits cardiac fibroblast growth through both subtypes of estrogen receptor. Biochem. Biophys. Res. Commun. 311:454–9.
Stewart JAJr, Cashatt DO, Borck AC, Brown JE, Carver WE. (2006) 17beta-estradiol modulation of angiotensin II-stimulated response in cardiac fibroblasts. J. Mol. Cell. Cardiol. 41:97–107.
Mercier I, Colombo F, Mader S, Calderone A. (2002) Ovarian hormones induce TGF-beta(3) and fibronectin mRNAs but exhibit a disparate action on cardiac fibroblast proliferation. Cardiovasc. Res. 53:728–39.
Wang YJ, Lin MW, Wu SN, Sung RJ. (2007) The activation by estrogen receptor agonists of the BK(Ca)-channel in human cardiac fibroblasts. Biochem. Pharmacol. 73:1347–57.
Hoetzer GL et al. (2007) Gender differences in circulating endothelial progenitor cell colony-forming capacity and migratory activity in middle-aged adults. Am. J. Cardiol. 99:46–8.
Strehlow K et al. (2003) Estrogen increases bone marrow-derived endothelial progenitor cell production and diminishes neointima formation. Circulation. 107:3059–65.
Foresta C et al. (2007) Oestrogen stimulates endothelial progenitor cells via oestrogen receptor-alpha. Clin. Endocrinol. (Oxf). 67:520–5.
Ciulla MM et al. (2006) Endothelial colony forming capacity is related to C-reactive protein levels in healthy subjects. Curr. Neurovasc. Res. 3:99–106.
Iwakura A et al. (2006) Estradiol enhances recovery after myocardial infarction by augmenting incorporation of bone marrow-derived endothelial progenitor cells into sites of ischemia-induced neovascularization via endothelial nitric oxide synthase-mediated activation of matrix metalloproteinase-9. Circulation. 113:1605–14.
Heissig B et al. (2002) Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 109:625–37.
Imanishi T, Kobayashi K, Hano T, Nishio I. (2005) Effect of estrogen on differentiation and senescence in endothelial progenitor cells derived from bone marrow in spontaneously hypertensive rats. Hypertens. Res. 28:763–72.
Imanishi T, Hano T, Nishio I. (2005) Estrogen reduces endothelial progenitor cell senescence through augmentation of telomerase activity. J. Hypertens. 23:1699–706.
Foresta C et al. (2006) Reduced number of circulating endothelial progenitor cells in hypogonadal men. J. Clin. Endocrinol. Metab. 91:4599–602.
Foresta C et al. (2007) Androgens stimulate endothelial progenitor cells through an androgen receptor-mediated pathway. Clin. Endocrinol. (Oxf). 68:284–9.
D’Eon TM et al. (2005) Estrogen regulation of adiposity and fuel partitioning. Evidence of genomic and non-genomic regulation of lipogenic and oxidative pathways. J. Biol. Chem. 280:35983–91.
Jaubert AM et al. (2007) Nongenomic estrogen effects on nitric oxide synthase activity in rat adipocytes. Endocrinology. 148:2444–52.
Enerback S, Gimble JM. (1993) Lipoprotein lipase gene expression: physiological regulators at the transcriptional and post-transcriptional level. Biochim. Biophys. Acta. 1169:107–25.
Mayes JS, Watson GH. (2004) Direct effects of sex steroid hormones on adipose tissues and obesity. Obes. Rev. 5:197–216.
Joyner JM, Hutley LJ, Cameron DP. (2001) Estrogen receptors in human preadipocytes. Endocrine. 15:225–30.
Shinozaki S et al. (2007) Site-specific effect of estradiol on gene expression in the adipose tissue of ob/ob mice. Horm. Metab. Res. 39:192–6.
Bjorntorp P. (1991) Metabolic implications of body fat distribution. Diabetes Care. 14:1132–43.
Xu X, De Pergola G, Bjorntorp P. (1990) The effects of androgens on the regulation of lipolysis in adipose precursor cells. Endocrinology. 126: 1229–34.
Dieudonne MN, Pecquery R, Boumediene A, Leneveu MC, Giudicelli Y. (1998) Androgen receptors in human preadipocytes and adipocytes: regional specificities and regulation by sex steroids. Am. J. Physiol. 274:C1645–52.
James RG, Krakower GR, Kissebah AH. (1996) Influence of androgenicity on adipocytes and precursor cells in female rats. Obes. Res. 4:463–70.
Bouloumie A, Valet P, Dauzats M, Lafontan M, Saulnier-Blache JS. (1994) In vivo upregulation of adipocyte alpha 2-adrenoceptors by androgens is consequence of direct action on fat cells. Am. J. Physiol. 267:C926–31.
Garcia E, Lacasa M, Agli B, Giudicelli Y, Lacasa D. (1999) Modulation of rat preadipocyte adipose conversion by androgenic status: involvement of C/EBPs transcription factors. J. Endocrinol. 161:89–97.
Grumbach MM. (2000) Estrogen, bone, growth and sex: a sea change in conventional wisdom. J. Pediatr. Endocrinol. Metab. 13 Suppl 6:1439–55.
Modrowski D, Miravet L, Feuga M, Marie PJ. (1993) Increased proliferation of osteoblast precursor cells in estrogen-deficient rats. Am. J. Physiol. 264:E190–6.
Di Gregorio GB et al. (2001) Attenuation of the self-renewal of transit-amplifying osteoblast progenitors in the murine bone marrow by 17 beta-estradiol. J. Clin. Invest. 107:803–12.
Oreffo RO, Kusec V, Romberg S, Triffitt JT. (1999) Human bone marrow osteoprogenitors express estrogen receptor-alpha and bone morphogenetic proteins 2 and 4 mRNA during os-teoblastic differentiation. J. Cell. Biochem. 75:382–92.
Gao Y et al. (2004) Estrogen prevents bone loss through transforming growth factor beta signaling in T cells. Proc. Natl. Acad. Sci. U. S. A. 101: 16618–23.
Cenci S et al. (2003) Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN-gamma-induced class II transactivator. Proc. Natl. Acad. Sci. U. S. A. 100: 10405–10.
Ohmori S, Kanda K, Kawano S, Kambe F, Seo H. (2001) Effects of estrogen on tail suspension-induced disuse atrophy in ovariectomized rats: evaluation of the expression of interleukin-6 mRNAin the femur. Environ. Med. 45:12–4.
Masiukiewicz US, Mitnick M, Gulanski BI, Insogna KL. (2002) Evidence that the IL-6/IL-6 soluble receptor cytokine system plays a role in the increased skeletal sensitivity to PTH in estrogen-deficient women. J. Clin. Endocrinol. Metab. 87:2892–8.
Jilka RL et al. (1992) Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science. 257:88–91.
Ryan MR et al. (2005) An IL-7-dependent rebound in thymic T cell output contributes to the bone loss induced by estrogen deficiency. Proc. Natl. Acad. Sci. U. S. A. 102:16735–40.
Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R. (2002) Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J. Clin. Invest. 110:1643–50.
Kodama I et al. (2004) Estrogen regulates the production of VEGF for osteoclast formation and activity in op/op mice. J. Bone Miner. Res. 19:200–6.
Kang HY et al. (2004) Nongenomic androgen activation of phosphatidylinositol 3-kinase/Akt signaling pathway in MC3T3-E1 osteoblasts. J. Bone Miner. Res. 19:1181–90.
Kousteni S et al. (2001) Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 104:719–30.
Migliaccio A et al. (2000) Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. Embo. J. 19:5406–17.
Gori F, Hofbauer LC, Conover CA, Khosla S. (1999): Effects of androgens on the insulin-like growth factor system in an androgen-responsive human osteoblastic cell line. Endocrinology. 140:5579–86.
Chen Q, Kaji H, Kanatani M, Sugimoto T, Chihara K. (2004) Testosterone increases osteoprotegerin mRNA expression in mouse osteoblast cells. Horm. Metab. Res. 36:674–8.
Hofbauer LC, Hicok KC, Chen D, Khosla S. (2002) Regulation of osteoprotegerin production by androgens and anti-androgens in human osteoblastic lineage cells. Eur. J. Endocrinol. 147:269–73.
Brannvall K, Korhonen L, Lindholm D. (2002) Estrogen-receptor-dependent regulation of neural stem cell proliferation and differentiation. Mol. Cell. Neurosci. 21:512–20.
Murashov AK, Pak ES, Hendricks WA, Tatko LM. (2004) 17beta-Estradiol enhances neuronal differentiation of mouse embryonic stem cells. FEBS Lett. 569:165–8.
Kishi Y et al. (2005) Estrogen promotes differentiation and survival of dopaminergic neurons derived from human neural stem cells. J. Neurosci. Res. 79:279–86.
Liao SL, Chen WY, Chen CJ. (2002) Estrogen attenuates tumor necrosis factor-alpha expression to provide ischemic neuroprotection in female rats. Neurosci. Lett. 330:159–62.
Segars JH, Driggers PH. (2002) Estrogen action and cytoplasmic signaling cascades. Part I: membrane-associated signaling complexes. Trends Endocrinol. Metab. 13:349–54.
Brannvall K, Bogdanovic N, Korhonen L, Lindholm D. (2005) 19-Nortestosterone influences neural stem cell proliferation and neurogenesis in the rat brain. Eur. J. Neurosci. 21:871–8.
Acknowledgments
This work was supported in part by NIH R01GM070628, NIH R01HL085595, NIH K99/R00 HL0876077, NIH F32HL085982, AHA Grant-in-aid, and AHA Post-doctoral Fellowship 0725663Z.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ray, R., Novotny, N.M., Crisostomo, P.R. et al. Sex Steroids and Stem Cell Function. Mol Med 14, 493–501 (2008). https://doi.org/10.2119/2008-00004.Ray
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2008-00004.Ray