
Abstract
Background
Interpretation of rituximab efficacy for refractory idiopathic inflammatory myopathies (IIM) is hampered by the absence of a uniform definition of refractory myositis and clinical response. Therefore, rigorous criteria of refractoriness, together with a homogenous definition of clinical improvement, were used to evaluate rituximab one-year response.
Methods
A retrospective cohort study including 43 IIM (15 antisynthetase syndrome, 16 dermatomyositis, 12 polymyositis) was conducted. All patients had refractory disease (inadequate response to at least two immunosuppressives/immunomodulatories and no less than three months sequentially or concomitantly glucocorticoid tapering) criteria. Clinical/laboratory improvement at one-year was based on modified International Myositis Assessment & Clinical Studies Group (IMACS) core set measures. The patients received two infusions of rituximab (1 g each) at baseline, followed by repeated dose after 6 months. Baseline immunosuppressive therapy was maintained and glucocorticoid dose was tapered according to clinical/laboratory parameters.
Results
Five patients had side effects at the first rituximab application and were excluded. Therefore, 38 out of 43 patients completed the one-year follow up. Almost 75% of the patients attained clinical and laboratory response after one-year. A significant reduction in median glucocorticoid dose (18.8 vs. 6.3 mg/day) was achieved and 42% patients were able to discontinue prednisone. In contrast, young individuals and patients with dysphagia had a tendency to be non-responders to rituximab. No severe infections were observed.
Conclusion
This study provides convincing evidence that rituximab is an effective and safe therapy for refractory IIM.
Similar content being viewed by others


Background
Idiopathic inflammatory myopathies (IIM) constitute a heterogeneous group of chronic systemic autoimmune diseases with a high rate of morbidity and disability [1,2,3]. Based on their clinical, laboratory, histopathological and progression features, IIM can be classified as polymyositis (PM), dermatomyositis (DM), antisynthetase syndrome (ASS), inclusion body myositis, and others [2, 3].
A number of studies have suggested rituximab efficacy for refractory IIM, with response rates ranging from 61 to 83% [4,5,6,7,8,9,10,11]. This high and wide range of response rate is partly explained by the lack of a standardized definition for refractoriness and/or use of heterogeneous response parameters. In fact, refractory myositis has several definitions including intolerance to or an inadequate response to glucocorticoids and at least one other immunosuppressive agent, but few studies provide a clear description of whether the maximum tolerated therapeutic dose was achieved [5,6,7,8,9,10,11].
With regard to rituximab response parameters, most reports are limited to serum level of creatine phosphokinase and muscle strength improvements [6,7,8,9,10,11]. However, creatine phosphokinase may be not the best parameter, particularly if the evaluation includes different types of myositis, such as DM and ASS, in which other target organ involvement is more relevant than muscular involvement [2]. Of note, the disease activity core set measures validated by the International Myositis Assessment & Clinical Studies Group (IMACS) [12, 13] have not been previously used to evaluate refractory IIM response to therapy. These measures defined response as a > 20% improvement on three out of any 6 of the following core set measures: Health Assessment Questionnaire (HAQ); Manual Muscle Testing-8 (MMT-8); Physician Global Activity - Visual Analogue Scale (VAS); Patient Global Activity - VAS; serum muscle enzymes; Myositis Disease Activity Assessment Tool (MDAAT); with no more than two core set measures worsening by > 25%, which cannot include MMT.
Therefore, the aim of the present study was to evaluate the efficacy and predictors of clinical improvement of rituximab in a homogeneous population of refractory IIM cases, using a rigorous definition of refractory disease and modified IMACS core set measures to evaluate long-term response.
Methods
Study design
This retrospective single-center cohort study conducted from 2011 to 2016 included 43 consecutive adult patients with refractory IIM: 15 ASS (defined as myositis, arthritis, pulmonary disease, positive antisynthetase antibody, with or without mechanic’s hands, fever and/or Raynaud’s phenomenon) [14]; 16 DM and 12 PM according to the criteria of Bohan and Peter [15].
Patient data
Patients with clinically amyopathic DM, overlap myositis, neoplasia associated myositis, necrotizing myopathies, acute and/or chronic infections were excluded.
Data were included in an ongoing electronic database protocol. Demographic, clinical, laboratory and therapeutic data were obtained by electronic medical records, containing previously standardized and parameterized data. The following parameters were analyzed: current age, gender, ethnicity, time between diagnosis and symptom onset, disease duration, gastrointestinal (upper dysphagia), pulmonary (moderate dyspnea or computed tomography disclosing evidence of interstitial pneumopathy and/or “ground-glass” pneumopathy), joint (arthralgia and/or arthritis), previous and current drug treatment.
Refractory myositis was defined as an inadequate response to at least two immunosuppressant/immunomodulatory drugs (cyclophophamide, azathioprine, methotrexate, cyclosporine, leflunomide, mycophenolate mofetil and/or intravenous human immunoglobulin, in their full-dose, for a minimum period of 3 months) given sequentially or concomitantly, hampering glucocorticoid tapering. Upper dysphagia and pulmonary involvement were considered as disease severity parameters. Severe infection was defined as requiring hospitalization and/or intravenous antibiotic therapy.
Rituximab schedule
Rituximab treatment consisted of two infusions (1 g each, 2 weeks apart) and this same scheme was repeated 6 months after the first dose for patients showing no response or stable disease. The 6-month second dose was contraindicated for patients with recrudescent disease, hypogammaglobulinemia, side effects at first rituximab infusion and recurrent or severe infections. After starting on rituximab only one immunosuppressant was maintained at full-dose, and glucocorticoid tapering was started 2 months after initial rituximab treatment.
Disease activity
At the one-year evaluation, clinical and laboratory improvements were defined as > 20% improvement in at least three of the following modified IMACS core set measures: MMT-8 [12], physician’ and patient’ VAS [13], HAQ [16] and serum levels of muscle enzymes; with no more than two previous core set measures worsening by > 25%, which cannot include MMT.
Serum levels of creatine phosphokinase (normal range: 24–173 U/L) and aldolase (1.0–7.5 U/L) were evaluated. The following autoantibodies were investigated in this study: antinuclear factor (Hep2) and also anti-Jo-1, anti-OJ, anti-EJ, anti-PL-7, anti-PL-12, anti-Mi-2 and anti-SS-A/Ro-52. For the myositis-specific and myositis-associated autoantibodies’ assessment, a commercially available line blot test kit (Myositis Profile Euroline Blot test kit, Euroimmun, Lübeck, Germany) was used according to the manufacturer’s protocol and to the previously published study [17]. Reaction positivity was also defined according to a previously study [17].
Statistical analysis
The Kolmogorov-Smirnov test was used to evaluate the distribution of each parameter. The demographic and clinical features are expressed as mean ± standard deviation (SD) for the continuous variables or frequency (%) for the categorical variables. The median (25th - 75th interquartile range) was calculated for the continuous variables that were not normally distributed. Comparisons between different clinical, laboratory and treatment parameters at baseline and 12 months after rituximab infusion were performed using Student’s t-test or the Mann-Whitney U-test for continuous variables, whereas the Chi-squared test or Fisher’s exact test was used to evaluate the categorical variables. The 95% confidence interval (95% CI) of percentage was calculated by a binomial distribution. Age at Rituximab application sensitivity and specificity for identifying therapy responder were calculated, and a receiver operating characteristic (ROC) curve was constructed. P < 0.05 was considered significant. All of the analyses were performed using the SPSS 15.0 statistics software (Chicago, USA).
Results
Five of the initial 43 patients were later excluded due to moderate allergic reactions to the first rituximab infusion (N = 4) or lost to follow-up (N = 1). There were no cases of patients with recrudescent disease, hypogammaglobulinemia or severe infections. Therefore, 38 patients remained in the study for 1 year: 15 (39.5%) patients with DM, 10 (26.3%) with PM, and 13 (34.2%) with ASS (Table 1).
Among the 38 patients assessed, mean current age was 42.6 ± 10.9 years, 84.2% were female gender and 68.4% had white ethnicity. Median disease time was 3.0 years, whereas median time between disease diagnosis and symptom onset was 4.5 months.
The antinuclear factor was present in 81.6% of patients with the following autoantibodies specificities: anti-Ro-52 (42.1%), anti Jo-1 (34.2%), anti-Mi-2 (10.5%), and no cases of anti-OJ, anti-EJ, anti-PL-7 or anti-PL-12 autoantibodies.
All 38 patients were in concomitant use of at least two immunosuppressive / immunomodulatory drugs, in their full-dose, for a minimum period of 3 months, hampering glucocorticoid tapering. Due to disease severity, 34 (89.5%) patients had also received methylprednisolone pulse therapy 1 g/day, for three consecutive days, and/or intravenous human immunoglobulin (1 g/kg/day, for 2 days, for two consecutive days). Moreover, immediately before the first dose of rituximab, 23 (60.5%) of 38 patients received again this same scheme (methylprednisolone and intravenous human immunoglobulin pulse therapies). At the time of rituximab application, median dose of prednisone was 18.8 mg/day.
Comparison of therapies at study entry vs. 12 months after rituximab application revealed a reduction in median glucocorticoid dose (18.8 vs. 6.3 mg/day; P < 0.001) (Table 2) and complete discontinuation of prednisone in 16 (42.1%) of the 38 patients.
Twenty-nine (72.5%) of the 38 patients achieved overall progress according to the modified core set of IMACS after 12 months of rituximab treatment.
With regard to adverse events in the 38 patients at one-year follow-up, none had severe infection, two (5.3%) patients had mild allergic reactions and one (2.6%) patient was diagnosed with non-Hodgkin’s lymphoma (Table 2).
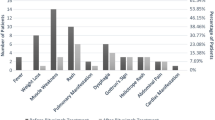
Further analysis of responders vs. non-responders at baseline identified younger age (P = 0.008) and higher frequency of dysphagia (P = 0.038) in non-responders (Table 3). The area under the ROC curve was 0.669 and age at 32 had 72% sensitivity and 67% specificity.
Female gender, ethnicity, disease duration and time between diagnosis and symptom onset were comparable between responder and non-responder groups. There was also no differences in myositis type (DM, PM or ASS), joint and pulmonary clinical symptoms, initial serum level of muscle enzymes, autoantibodies, or pre-treatment with methylprednisolone and intravenous human immunoglobulin pulse therapies (P > 0.05).
Discussion
In the present one-year study, long-term rituximab efficacy in refractory patients with IIM was demonstrated.
Rigorous criteria of refractoriness and also the modified IMACS disease activity response parameters were adopted in this research. Notably, due to disease severity, more than half of the patients also needed to receive methylprednisolone associated with intravenous human immunoglobulin pulse therapy to induce disease remission. In contrast, a less strict criterion of refractoriness was observed in previous studies and data on severe symptoms such as dysphagia were not reported hampering comparison with the present analysis [4,5,6,7,8,9,10,11]. In fact, among patients evaluated in the present study, more than two-thirds had dysphagia, a known serious problem in patients with IIM that can be associated with nutritional deficiency, aspiration pneumonia and poor prognosis [18].
The rituximab protocol was a more aggressive approach than others previously reported [4,5,6,7,8,9,10,11], and included pre-infusion of methylprednisolone and intravenous human immunoglobulin for the majority of the patients. In addition, the rituximab fixed dose retreatment protocol was chosen as opposed to on-demand retreatment [4,5,6,7,8,9,10,11], taking into account refractoriness.
The long-term IMACS modified response rate obtained with the present protocol was comparable to that reported for the RIM trial [5], a remarkable result taking into consideration the disease severity and refractoriness of the patients selected. Our data reveals that this outcome occurred for all patients at 6 months and the improvement persisted at 1 year. In this regard, a highly successful prednisone taper was obtained, with a significant early mean dose reduction at 6 months and a substantial number of patients (42%) able to completely discontinue prednisone at 12 months. Reinforcing these results, a parallel improvement in MMT-8, HAQ, physician and patient’ VAS, as well as in muscle enzymes occurred at 12 months.
Autoantibodies, especially anti-Jo-1 and anti-Mi-2, proved predictors of clinical improvement in a cohort of rituximab-treated myositis’ patients, whereas at lack of definable autoantibodies was a predictor of no improvement [5]. Although this association was not found in the present study, the majority of patients with anti-Jo-1 and all with anti-Mi-2 autoantibodies, responded to a rituximab.
During the follow-up, rituximab was well-tolerated with few adverse reactions. The most common side effects in literature are infections (mainly respiratory tract infections), of which 5% were severe, requiring hospitalization. Infusion reactions rarely occurred and these were often mild and easily controlled with glucocorticoid. Notably, there were no cases of severe infections requiring hospitalization in the present study. The intravenous human immunoglobulin pre-rituximab may be contributed for these data. However, sustained clinical and laboratory improvement may be due to rituximab, since there was no difference between responders and non-responders regarding previous use of intravenous human immunoglobulin.
As a limitation of the present study, a small sample was included, given the rarity of the IIM and the strict inclusion and exclusion criteria applied. Moreover, a sequential analysis of the dysphagia (i.e.: manometry) and pulmonary function were not performed. Finally, it should be emphasized that the concomitant use of methylprednisolone associated with intravenous human immunoglobulin for the majority of the patients might have affected the outcomes.
Conclusions
The present study provides convincing evidence that rituximab treatment is an effective and safe therapy for refractory IIM with a sustained 1 year response and significant tapering/discontinuation of glucocorticoid therapy. Moreover, young individuals and patients with dysphagia have a tendency to be more refractory to rituximab.
Abbreviations
- ASS:
-
Antisynthetase syndrome
- CI:
-
Confidence interval
- DM:
-
Dermatomyositis
- HAQ:
-
Health Assessment Questionnaire
- IIM:
-
Idiopathic inflammatory myopathies
- IMACS:
-
International Myositis Assessment & Clinical Studies Group
- MDAAT:
-
Myositis Disease Activity Assessment Tool
- MMT:
-
Manual muscle testing
- PM:
-
Polymyositis
- ROC:
-
Receiver operating characteristic
- SD:
-
Standard deviation
- VAS:
-
Visual analogue scale
References
Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371:2201–12.
Dalakas MC. Polymyositis, dermatomyositis and inclusion-body myositis. N Engl J Med. 1991;325:1487–98.
Fasano S, Gordon P, Hajji R, Loyo E, Isenberg DA. Rituximab in the treatment of inflammatory myopathies: a review. Rheumatology (Oxford). 2017;56:26–36.
Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65:314–24.
Aggarwal R, Bandos A, Reed AM, Ascherman DP, Barohn RJ, Feldman BM, et al; RIM Study Group, Oddis CV. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheum. 2014;66:740–9.
Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to anti-synthetase syndrome. Respir Med. 2012;106:581–7.
Levie TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601–7.
Noss EH, Hausner-Sypek DL, Weinblatt ME. Rituximab as therapy for refractory polymyositis and dermatomyositis. J Rheumatol. 2006;33:1021–6.
Brulhart L, Waldburger JM, Gabay C. Rituximab in the treatment of antisynthetase syndrome. Ann Rheum Dis. 2006;65:974–5.
Dinh HV, McCormack C, Hall S, Prince HM. Rituximab for the treatment of the skin manifestations of dermatomyositis: a report of 3 cases. J Am Acad Dermatol. 2007;56:148–53.
Frikha F, Rigolet A, Behin A, Fautrel B, Herson S, Benveniste O. Efficacy of rituximab in refractory and relapsing myositis with anti-Jo-1 antibodies: a report of two cases. Rheumatology. 2009;48:1166–8.
Rider LG, Koziol D, Giannini EH, Jain MS, Smith MR, Whitney-Mahoney K, et al. Validation of manual muscle testing and a subset of eight muscles for adult and juvenile idiopathic inflammatory myopathies. Arthritis Care Res (Hoboken). 2010;62:465–72.
Sultan SM, Allen E, Oddis CV, Kiely P, Cooper RG, Lundberg IE, et al. Reliability and validity of the myositis disease activity assessment tool. Arthritis Rheum. 2008;58:3593–9.
Mahler M, Miller FW, Fritzler MJ. Idiopathic inflammatory myopathies and the anti-synthetase syndrome: a comprehensive review. Autoimmun Rev. 2014;13:367–71.
Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344–7.
Bruce B, Fries JF. The Stanford health assessment questionnaire: dimensions and practical applications. Health Qual Life Outcomes. 2003;1:20.
Cruellas MG, Viana V dos S, Levy-Neto M, Souza FH, Shinjo SK. Myositis-specific and myositis-associated autoantibody profiles and their clinical associations in a large series of patients with polymyositis and dermatomyositis. Clinics. 2013;68:909–14.
Daković Z, Vesić S, Tomović M, Vuković J. Oropharyngeal dysphagia as dominant and life-threatening symptom in dermatomyositis. Vojnosanit Pregl. 2009;66:671–4.
Acknowledgments
Not applicable.
Funding
Federico Foundation to EB and SKS; Fundação Faculdade de Medicina to SKS.
Availability of data and materials
Please contact author for data requests.
Author information
Authors and Affiliations
Contributions
All authors contributed to write and review the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the local Ethics Committee (HCFMUSP, CAPPesq Number #0324/11).
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
de Souza, F.H.C., Miossi, R., de Moraes, J.C.B. et al. Favorable rituximab response in patients with refractory idiopathic inflammatory myopathies. Adv Rheumatol 58, 31 (2018). https://doi.org/10.1186/s42358-018-0030-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42358-018-0030-z