
Abstract
Background
All forms of shock, particularly septic shock, that involve insufficient blood supply to tissues to meet metabolic demands are main causes of morbidity and mortality worldwide and develop in one third of patients admitted to the intensive care unit (ICU). This alarmingly high incidence underlines the importance of further research in this field. Uncertainty remains about the undeceiving parameters for guidance of fluid resuscitation to avoid acute kidney injury (AKI) and renal replacement therapy. However, there are consequences of fluid overload (e.g. organ oedema and high interstitial pressures). This proposed study aims to establish a new framework of parameters for the guidance of fluid resuscitation in shock therapy focusing on the first 72 h, amending the currently used parameters (i.e. cardiac output, heart rate, blood pressure, central venous pressure) with an array of additional measurements including assessment of total body water (TBW), renal vascular resistance (renal resistive index (RRI)), intra-abdominal pressure (IAP) and microcirculatory blood flow (MBF).
Methods
This observational cohort study includes patients in shock presenting to the ICU. In addition to routine parameters (i.e. cardiac output, heart rate, blood pressure, central venous pressure) for volume resuscitation, MBF will be recorded using sublingual incident dark field microscopy, IAP, RRI seen in duplex-sonography and TBW will be estimated using bio-impedance analysis for correlation and eventually signalling of AKI and need for renal replacement therapy. Follow-up measurements will be taken at set intervals up to 72 h post admittance. To investigate the association between the newly investigated parameters and the occurrence of an AKI, we will compare the difference between measurements taken after admittance to the ICU and baseline values in patients with and without AKI using a two-sample t test. A linear regression model will be constructed to investigate the first and secondary outcome.
Results
Between January 2016 and August 2017, a total of 33 patients were recruited. The final results are expected in the second half of 2018.
Conclusions
The VoluKid study will investigate a new approach to assess volaemia and parameters indicating AKI in patients in shock who undergo volume resuscitation in the ICU.
Trial registration
The study was registered at ClinicalTrials.gov (Identifier: NCT02666404) on January 18, 2016, and at the Swiss National Clinical Trial Portal (SNCPT; Identifier: SNCTP000001693) on February 13, 2016.
Similar content being viewed by others

Background
Although the application of intravenous crystalloid fluids, red blood cell concentrates in anaemia and catecholamines or inotropes are the mainstay of shock therapy [1, 2], no clear endpoints for the fluid resuscitation or the haemodynamic endpoints for catecholamine therapy have been established so far [3]. Static parameters such as central venous or arterial pressure, cardiac output or plasma lactate fail to guide the amount of fluid administered [4]. The administration of intravenous fluids in response to so-called dynamic tests, such as stroke volume variation in response to fluid bolus administration, also did not show an influence on organ dysfunction and mortality due to shock [5]. Acute kidney injury (AKI) is the most frequent organ dysfunction in patients in shock [6]. Despite a more aggressive early fluid resuscitation and correction of arterial blood pressure, the incidence of AKI does not seem to decrease. One possible reason is that the excessive amount of fluid administered to these patients for haemodynamic stabilisation and maintenance of urinary output harms kidney function. Indeed, a correlation between total amount of fluid administered, high central venous pressure, organ dysfunction and mortality has been shown in patients with severe sepsis and septic shock. Established static and dynamic haemodynamic parameters are not influenced by the severity of capillary leakage and microcirculatory impairment due to inflammation-induced injury of capillaries [6,7,8]. The relatively low perfusion pressure together with interstitial oedema, microcirculatory injury and high outflow pressure (e.g. central venous pressure) may harm renal function by a decreased glomerular urine excretion. Intra-abdominal pressure (IAP), total body water (TBW) and central venous pressure together with the renal resistive index (RRI) may be additionally measured and integrated to reduce the rate of AKI and to guide fluid therapy in shock.
Primary objective
In patients with shock, AKI is the most common organ failure resulting in severe morbidity and mortality [9]. Because classical haemodynamic measurements [10,11,12] have failed to monitor and improve the risk of AKI, we plan to prospectively evaluate the correlation of regional parameters, such as IAP, RRI, microcirculation and TBW, for occurrence of AKI and renal replacement therapy.
We expect both IAP and the RRI to be higher in patients with or at risk for AKI compared to those without. We also expect to find consistent differences in all parameters between patients with and without AKI, as well as in patients with and without increased lactate values. In contrast to established haemodynamic parameters of preload and flow, like central venous pressure or cardiac output and blood pressure, IAP and RRI are influenced by the severity of capillary leakage and microcirculatory impairment.
We hypothesise that by integrating IAP and RRI for guidance of fluid resuscitation in shock, we will be able to reduce incidence of AKI and the consecutive need for renal replacement therapy as well as diminish fluid overload and its sequelae, but eventually patient morbidity and mortality.
The goal of the present study is to refine fluid resuscitation endpoints with additional regional measurements to administer the optimal amount of fluid with the smallest possible adverse effects.
Methods
Trial design
This is an observational study of patients in shock to explore values for intra-abdominal pressure, renal resistive index (RRI), sublingual microcirculation and measurement of total body water estimated using bio-impedance analysis that correlate and eventually signal AKI and need for renal replacement therapy. We would like to recruit 100 patients right after the diagnosis of shock.
Approvals
Approval to conduct this study was granted by the Ethics Committee of Northwestern and Central Switzerland (EKNZ 2015-401) in September 2016. The study is registered at ClinicalTrials.gov (identifier NCT02666404) and at the Swiss National Clinical Trial Portal (SNCPT; identifier SNCTP000001693).
Setting
Study design
This Observational Clinical Trial will be conducted at the Surgical Intensive Care Unit of the University Hospital Basel, Switzerland. Patient recruitment has already begun.
Sample size
For the VoluKid pilot study, we will enrol a total of 100 patients in the state of shock. Assuming an incidence of AKI of 40–60%, this sample size would allow us to build a multivariable model for AKI including the most relevant factors according to a rule-of-thumb of 10 events per variable in a logistic regression model [13].
Eligibility criteria
Patient screening and recruitment
All patients admitted to the ICU are automatically evaluated for study eligibility. The diagnosis of shock is mainly clinical, based on evidence of standardised and internationally established shock criteria.
We will screen every admitted ICU patient for participation in the study until we reach the targeted sample size.
Inclusion criteria
Participants fulfilling the following inclusion criteria are eligible for the study:
-
Adult patients (aged ≥ 18 years)
-
Patient is in a state of shock (any type, fluid dependent) upon admittance to the ICU
Exclusion criteria
Participants meeting the following criteria are excluded from the study:
-
Patients aged < 18 years
-
Non-shock state
-
Terminal state
-
Chronic atrial fibrillation
-
Pregnancy
-
Jehovah’s Witnesses
-
Chronic kidney disease
Criteria for withdrawal/discontinuation of participants
Participants will be withdrawn from the study if one of the following incidents occurs:
-
Rapid recovery
-
On demand of the patient or his representative (next of kin/nominated consultee)
-
Missing baseline values
After withdrawal/discontinuation of a participant, data of this specific patient will not be used, and the participant will be replaced by a new one to reach our goal of 100 complete data sets. In case of ex-post study withdrawal, patient data will be destroyed.
Trial assessments
Personal characteristics and demographic data
The following patient characteristics and demographic data will be collected from patient files or assessed upon admittance to the ICU:
-
Past medical history (focused on coronary heart disease, peripheral artery occlusive disease, diabetes, kidney disease)
-
Intensive care severity scores: SOFA (Sequential Organ Failure Assessment, Appendix 1 [14]) score (daily), SAPS (Simplified Acute Physiology, see Appendix 2 [15]) II score, CACI (Age-adjusted Charlson Comorbidity Index, see Appendix 3 [16])
-
Administered pre-study blood products and intravenous fluids
-
Type of shock
-
Nature of initial surgical procedure(s) and treatments
Established parameters
-
Demographic data (age, gender)
-
Haemodynamic parameters (cardiac index [10], central venous pressure [10, 11], blood pressure [10, 12, 17], heart rate, central/mixed venous oxygen saturation = ScvO2/SvO2, pulmonary capillary occlusion pressure (PAOP) [10])
-
Use of inotropic [18, 19] and vasoactive [19] drugs (epinephrine, norepinephrine, milrinone, dobutamine and levosimendan)
-
Laboratory parameters (blood gas analysis including lactate [19,20,21,22], HCO3−, sodium [23], potassium, blood sugar concentration [24] and need for insulin therapy [25] with respect to need for immunosuppressive therapy [26])
-
Parameters of renal function (creatinine, creatinine clearance, blood urea nitrogen, cystatine C, urine output, potassium, urinary protein/albumin, urinary sediment analysis, sodium excretion fraction, need for renal replacement therapy) [17, 27,28,29,30,31,32,33,34,35,36]
-
Creatinine clearance after 24, 48 and 72 h
-
Urinary sediment analysis at 72 h
-
Urinary protein screening after 72 h
-
FLUID administration during study period (72 h)
-
Total amount of fluids (mL) administered [1,2,3, 24, 37,38,39,40,41] with consideration of total sodium and chloride administered
-
Total amount of crystalloids (mL) administered after onset of shock symptoms [28, 42]
-
Total amount of colloids (mL) administered [27, 28, 43,44,45,46]: Voluven, Gelofundin, amount of albumin infusion (ml of albumin 5 or 20%) [3]
-
Amount of red blood cell [47,48,49], thrombocyte and plasma transfusion (ml)
Alternative parameters influenced by severity of capillary leakage and microcirculatory injury
-
Renal artery resistive index (Doppler analysis)
-
IAP (bladder pressure measurement)
-
TBW (bio-impedance analysis)
-
MBF [50] (CytoCam incident dark-field illumination)
Trial measurements
Data sources and study measurements
Time points and techniques for data collection
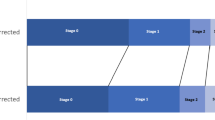
Figure 1 shows an overview of the practical techniques, samples taken and the data collected at the four time points of measurement.

Study period overview
Measurements are performed at the following time points:
-
1.
As soon as patient is stable and measurements are feasible
-
2.
24 h after admission
-
3.
48 h after admission
-
4.
72 h after admission
Experimental measurements
Bio-impedance analysis is a commonly used method for assessment of body composition. It actually determines the electrical impedance (opposition to the flow of an electric current through body tissues which can then be used to calculate an estimate of TBW). TBW is used to estimate fat-free body mass and, by difference with body weight, body fat. In our study, we use the following device: BIA 101 Anniversary Sport Edition, AKERN, serial no ASE201305001.
Intra-abdominal pressure measurement: Elevated IAP can be seen in a variety of critically ill patients. The threat of intra-abdominal compartment syndrome renders diagnosis and management of elevated IAP of high importance for improvement of patient outcome. IAP represents the pressure within the abdominal cavity and is influenced by numerous factors: breathing (IAP increases with inspiration and decreases with expiration), volume of solid organs or hollow viscera, ascites, blood, tumours, gravid uterus and conditions that limit abdominal wall expansion (scars, oedema).
Normal IAP is estimated at 5–7 mmHg in the critically ill; however, this varies according to disease severity with an IAP of 20–30 mmHg frequently found in patients with severe sepsis or an acute abdomen [51]. An IAP above 15 mmHg usually is associated with significant end-organ dysfunction and failure in consistence with grade II intra-abdominal hypertension (IAH) as defined by the International Conference of Experts on Intra-Abdominal Hypertension and Abdominal Compartment Syndrome.
Analogous to the widely accepted concept of cerebral perfusion pressure (CPP = mean arterial pressure minus intracranial pressure), abdominal perfusion pressure (APP, accordingly calculated as mean arterial pressure (MAP) minus IAP) has been proposed as a more accurate predictor of visceral perfusion and as an endpoint for resuscitation [51,52,53]. One must note that APP, by considering both arterial inflow (MAP) and restrictions to venous outflow (IAP), could be demonstrated to be statistically superior to MAP or IAP alone as well as to other common resuscitation endpoints (e.g. arterial pH, base deficit, arterial lactate and hourly urinary output) for survival prediction from IAH/abdominal compartment syndrome (ACS) (Fig. 1). A target APP of 60 mmHg is recommended since it has been demonstrated to correlate with improved survival from IAH/ACS [52].
IAH has been defined as a sustained or repeated pathologic elevation of IAP > 12 mmHg [51, 52] and is graded as follows:
Grade I | IAP 12–15 mmHg |
Grade II | IAP 16–20 mmHg |
Grade III | IAP 21–25 mmHg |
Grade IV | IAP > 25 mmHg |
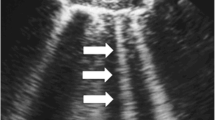
Doppler analysis for RRI: Renal resistive index (RRI) is a commonly used ultrasound method based on the Doppler effect to evaluate the peripheral resistance in the respective organ. Using colour Doppler sonography, we will locate interlobar arteries in the upper, middle and lower pole of each kidney. During examination at least two identical spectral waves will be examined to determine RRI. The RRI of an interlobar artery is defined as the difference between peak systolic and end-diastolic blood velocities divided by peak systolic velocity [54]. A mean of the six RRI values is calculated resulting in one value per examination and patient. In healthy volunteers, activation of auto-regulatory mechanisms through compression of renal vessels can be assessed by a fall of RRI [55]. There seems to be a strong correlation between RRI and renal functional reserve (RFR) which is defined as the capacity to increase the glomerular filtration rate after a stress test (e.g. protein load). Lack of RFR also is suggested to be a risk factor for AKI. The course of RRI over time may therefore be considered as a surrogate of renal functional reserve indicating the degree of kidney injury [56].
CytoCam incident darkfield illumination: Oxygen is delivered to tissue cells exclusively by microcirculation, which involves a complex interaction of the cellular systems, including red and white blood cells, endothelial, smooth muscle and parenchymal cells. The function of the organs is directly dependent on the function of their respective microcirculation. Therefore, achieving sufficient microcirculatory function can be considered to be the main purpose of the cardiovascular system and bears particular importance to critically ill patients, especially those in shock. Particularly in septic patients, studies [57] have demonstrated adverse or persistent alterations in microcirculation unresponsive to therapy. In addition, these microcirculatory alterations have been shown in various studies to be independent of systemic hemodynamic variables, making the observation of microcirculation a potentially important extension of the conventional systemic hemodynamic monitoring of critically ill patients which is part of the rationale for our study-specific measurements [58,59,60].
CytoCam-IDF imaging can be regarded as a handheld microscope using a pen-like probe incorporating IDF illumination for examination of the microcirculation [61].
Rationale for study measurements: We would like to integrate CytoCam-IDF imaging of microcirculation in our study to evaluate coherence between intra-abdominal pressure and microcirculation of sublingual tissue. We could reason to stop fluid resuscitation at the level of intra-abdominal pressure where we measure full resuscitation of sublingual microcirculation.
The aim of our study is to define an IAP value associated with imminent kidney dysfunction and failure and put it in context with the amount of fluids administered for volume resuscitation (i.e. TBW). We hope to establish a clear definition to indicate the RRI levels at which normal/sufficient kidney function is in danger, given the highly probable connection between RRI and AKI.
Outcome measures
Primary outcome measure
The primary outcome measure is the incidence of acute kidney injury (AKI).
Secondary outcome measures
Need for renal replacement therapy (first secondary outcome; KDIGO staging for severity of AKI, see Appendix 4 [62]), length of ICU stay (hours), changes in SOFA score over time (daily assessment of SOFA score), length of hospital stay (days), need for mechanical ventilation, in-hospital and 30- and 90-day mortality.
Definitions/conditions
Inclusion criteria
Criteria for diagnosis of shock state:
-
Evidence of insufficient tissue perfusion (obtundation, oliguria, peripheral cyanosis and skin mottling)
-
Signs of compensatory mechanisms (tachycardia, tachypnoea, diaphoresis)
-
Fluid dependency based on fluid responsiveness, defined as increase in cardiac output or stroke volume following a preload challenge [63]
-
Specific criteria:
◦ Altered mental status
◦ Heart rate > 100
◦ Respiratory rate > 22 [64]
◦ Hypotension (systolic BP < 90 mmHg) [65, 66] or a 30-mmHg fall in baseline BP
◦ Urine output < 0.5 mL/kg/h
-
Laboratory findings:
◦ Base deficit < − 4 mEq/L [64]
◦ PaCO2 < 32 mmHg
Exclusion criteria
Age < 18 years: We will only include adult patients in our study.
Non-shock state: Patients who do not fulfil the abovementioned criteria for the status of shock will not be included in our study.
Terminal state: Patients suffering from an incurable disease and who have a terminal illness will not be included in our study.
Chronic atrial fibrillation: We define chronic atrial fibrillation by an abnormal heart rhythm that continuous for more than a week.
Pregnancy: A negative pregnancy test will be necessary for study inclusion in women aged ≤ 45 years.
Jehovah’s Witnesses: We will not include adult patients that are members of this religious community.
Chronic kidney disease: We will exclude patients with known deteriorated kidney function prior to ICU admission.
Primary outcome measure
We define AKI by KDIGO stage 1 [62] or a need for renal replacement therapy [27]. The necessity for RRT is defined as cumulative positive fluid balance > 0.1 l/kg body weight AND
-
Urin output < 0.5 ml/kg for 12 h OR
-
Serum potassium level > 5.5 mmol/l OR
-
paO2 < 10 kPa with PEEP levels > 12 mbar
Serious adverse event
Serious adverse events (SAEs) are classified as any medical occurrence that results in death, is life-threatening, requires prolongation of existing hospitalisation, or results in persistent or significant disability/incapacity.
Study period overview
The study period consists of enrolment and three additional scheduled measurement points after 24, 48, and 72 h (Fig. 1). Enrolment is defined as the point of time the shock state is being detected.
Screening
As mentioned above, all patients admitted to the ICU are automatically being evaluated for study eligibility (Fig. 2).

Steps of evaluation of study eligibility
Data management
Data collection methods
The collected data will be summarised and stored in an electronic Case Report Form established by the Clinical Trials Unit (CTU) Basel.
Data handling
In agreement with GCP guidelines for anonymization of patient data, our Case Report Form will include only the patient’s surname and year of birth, which together will be used to issue a unique file number. As we will perform further measurements in addition to the established parameters currently used for guidance of fluid resuscitation of patients in shock, the investigators will not need to be blinded. The treating board-certified intensivist will not have access to the data. All data from this multicentre study will be kept within the Investigator Site File, and only the study team will have access to it.
All study data will be archived for a minimum of 10 years after study termination or premature termination of the clinical trial.
Data collection and follow-up of withdrawn participants
An individual participant will be excluded from the study if any of the following occur in the participant in question:
-
Withdrawal of consent by the patient or the independent physician
-
An adverse event occurs that, in the opinion of the sponsor, contraindicates further measurements (emergency setting)
Participants who could not be followed over the intended period and all designated points of assessments, regardless of reason, will not be followed. Unless consent for follow-up is withdrawn, participants will be followed for the full study period with all laboratory and clinical evaluations collected as defined in the protocol. We guarantee that our measurements will in no way delay therapy.
Data monitoring
No regular monitoring visits at the investigator’s site prior to the start and during the course of the study are planned by the Sponsor.
The source data/documents will remain accessible to monitors, and questions will be answered during any possible monitoring.
Data collection (e.g., assessment of RRI) will be closely supervised by the project partners and the sponsor of the study.
Confidentiality and data access
The investigator affirms and upholds the principle of the participant’s right to privacy and that they shall comply with applicable privacy laws. Anonymity of the participants will be expressly guaranteed when presenting the data at scientific meetings or publishing them in scientific journals.
Medical information of individual participants obtained as a result of this study will be considered confidential, and disclosure to third parties will be prohibited. If necessary, participant confidentiality will be further ensured by utilising participant identification code numbers to correspond to treatment data in the computer files. Only the study team will gain access to study-specific data.
For data verification purposes, authorised representatives of the Sponsor (Investigator), a competent authority (e.g. Swissmedic) or an ethics committee may require direct access to parts of the medical records relevant to the study, including each participant’s medical history.
Patient information and informed consent
As we will only include patients in shock in our study, whenever possible, we will explain the nature of the study, its purpose, the procedures involved, the expected duration, the potential risks and benefits and any discomfort it may entail to each participant. Each participant will be informed that the participation in the study is voluntary and that he/she may withdraw from the study at any time and that withdrawal of consent will not affect his/her subsequent medical assistance and treatment.
If anyhow possible, the participant or their representative will be informed that the patient’s medical records may be examined by authorised individuals other than their treating physician.
All participants in the study will be provided a participant information sheet and a consent form describing the study and providing sufficient information for participant to make an informed decision about their participation in the study. Depending on the participant’s state of health, there might be no time for the enrolment decision.
The patient information sheet and the consent form will be submitted to the CEC to be reviewed and approved. If possible, the formal consent of a participant, using the approved consent form, must be obtained before the participant is submitted to any study procedure.
If possible, the participant or their representative should read and consider the statement before signing and dating the informed consent form and should be given a copy of the signed document. The consent form must also be signed and dated by the investigator (or his designee), and it will be retained as part of the study records.
If the patient’s general condition does not allow informed consent, an independent physician (not member of the study group) will evaluate this specific patient’s enrolment in our study and sign the informed consent form in the patient’s name as his representative. After recovery, the patient will be informed about his/her participation in the trial and he/she will have the possibility to withdraw his data from the study. In case of ex-post study withdrawal, patient data will be destroyed.
The sponsor/investigator may terminate the study prematurely according to certain circumstances, for example:
-
Ethical concerns
-
Insufficient participant recruitment
-
When the safety of the participants is doubtful or at risk
-
When there are alterations in accepted clinical practice that make the continuation of the study unwise
In case of preterm study termination, the collected data will not be published.
Ethics and dissemination
Ethical conduct
The study will be carried out in accordance to the protocol and with principles detailed in the current version of the Declaration of Helsinki, the guidelines of Good Clinical Practice (GCP) issued by ICH, in case of medical device: the European Directive on medical devices 93/42/EEC and the ISO Norm 14155 and ISO 14971, the Swiss Law and Swiss regulatory authority’s requirements. The CEC and regulatory authorities will receive annual safety and interim reports and will be informed about study termination/end in agreement with local requirements.
Research ethics approval
The Sponsor is responsible for implementing and maintaining quality assurance and quality control systems with written SOPs and Working Instructions at all sites in this multicentre study. The principal investigator is responsible for proper training of all involved study personnel.
The responsible investigator ensures that approval from an appropriately constituted Competent Ethics Committee (CEC; EKNZ in Switzerland) is sought for the clinical study.
Since all our measurements are non-invasive, serious adverse events are not to be expected in our study.
The sponsor and principal investigator are responsible to report all changes in the research activity and all unanticipated problems involving risks to humans, this includes planned or premature study end and the final report. The principal investigator guarantees that no changes will be made to the protocol without prior sponsor and CEC approval, except where necessary to eliminate apparent immediate hazards to study participants.
Premature study end or interruption of the study will be reported within 15 days. The regular end of the study will be reported to the CEC within 90 days, and the final study report will be submitted within 1 year after study end.
Protocol amendments
Substantial amendments will only be implemented after approval of the CEC and Competent Authority (CA), respectively. Under emergency circumstances, deviations from the protocol to protect the rights, safety and well-being of patients may proceed without prior approval of the sponsor and the CEC/CA. Such deviations will be documented and reported to the sponsor and the CEC/CA as soon as possible.
All non-substantial amendments will be communicated to the CA as soon as possible if applicable and to the CEC within the Annual Safety Report (ASR).
Patient safety
As we are only performing non-invasive measurements in additional to interventions, pharmacotherapy and current routine measurements in patients in shock, there will be no contributing risk to patients by study enrolment. By using study-specific measurements (bio-impedance analysis, Doppler analysis, CytoCam incident dark-field illumination), we expect to develop a more precise way to guide fluid resuscitation in patients in shock. This study has a strong potential of benefits for future ICU patients.
Methods of minimising bias
Patients in shock require intravascular volume resuscitation. On our case report form, we will document the amount and type of volume administered, perform the foreseen measurements and analyse the predictive value for AKI post hoc. We hope to use our dataset to establish new endpoints to guide fluid resuscitation in patients in shock.
Due to the pure observational approach, our study has a very low risk for bias. We only are looking for correlations between the measured parameters, amount and type of fluid administered and the incidence of AKI. From this data, no causality should be generated.
Audits and inspections
No audit of trial conduct is planned. The study documentation and the source data/documents will remain accessible to auditors/inspectors (also Competent Ethics Committee and Competent Authorities), and questions will be answered during inspections. All involved parties will keep the participant data strictly confidential.
Declaration of interests
This study protocol was composed and approved by both the sponsor and principal investigator. No part of this study will be submitted elsewhere either in total or in part. Each of the authors declares that there are no conflicts of interest.
Ancillary and post-trial care
Not applicable. No financial or other form of compensation for study participation will be provided.
Publication and dissemination policy
Study results will be communicated to patients based on expected early convalescence from shock. There will be no public access to our data during the study and until publication.
We plan to publish the data in a major peer-reviewed clinical journal.
Statistics
Sample size calculation
Since no pilot study has been done to determine the standard deviation of our population, our sample size calculation has been based on the assumption of AKI incidence between 40 and 60%. Thereby, a random sample of 100 patients with a sample proportion of 0.5 and a confidence interval of 95% (i.e. z* value of 1.96) reveals a margin of error of +/− 1.96 × 0.05 = 0.098, or 9.8%. So our 95% confidence interval for the percentage of times our patients will develop AKI is 0.5 (or 50%), +/− 0.098 (rounded to 0.1 or 10%). In other words, using a confidence level of 95% and a confidence interval of 10 with the assumption that 50% (40–60%) develop AKI, the calculated sample size would be 96.
Statistical analysis
Baseline characteristics will be summarised for patients with and without AKI using mean (standard deviation), median (interquartile range) or number (percentages), as appropriate.
To investigate the association of the new parameters with the occurrence of an AKI, we will compare the difference between measurements taken after admittance to the ICU with baseline values in patients with and without AKI using a two-sample t test. In addition, we will build a logistic regression model including the differences between these same measurements for the following parameters: volume of infusion, IAP, RRI and cardiac output. We will repeat this model using the differences between measurements taken 24 h after admittance to the ICU and the baseline values.
More precisely, we will use the simple logistic regression with AKI as the dependent variable to separately estimate the influence of the independent variables (i.e. volume of infusion, RRI, IAP and cardiac output). With the multiple logistic regression with the same dependent and independent variables, we will investigate the combined influence of volume of infusion, RRI, IAP and cardiac output on AKI in varying combinations of the dependent variables depending on the results of the simple logistic regression.
In addition, we will plot the course of IAP, RRI, cardiac output and blood pressure on a time scale and mark the time-point at which AKI was detected to find a possible correlation between the extent of change of values and the anticipation/early detection of AKI.
Secondary outcomes (need for renal replacement therapy (KDIGO staging for severity of AKI), length of ICU stay, length of hospital stay, need for mechanical ventilation in-hospital and 30- and 90-day mortality) will be explored descriptively. The course of IAP and RRI together with cardiac output and blood pressure will be plotted over time in patients with and without AKI and in patients with and without decrease in lactate.
All analyses and graphs will be performed using Intercooled Stata Version 13.1 for Macintosh (StataCorp, College Station, TX, USA).
There will be no interim analyses or stopping guidelines for the study except in the event of a patient’s death. Even if there is any indication that integration of the additional parameters lowers the incidence of AKI and renal replacement therapy in the planned prospective evaluation of fluid volume resuscitation management guided by IAP and RRI, we will continue and finish the study as planned. No fixed interim analysis is planned at this point.
Results
Participant recruitment began in January 2016, and completion is estimated for end 2018. All patients admitted to the Surgical Intensive Care Unit, University Hospital Basel, Switzerland, are screened for study eligibility.
Discussion
Trial rationale
Complexity of therapy in severe sepsis and septic shock as well as in other shock states was the topic of numerous studies showing no benefit of advanced macrocirculatory parameters for guidance of fluid resuscitation in shock. In our study, we follow a new approach due to prospective analysis of the study-specific measurements. Independent from our objective to establish new endpoints for guidance in shock therapy, our research is planned to be expanded to the evaluation of future therapies [67], such as renal biomarkers (e.g. liver fatty acid binding protein (L-FABP), (NGAL), kidney injury molecule-1 (KIM-1) and N-acetyl-β-D glucosaminidase (NAG)) [68, 69].
After establishment of more regional volume resuscitation endpoints, we plan to evaluate these endpoints in a randomised, controlled setting against established of haemodynamically guided shock therapy. Evidence from these regional parameters combined with the measurement of renal biomarkers might help to detect patients who would or would not benefit further from volume resuscitation.
Conclusion
The study’s main strength is the innovative approach of implementation of new methods to assess volaemia and parameters indicating AKI in patients in shock who undergo volume resuscitation in the ICU. The aim is to anticipate or evidence AKI earlier based on the detection of a pattern change of the patient’s volume status. The VoluKid study will recruit from the surgical intensive care unit, University Hospital Basel, Switzerland. The study is limited by the heterogeneous general condition and past medical history of critically ill patients who suffer from different types of shock.
References
Carlsen S, Perner A. East Danish Septic Shock Cohort I. Initial fluid resuscitation of patients with septic shock in the intensive care unit. Acta Anaesthesiol Scand. 2011;55(4):394–400. https://doi.org/10.1111/j.1399-6576.2011.02399.x.
Rochwerg B, Alhazzani W, Gibson A, et al. Fluid type and the use of renal replacement therapy in sepsis: a systematic review and network meta-analysis. Intensive Care Med. 2015; https://doi.org/10.1007/s00134-015-3794-1.
Leone M. Septic shock resuscitation: assembling the puzzle*. Crit Care Med. 2014;42(10):2294–5. https://doi.org/10.1097/CCM.0000000000000536.
Cherpanath TGV, Geerts BF, Lagrand WK, et al. Basic concepts of fluid responsiveness. Neth Hear J. 2013;21(12):530–6. https://doi.org/10.1007/s12471-013-0487-7.
Xu Q, Yan J, Cai G, et al. Effect of two volume responsiveness evaluation methods on fluid resuscitation and prognosis in septic shock patients. Chin Med J. 2014;127(3):483–7.
Ergin B, Kapucu A, Demirci-Tansel C, et al. The renal microcirculation in sepsis. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2015;30(2):169–77. https://doi.org/10.1093/ndt/gfu105 [published Online First: 2014/05/23].
Tatara T. Context-sensitive fluid therapy in critical illness. J Intensive Care. 2016;4:20. https://doi.org/10.1186/s40560-016-0150-7.
Saugel B, Huber W, Nierhaus A, et al. Advanced hemodynamic management in patients with septic shock. Biomed Res Int. 2016;2016:8268569. https://doi.org/10.1155/2016/8268569.
Hoste EA, De Corte W. Epidemiology of AKI in the ICU. Acta Clin Belg. 2007;62(Suppl 2):314–7. [published Online First: 2008/02/21]
Mullens W, Abrahams Z, Francis GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53(7):589–96. https://doi.org/10.1016/j.jacc.2008.05.068.
Boyd JH, Forbes J, Nakada TA, et al. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med. 2011;39(2):259–65. https://doi.org/10.1097/CCM.0b013e3181feeb15.
Asfar P, Meziani F, Hamel JF, et al. High versus low blood-pressure target in patients with septic shock. N Engl J Med. 2014;370(17):1583–93. https://doi.org/10.1056/NEJMoa1312173.
Peduzzi P, Concato J, Kemper E, et al. A simulation study of the number of events per variable in logistic regression analysis. J Clin Epidemiol. 1996;49(12):1373–9.
Vincent JL, Moreno R, Takala J, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22(7):707–10.
Le Gall JR, Lemeshow S, Saulnier F. A new Simplified Acute Physiology Score (SAPS II) based on a European/North American multicenter study. JAMA. 1993;270(24):2957–63.
Charlson M, Szatrowski TP, Peterson J, et al. Validation of a combined comorbidity index. J Clin Epidemiol. 1994;47(11):1245–51.
Leone M, Asfar P, Radermacher P, et al. Optimizing mean arterial pressure in septic shock: a critical reappraisal of the literature. Crit Care. 2015;19:101. https://doi.org/10.1186/s13054-015-0794-z.
Bozza FA, Carnevale R, Japiassu AM, et al. Early fluid resuscitation in sepsis: evidence and perspectives. Shock. 2010;34(Suppl 1):40–3. https://doi.org/10.1097/SHK.0b013e3181e7e668.
Vincent JL, De Backer D. Circulatory shock. N Engl J Med. 2013;369(18):1726–34. https://doi.org/10.1056/NEJMra1208943.
Jansen TC, van Bommel J, Schoonderbeek FJ, et al. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med. 2010;182(6):752–61. https://doi.org/10.1164/rccm.200912-1918OC.
Lilly CM. The ProCESS trial––a new era of sepsis management. N Engl J Med. 2014;370(18):1750–1. https://doi.org/10.1056/NEJMe1402564.
Capp R, Horton CL, Takhar SS, et al. Predictors of patients who present to the emergency department with sepsis and progress to septic shock between 4 and 48 hours of emergency department arrival. Crit Care Med. 2015;43(5):983–8. https://doi.org/10.1097/CCM.0000000000000861.
Van de Louw A, Shaffer C, Schaefer E. Early intensive care unit-acquired hypernatremia in severe sepsis patients receiving 0.9% saline fluid resuscitation. Acta Anaesthesiol Scand. 2014;58(8):1007–14. https://doi.org/10.1111/aas.12368.
Martensson J, Bellomo R. Prevention of renal dysfunction in postoperative elderly patients. Curr Opin Crit Care. 2014;20(4):451–9. https://doi.org/10.1097/MCC.0000000000000107.
Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008;358(2):125–39. https://doi.org/10.1056/NEJMoa070716.
Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13(3):260–8. https://doi.org/10.1016/S1473-3099(13)70001-X.
Schortgen F, Lacherade JC, Bruneel F, Cattaneo I, Hemery F, Lemaire F, Brochard L. Effects of hydroxyethylstarch and gelatin on renal function in severe sepsis: a multicentre randomised study. Lancet. 2001;357(9260):911–6.
Bayer O, Reinhart K, Sakr Y, et al. Renal effects of synthetic colloids and crystalloids in patients with severe sepsis: a prospective sequential comparison. Crit Care Med. 2011;39(6):1335–42. https://doi.org/10.1097/CCM.0b013e318212096a.
Amery CE, Forni LG. Renal disease presenting as acute kidney injury: the diagnostic conundrum on the intensive care unit. Curr Opin Crit Care. 2014;20(6):606–12. https://doi.org/10.1097/MCC.0000000000000155.
Zarbock A, Gomez H, Kellum JA. Sepsis-induced acute kidney injury revisited: pathophysiology, prevention and future therapies. Curr Opin Crit Care. 2014;20(6):588–95. https://doi.org/10.1097/MCC.0000000000000153.
Druml W. Renal dysfunction in heart failure and hypervolumenia: importance of congestion and backward failure. Med Klin Intensivmed Notfmed. 2014;109(4):252–6. https://doi.org/10.1007/s00063-013-0323-2.
Chawla LS, Eggers PW, Star RA, et al. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371(1):58–66. https://doi.org/10.1056/NEJMra1214243.
Muller RB, Haase N, Lange T, et al. Acute kidney injury with hydroxyethyl starch 130/0.42 in severe sepsis. Acta Anaesthesiol Scand. 2015;59(3):329–36. https://doi.org/10.1111/aas.12453.
Guirgis FW, Williams DJ, Hale M, et al. The relationship of intravenous fluid chloride content to kidney function in patients with severe sepsis or septic shock. Am J Emerg Med. 2015;33(3):439–43. https://doi.org/10.1016/j.ajem.2014.12.013.
Damman K, Testani JM. The kidney in heart failure: an update. Eur Heart J. 2015;36(23):1437–44. https://doi.org/10.1093/eurheartj/ehv010.
Gomez H, Ince C, De Backer D, et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41(1):3–11. https://doi.org/10.1097/SHK.0000000000000052.
Trof RJ, Sukul SP, Twisk JW, et al. Greater cardiac response of colloid than saline fluid loading in septic and non-septic critically ill patients with clinical hypovolaemia. Intensive Care Med. 2010;36(4):697–701. https://doi.org/10.1007/s00134-010-1776-x.
Micek ST, McEvoy C, McKenzie M, et al. Fluid balance and cardiac function in septic shock as predictors of hospital mortality. Crit Care. 2013;17(5):R246. https://doi.org/10.1186/cc13072.
Sirvent JM, Ferri C, Baro A, et al. Fluid balance in sepsis and septic shock as a determining factor of mortality. Am J Emerg Med. 2015;33(2):186–9. https://doi.org/10.1016/j.ajem.2014.11.016.
Marik PE. The demise of early goal-directed therapy for severe sepsis and septic shock. Acta Anaesthesiol Scand. 2015;59(5):561–7. https://doi.org/10.1111/aas.12479.
Bihari S, Prakash S, Bersten AD. Post resusicitation fluid boluses in severe sepsis or septic shock: prevalence and efficacy (price study). Shock. 2013;40(1):28–34. https://doi.org/10.1097/SHK.0b013e31829727f1.
Bark BP, Oberg CM, Grande PO. Plasma volume expansion by 0.9% NaCl during sepsis/systemic inflammatory response syndrome, after hemorrhage, and during a normal state. Shock. 2013;40(1):59–64. https://doi.org/10.1097/SHK.0b013e3182986a62.
Trof RJ, Groeneveld AB. Use of synthetic colloids in sepsis: a critical review on efficacy, safety and patient benefits. Minerva Anestesiol. 2011;77(12):1216–23.
Mutter TC, Ruth CA, Dart AB. Hydroxyethyl starch (HES) versus other fluid therapies: effects on kidney function. The Cochrane database of systematic reviews. 2013;7:CD007594. https://doi.org/10.1002/14651858.CD007594.pub3.
Dart AB, Mutter TC, Ruth CA, et al. Hydroxyethyl starch (HES) versus other fluid therapies: effects on kidney function. The Cochrane database of systematic reviews. 2010;1:CD007594. https://doi.org/10.1002/14651858.CD007594.pub2.
Dubin A, Pozo MO, Casabella CA, et al. Comparison of 6% hydroxyethyl starch 130/0.4 and saline solution for resuscitation of the microcirculation during the early goal-directed therapy of septic patients. J Crit Care. 2010;25(4):659 e1-8. https://doi.org/10.1016/j.jcrc.2010.04.007.
Perner A, Smith SH, Carlsen S, et al. Red blood cell transfusion during septic shock in the ICU. Acta Anaesthesiol Scand. 2012;56(6):718–23. https://doi.org/10.1111/j.1399-6576.2012.02666.x.
Park DW, Chun BC, Kwon SS, et al. Red blood cell transfusions are associated with lower mortality in patients with severe sepsis and septic shock: a propensity-matched analysis*. Crit Care Med. 2012;40(12):3140–5. https://doi.org/10.1097/CCM.0b013e3182657b75.
Hebert PC, Carson JL. Transfusion threshold of 7 g per deciliter––the new normal. N Engl J Med. 2014;371(15):1459–61. https://doi.org/10.1056/NEJMe1408976.
Schmid-Schonbein GW. Biomechanics of microcirculatory blood perfusion. Annu Rev Biomed Eng. 1999;1:73–102. https://doi.org/10.1146/annurev.bioeng.1.1.73.
Malbrain ML, Cheatham ML, Kirkpatrick A, et al. Results from the international conference of experts on intra-abdominal hypertension and abdominal compartment syndrome. I. Definitions. Intensive Care Med. 2006;32(11):1722–32. https://doi.org/10.1007/s00134-006-0349-5.
Cheatham ML, Malbrain ML, Kirkpatrick A, et al. Results from the international conference of experts on intra-abdominal hypertension and abdominal compartment syndrome. II. Recommendations. Intensive Care Med. 2007;33(6):951–62. https://doi.org/10.1007/s00134-007-0592-4.
Cheatham ML, Malbrain ML. Cardiovascular implications of abdominal compartment syndrome. Acta Clin Belg. 2007;62(Suppl 1):98–112. https://doi.org/10.1179/acb.2007.62.s1.013.
Boddi M, Natucci F, Ciani E. The internist and the renal resistive index: truths and doubts. Intern Emerg Med. 2015; https://doi.org/10.1007/s11739-015-1289-2.
Samoni S, Nalesso F, Meola M, et al. Intra-parenchymal renal resistive index variation (IRRIV) describes renal functional reserve (RFR): pilot study in healthy volunteers. Front Physiol. 2016;7:286. https://doi.org/10.3389/fphys.2016.00286.
Tublin ME, Bude RO, Platt JF. Review. The resistive index in renal Doppler sonography: where do we stand? AJR Am J Roentgenol. 2003;180(4):885–92. https://doi.org/10.2214/ajr.180.4.1800885.
Al-Ashry H, Abuzaid A, Asim M, et al. Microcirculation alteration and biomarker dilemma in early septic shock diagnosis and treatment. Curr Vasc Pharmacol. 2016;14(4):330–44.
Kara A, Akin S, Ince C. Monitoring microcirculation in critical illness. Curr Opin Crit Care. 2016;22(5):444–52. https://doi.org/10.1097/MCC.0000000000000335.
Miranda M, Balarini M, Caixeta D, et al. Microcirculatory dysfunction in sepsis: pathophysiology, clinical monitoring, and potential therapies. Am J Phys Heart Circ Phys. 2016;311(1):H24–35. https://doi.org/10.1152/ajpheart.00034.2016.
Colbert JF, Schmidt EP. Endothelial and microcirculatory function and dysfunction in sepsis. Clin Chest Med. 2016;37(2):263–75. https://doi.org/10.1016/j.ccm.2016.01.009.
Aykut G, Veenstra G, Scorcella C, et al. Cytocam-IDF (incident dark field illumination) imaging for bedside monitoring of the microcirculation. Intensive Care Med Exp. 2015;3(1):40. https://doi.org/10.1186/s40635-015-0040-7.
Roy AK, Mc Gorrian C, Treacy C, et al. A comparison of traditional and novel definitions (RIFLE, AKIN, and KDIGO) of acute kidney injury for the prediction of outcomes in acute decompensated heart failure. Cardiorenal Med. 2013;3(1):26–37. https://doi.org/10.1159/000347037.
Frazee E, Kashani K. Fluid management for critically ill patients: a review of the current state of fluid therapy in the intensive care unit. Kidney diseases (Basel, Switzerland). 2016;2(2):64–71. https://doi.org/10.1159/000446265 [published Online First: 2016/08/19].
Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–10. https://doi.org/10.1001/jama.2016.0287.
Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992;101(6):1644-1655. [published Online First: 1992/06/01].
Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31(4):1250–6. https://doi.org/10.1097/01.CCM.0000050454.01978.3B.
Martin-Loeches I, Levy MM, Artigas A. Management of severe sepsis: advances, challenges, and current status. Drug Des Devel Ther. 2015;9:2079–88. https://doi.org/10.2147/DDDT.S78757.
Legrand M, Mebazaa A, Ronco C, et al. When cardiac failure, kidney dysfunction, and kidney injury intersect in acute conditions: the case of cardiorenal syndrome. Crit Care Med. 2014;42(9):2109–17. https://doi.org/10.1097/CCM.0000000000000404.
Xu Y, Xie Y, Shao X, et al. L-FABP: a novel biomarker of kidney disease. Clin Chim Acta; Int J Clin Chem. 2015;445:85–90. https://doi.org/10.1016/j.cca.2015.03.017.
Acknowledgements
We thank Allison Dwileski for the language revision of the manuscript.
Funding
This research project has received financial support by two independent foundations as declared:
– Gottfried and Julia Bangerter-Rhyner foundation Basel CHF 30,000
– Freiwillige Akademische Gesellschaft foundation Basel CHF 15,000
The grants were used to buy the CytoCam (Braedius Medical), for the establishment of the electronic Case Report Form and for coverage of personnel costs (e.g. statistician).
Sponsor of the study:
PD Dr. Martin Siegemund, Deputy Head
Surgical Intensive Care Unit
Department for Anesthesia, Surgical Intensive Care,
Prehospital Emergency Medicine and Pain Therapy
University Hospital Basel
Tel. + 41-61-328-6414
E-mail: martin.siegemund@usb.ch
Research support
Martin Siegemund, MD has received speaker honoraria from Fresenius Kabi Switzerland.
Availability of data and materials
Due to ongoing data collection, currently, no data is publicly available. The results will be submitted to peer-reviewed journals for publication and will be presented at relevant academic conferences.
Author information
Authors and Affiliations
Contributions
The authors have contributed, are contributing and will contribute to this manuscript/study conductance/publication of study results as follows: conception or design of the work (MS, MD, JK, JvB and AH), data collection (LG, FJ, TS, KL, JDS and MA), data analysis and interpretation (MS, MD, JK, JvB, MA and AH), drafting the article (AH and MS) and critical revision of the article (all declared authors). All authors declared final approval of the version to be published.
Corresponding author
Ethics declarations
Authors’ information
The sponsor-investigators are Martin Siegemund, MD, Deputy Chief Physician, and Alexa Hollinger, MD, Medical Resident, Critical Care Fellow. The co-investigators are Franziska Jockers, MD, Thomas Schweingruber, MD, Lukas Gantner, MD, Katrin Ledergerber, MD, and Jonas Dominic Scheuzger, MD. Michael Dickenmann, MD, Markus Aschwanden, MD, Johann Knotzer, MD, and Jasper van Bommel, MD, are the project partners, serve as consultants and/or assist in data acquisition.
Ethics approval and consent to participate
This study is conducted in compliance with the protocol, the current version of the Declaration of Helsinki, the ICH-GCP or ISO EN 14155 (as far as applicable) as well as all national legal and regulatory requirements. Research Ethics Committee approval has been granted for this study by EKNZ (Ethikkommission Nordwest-und Zentralschweiz), Switzerland (reference 2015-401). As shock will be the common general condition among patients, informed consent by the patient him-/herself will not be possible in most cases. In this case, a nominated (study-independent physician) and, if available, a personal consultee (next of kin) will evaluate the patient’s enrolment in our study and sign as a representative. As soon as the patient regains the capacity, his/her informed consent will be sought.
Dryad identifier: doi: https://doi.org/10.5061/dryad.f6d42; https://doi.org/10.5061/dryad.f6d42
Protocol version: Version 2, 06.12.2015
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendices
Appendix 1
Appendix 2
Appendix 3
Appendix 4
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hollinger, A., Gantner, L., Jockers, F. et al. Impact of amount of fluid for circulatory resuscitation on renal function in patients in shock: evaluating the influence of intra-abdominal pressure, renal resistive index, sublingual microcirculation and total body water measured by bio-impedance analysis on haemodynamic parameters for guidance of volume resuscitation in shock therapy: a protocol for the VoluKid pilot study–an observational clinical trial. Ren Replace Ther 4, 14 (2018). https://doi.org/10.1186/s41100-018-0156-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41100-018-0156-9