
Abstract
Hypertension is a prevalent condition worldwide and is the key risk factor for fatal cardiovascular complications, such as stroke, sudden cardiac death and heart failure. Reduced bioavailability of nitric oxide (NO) in the endothelium is an important precursor for impaired vasodilation and hypertension. In the heart, NO deficiency deteriorates the adverse consequences of pressure-overload and causes cardiac hypertrophy, fibrosis and myocardial infarction which lead to fatal heart failure and sudden cardiac death. Recent consensus is that both endothelial and neuronal nitric oxide synthases (eNOS or NOS3 and nNOS or NOS1) are the constitutive sources of NO in the myocardium. Between the two, nNOS is the predominant isoform of NOS that controls intracellular Ca2+ homeostasis, myocyte contraction, relaxation and signaling pathways including nitroso-redox balance. Notably, our recent research indicates that cardiac eNOS protein is reduced but nNOS protein expression and activity are increased in hypertension. Furthermore, nNOS is induced by the interplay between angiotensin II (Ang II) type 1 receptor (AT1R) and Ang II type 2 receptor (AT2R), mediated by NADPH oxidase and reactive oxygen species (ROS)-dependent eNOS activity in cardiac myocytes. nNOS, in turn, protects the heart from pathogenesis via positive lusitropy in hypertension. Soluble guanylate cyclase (sGC)-cGMP/PKG-dependent phosphorylation of myofilament proteins are novel targets of nNOS in hypertensive myocardium. In this short review, we will endeavor to overview new findings of the up-stream and downstream regulation of cardiac nNOS in hypertension, shed light on the underlying mechanisms which may be of therapeutic value in hypertensive cardiomyopathy.
Similar content being viewed by others


Background
According to World Health Organization, over 1 billion people are associated with hypertension worldwide and the number of subjects with such disorder rises significantly in aged group [1–4]. Importantly, population with hypertension is in an upsurge in young generation. Attention should be drawn to this trend because the prevalent exposure to the risk factors for hypertension during early stage of the lifespan is likely to expend to the trajectory of later life of their adulthood [5] (“programming” for hypertension). Furthermore, hypertension-associated complication in the cardiovascular system is one of the most important causes of premature death worldwide (up to 45 % of death due to heart diseases). As such, the global economic burden of healthcare expenditure due to hypertension maintains a steady increase. So far, effective regime for hypertension or hypertension-related therapy for heart diseases is lacking.
Sustained pressure-overload in conjunction with systematic inflammation, oxidative stress and endothelial dysfunction in hypertension stimulates a series of adverse outcomes of the heart, including ventricular hypertrophy [6], collagen deposition or fibrosis [7], dilatation [8] and coronary artery stiffening or rarefaction [9, 10]. The fundamental mechanisms those cause these changes involve altered ion channels and transporters in the plasmalemmal membrane of cardiac myocytes; abnormal calcium handling in cardiac myocytes; metabolic derangement; sarcomere disorganization and changes in the wide spectrum of intracellular signaling pathways [6, 11–17]. Concomitant to the progression of hypertension-induced adverse remodeling of myocardium, a number of endogenous defense mechanisms can be triggered, which are involved in delaying and preventing the pathological processes. These defense mechanisms include: maintaining or stimulating cGMP pathway, e.g. nitric oxide [18], atrial natriuretic peptide [19, 20] and phosphodiesterase 5 inhibition [21]; antagonizing beta1-adrenergic pathway [22], e.g. beta3-adrenoreceptor- or muscarinic receptor dependent signaling pathways; AT2R, Ang 1–7 or 1–9 receptors and Mas receptor that antagonizes AT1R signaling [23–25]; metabolisms and AMPK signaling [26]. General consensus is that nNOS is an important cardiac protector in healthy and diseased hearts. In this review, we will highlight our novel findings in nNOS and its protective mechanisms of the heart in hypertension.
NO, eNOS and nNOS in the hypertensive myocardium
NO is an important autocrine and paracrine signaling molecule that plays crucial roles in regulating cardiovascular physiology and pathology. Deficiency in NO are associated with oxidative stress, diastolic dysfunction and most of adverse cardiovascular disorders including heart failure and cardiac arrhythmias [18]. Two constitutive NO synthases (cNOS) are responsible for the production of NO in healthy myocardium: eNOS and nNOS, which share similar structure and mechanism of activation.
In general, the active NOSs form a homodimer and convert the amino acid L-arginine to L-citrulline and NO (Fig. 1). NOS monomer contains a C-terminal reductase domain and a N-terminal oxygenase domain which are linked by calmodulin (CaM) binding region. The N-terminal oxidase domain contains the heme, tetrahydrobiopterin cofactors (BH4) and the binding site for the substrate arginine. Oxidase domain is the active site of NO synthesis. The reductase domain is composed of the flavin mononucleotide (FMN)-binding subdomain, the flavin adenine dinucleotide (FAD) and reduced nicotinamide-adenine dinucleotide phosphate (NADPH)-binding subdomains. During catalysis, the electrons are transferred from NADPH to FAD and FMN of one monomer to the heme domain of the opposite monomer [27]. Production of NO requires oxygen as the electron acceptor. NO diffuses freely across the plasma membrane, therefore, NO is generally known to be able to transport to the effector proteins in the cells or in the adjacent cells and exerts its effects (e.g. endothelial NO targets soluble guanylate cyclase, sGC, in smooth muscle to accomplish vasodilation).

Structure of an active nNOS protein. Schematic diagram shows the two monomers of nNOS (each contains an oxygenase domain (−COOH terminal) and a reductase domain (−NH2 terminal). Electrons transfer from NADPH in the reductase domain of one monomar (via FDA and FMN) to the heme iron in the oxygenase domain of the other monomer, facilitated by calmodulin (CaM), enable nNOS to catalyze the oxidation of L-arginine to L-citrulline and releases NO. Ca2+ activates CaM and nNOS activity
In fact, NO is highly reactive with short life-time (a few seconds). Therefore, the likelihood of NO to travel a distance to reach the target proteins in myocardium is low (especially given the oxidative environment of myocardium); instead, NO is produced from eNOS and nNOS in the designated compartments and affect downstream targets in confined location where the enzymes reside [27, 28]. The spatial regulation enhances the effectiveness of NO and the producing enzymes to fulfill specific tasks.
It should be noted that eNOS and nNOS are localized at discrete compartments and exert diverse functions in cardiomyocytes. E.g. eNOS is localized in the caveolae of cardiomyocytes and nNOS is in the cytosol with ryanodine receptors (RyR) in the sarcoplasmic reticulum (SR) in healthy heart. The clear distinction in compartmentalization between eNOS and nNOS are disarranged in diseased hearts. E.g. eNOS expression is often found to be diminished and nNOS translocates from SR to plasmalemmal membrane [29–31]. The changes in the location of nNOS enable it to affect different downstream targets for different functions.
The pathological stimuli may also change the property of eNOS or nNOS in the hypertension. eNOS has been found to be uncoupled in the myocardium of a mice model of transverse aortic constriction (TAC)-induced pressure overload and eNOS produces superoxide rather than NO [32–34]. Increased oxidative stress and depletion of BH4 due to the oxidation of BH4 to BH2 is one of the dominant mechanisms for eNOS uncoupling, consequently, eNOS becomes responsible for cardiac oxidative stress, left ventricular (LV) hypertrophy and dysfunction [32–34]. In its “coupled” state, eNOS-derived NO can be an important protective mechanism for TAC-induced LV hypertrophy [35–38]. In addition, eNOS is known to be an “endogenous beta-blocker” by restoring the sympatho-vagal balance, promoting vasodilatation and neoangiogenesis [39] and protects the heart under pathological stress.
In contrast, nNOS has consistently been shown to be upregulated in LV myocardium from the hearts of pressure-overload [40, 41]. Similarly, the activity of nNOS (phosphorylated:total nNOS ratio) is significantly increased in right ventricular (RV) myocardium of pulmonary hypertensive rats [42], confirming the upregulation of myocardial nNOS with increased pressure-overload. Accumulating evidence shows that nNOS up-regulation in diseased heart is cardiac protective: nNOS has been shown to inhibit xanthine oxidoreductase [43, 44] or NADPH oxidases [45, 46] and mitochondria production of ROS [47], as a result, reduces oxidative stress and suppresses adverse myocardial remodeling. In addition, nNOS facilitates the relaxation of LV myocytes and maintains lusitropy of the heart under pressure-overload. Since increased oxidative stress and diastolic dysfunction are often associated with structural and functional pathology of the myocardium, such as hypertrophy, arrhythmias and heart failure, nNOS modulation of various cardiac oxidases or improving diastolic function of the myocardium under pressure-overload enable nNOS to protect the heart from structural and functional remodeling those underlie fatal heart failure.
nNOS protein expression and activity in hypertensive myocardium
The mechanisms leading to nNOS up-regulation in diseased heart remain unclear. One of the suggestions is that the transcription and the translation of nNOS are oxygen-dependent. Hypoxia (8 %, 48 h) has convincingly show to increase the mRNA and protein expressions as well as the activity of nNOS in the systematic and local arteries (aorta, mesenteric arterioles and renal papilla) and in the brain by revealing a novel promotor region in exon 2 with increased translational efficiency [48]. Whether limited availability of O2 in hypertensive myocardium is essential in triggering nNOS up-regulation has not been elucidated yet.
Intriguingly, our recent results demonstrated that Ang II induces nNOS under the conditions where ROS are increased (Fig. 2). In isolated LV myocytes, Ang II treatment (3 h) significantly increased nNOS protein expression and activity via AT1R stimulation of NADPH oxidase activity and ROS production [45, 49]. In addition, nNOS in LV myocytes is increased in Ang II-induced hypertensive rats [50] (Fig. 2). In both cases, Ang II only stimulated nNOS but not eNOS because eNOS protein expression is either not changed (with acute Ang II treatment) or significantly reduced (in hypertension) [45, 50]. This is in contrast to the findings shown previously where Ang II treatment in vivo increased eNOS protein expression in the myocardium [51, 52]. Therefore, Ang II or AT1R has been implicated in the activation of constitutive NOS in cardiovascular system.

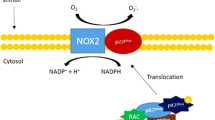
Schematic diagram of nNOS up-regulation by Ang II in cardiac myocytes and the potential target proteins, mechanism of regulation in cardiac myocytes from hypertension. Left. Ang II stimulates AT1R and NADPH oxidase in cardiac myocytes. AT1R and intracellular ROS activates Akt and phosphorylates endogeneous eNOS to produce NO. eNOS and NO-dependent S-nitrosation of AT2R lead to the translocation of AT2R to the plasma membrane and induces nNOS protein and increases NO production (data modified from Figure 8 of Basic Research in Cardiology, 2015, 110 (3):21). In hypertension, cardiac nNOS is up-regulated and facilitates myocyte relaxation. nNOS reduces Ca2+ influx via LTCC and promotes Ca2+ re-uptake via SERCA through PLN phosphorylation (secondary to PPase-dependent PKA phosphorylation) in healthy heart. In hypertension, nNOS inhibits LTCC and phosphorylates cMBP-C Ser273 and cTnI Ser23/24 via cGMP/PKG-dependent mechanism. As a result, myofilament Ca2+ sensitivity is reduced which accounts for nNOS-dependent positive lusitropy in hypertension
Mechanistic studies showed that NADPH oxidase is essential in mediating AT1R-dependent and ROS-stimulated signaling (Fig. 2). NADPH oxidase/ROS activates eNOS-phosphorylation and mediates eNOS-derived NO-dependent S-nitrosation of AT2R. Site specific mutagenesis analysis indicated that among 4 intracellular cysteine residues in AT2R, cysteine349 of AT2R is the key regulatory site for AT2R translocation to the plasma membrane [49]. Our results are in line with other reports showing the relationship between AT2R and nNOS. E.g. C21, an AT2R agonist, significantly increased nNOS expression in the paraventricular hypothalamic nucleus (PVN) and rostral ventrolateral medulla (RVLM) [53, 54] or inhibition of AT2R abolishes relaxin-induced nNOS protein expression and phosphorylation [55]. Furthermore, nNOS is unaffected in the infarcted myocardium from AT2R gene deficient mice [56], confirming the role of AT2R in nNOS up-regulation. In hypertensive myocardium, protein expressions of AT1R and AT2R are not changed comparing to those in sham rats (unpublished data). It is possible to extrapolate that ROS may function as an important trigger signal to nNOS up-regulation at least at early stage of myocardial stress. Subsequent eNOS/NO-dependent S-nitrosation of AT2R and its downstream signaling cascades are pivotal in the transcription and translation of nNOS proteins. Our results are the first to reveal the interplay between AT1R and AT2R or eNOS and AT2R in regulating nNOS protein expression in cardiac myocytes. Application of the knowledge to the therapeutic strategy towards hypertension and pressure-overload-induced adverse remodeling will be beneficial.
S-nitrosylation vs. soluble guanylate cyclase – cGMP pathway of nNOS in hypertensive myocardium
It is generally considered that the primary signaling pathway of NO is through the activation of soluble guanylyl cyclase (sGC) leading to the production of cyclic guanosine monophosphate (cGMP) and protein kinase G (PKG)-dependent phosphorylation of downstream proteins. However, it should be noted that the prototypic mechanism of NO involves direct modifications of a diverse range of proteins by S-nitrosylation (or S-nitrosation) of cysteine residues [57]. It turns out that the reaction between NO and target proteins are highly specific, requires substantially low concentration of NO and the process is actively regulated. The high specificity of NO-protein modification is achieved by NOSs being part of the signaling mechanism in protein complexes, in which S-nitrosylation or S-nitrosothiol (SNO) interaction occurs at the sites within the vicinity of NOSs.
NO activation of sGC/cGMP for PKG-dependent signaling requires high concentrations of NO [58]. I.e. NO activates sGC moderately via a classical heme-dependent mechanism at low NO concentration. However, at high concentration of NO, the activity of sGC can be increased by the order of 2 via heme-independent but via direct NO-cysteine interaction of sGC. Therefore, the concentration of NO near the vicinity of target proteins determine whether sGC is activated and cGMP/PKG-dependent pathway mediates the responses of NOSs.
It is well established that under normal conditions and in healthy heart, endothelial NO induces the relaxation via cGMP/PKG-dependent phosphorylation of myosin phosphatases in smooth muscle. In cardiac myocytes, however, the effect of nNOS on myocyte relaxation is independent of cGMP/PKG-dependent signaling, rather it is mediated by stimulating Ca2+ reuptake through Ca2+ ATPase in the SR (SERCA) via PKA-dependent phosphorylation of phospholamban at Ser [16] (PLN-p) [59]. The increased PKA-dependent phosphorylation is through nNOS-dependent inhibition of protein phosphatase 2A (PP2A) activity [59].
Interestingly, in LV myocytes treated with Ang II or from hypertensive hearts, increased nNOS activity facilitated myocyte relaxation via cGMP/PKG-dependent phosphorylations of PLN or cGMP/PKG-dependent phosphorylations of myosin binding protein C (cMyBPC-Ser273) and troponin I (cTnI-Ser23/24) (Fig. 2). As a result, nNOS reduces myofilament Ca2+ sensitivity and promotes myocyte relaxation in hypertension [50]. Inhibition of PKA-dependent signaling failed to prevent the effect of nNOS under these conditions. These results suggest that the downstream mediating mechanisms of nNOS-derived NO are shifted from sGC/cGMP-independent and PKA-dependent signaling pathway in healthy heart to cGMP/PKG-dependent mechanism in hypertensive hearts, presumably due to the greater bioavailability of NO from nNOS (Fig. 2). Similarly, cGMP/PKG-dependent signaling has been suggested to mediate the functions of nNOS following conditional nNOS over-expression in the myocardium in mice or lentiviral over-expression of nNOS in rats [60, 61]. Conversely, reducing myocardial nNOS and its activity in PMCA4b transgenic mice [62] or in the dystrophin deficiency model [63] decreased myocyte cGMP levels.
Recently, Kass’s group have shown that there are two pools of sGC in cardiac myocytes (caveolin-enriched membrane and nonlipid raft) with the former pool producing 2–3 fold more cGMP following NO stimulation [64]. Since nNOS up-regulation is known to be associated with translocation from SR to caveolae in the heart under stress [29–31] it is possible to postulate that nNOS up-regulation in diseased hearts or in response to pathogenic stimuli, e.g. Ang II, is associated with its translocation to the location where sGC is reachable with enhanced efficiency to produce greater amount of cGMP.
Therefore, the primary mechanism that mediates the effect of nNOS is S-nitrosylation or S-nitrosation of the cysteine residues of target proteins. Subsequently, the genre of downstream target proteins of NO determines the form of regulation in the signaling cascade. E.g. under the conditions of increased nNOS protein expression and activity, NO stimulates sGC (through S-nitrosation) and initiates cGMP/PKG-dependent signaling pathway. Alternatively, regulation of cardiac oxidases is able to affect downstream target proteins (e.g. Ca2+ handling proteins) to propagate the redox and Ca2+ signals, respectively.
Proteins targeted by nNOS in healthy and hypertensive heart
So far, the downstream target proteins of nNOS and its mode of regulation in healthy and diseased myocardium are not fully understood. Key excitation-contraction coupling proteins are known to be targeted by nNOS: L-type Ca2+ channels [65, 66], RyR [31], PLN [59], voltage-gated sodium channels (Nav1.5) [67] as well as structure proteins, such as syntrophin [67]. Recently, we have reported that myofilament proteins (cMyBPC and cTnI) are targeted by nNOS and nNOS-dependent alterations in the phosphorylation and change the myofilament Ca2+ sensitivity [50]. Although nNOS-dependent regulation of myofilament proteins is not clear at present, our preliminary results suggest that the myofilament proteins those are regulated by nNOS are different between sham and hypertension, implicating the significance of nNOS in regulating cardiac myofilament (Fig. 2).
The detailed signaling molecules those are targeted by nNOS and the differences in healthy and diseased hearts remain undetermined. Cardiac oxidases (including xanthine oxidoreductase, NADPH oxidase or mitochondrial cytochrome C oxidases or eNOS) are probably the key targets of nNOS in the heart under pressure-overload and regulation of which induces a plethora of responses involving oxidation, reduction, phosphorylation and various types of post-transcriptional regulations [68]. Other proteins such as sGC or PPs, whose activity are important in the hearts, are likely to play important roles mediating the effects of nNOS in hypertensive heart.
Conclusion
Hypertension is a common condition that leads to fatal cardiovascular complications such as myocardial infarction, stroke, sudden cardiac death and heart failure. Myocardial hypertrophy by sustained pressure-overload is the adverse remodeling in hypertension that predisposes to the apoptosis and fibrosis, malignant consequences upon pathogenic stimuli. During the pathological progression in the myocardium, nNOS is up-regulated and NO-derived pathway is associated with the prevention of cardiac hypertrophy by targeting oxidative stress, hypertrophic pathways and abnormal Ca2+ handlings. Our recent research reveals some of the important mechanisms of the upstream and downstream regulation of nNOS in hypertensive myocardium. Our results highlight that AT1R, NADPH oxidase/ROS and AT2R are important up-stream regulators of the transcription of nNOS in the myocardium under pressure-overload. In addition, nNOS-derived NO leads to cGMP/PKG-dependent phosphorylation of downstream target proteins including those in the SR and in the myofilament. Results from our research suggest that nNOS regulation of myofilament proteins is important in facilitating relaxation of the heart in hypertension. Our results reveal novel targets those possesses important clinical value in preventing the adverse remodeling with pressure-overload in the heart.
Abbreviations
- Ang II:
-
Angiotensin II
- AT1R:
-
Ang II type 1 receptor
- BH4:
-
Tetrahydrobiopterin
- CaM:
-
Calmodulin
- cGMP:
-
Cyclic guanosine monophosphate
- cMyBPC:
-
Myosin binding protein C
- cNOS:
-
Constitutive nitric oxide synthases
- cTnI:
-
Troponin I
- eNOS:
-
Endothelial nitric oxide synthases
- FAD:
-
Flavin adenine dinucleotide
- FMN:
-
Flavin mononucleotide
- LV:
-
Left ventricular
- nNOS or NOS1:
-
Neuronal nitric oxide synthases
- NO:
-
Nitric oxide
- PKG:
-
Protein kinase G
- PP2A:
-
Protein phosphatase 2A
- RV:
-
Right ventricular
- RyR:
-
Ryanodine receptors
- SERCA:
-
Ca2+ ATPase in the SR
- sGC:
-
Soluble guanylate cyclase
- SNO:
-
S-nitrosothiol
- SR:
-
Sarcoplasmic reticulum
- TAC:
-
Transverse aortic constriction
References
Alwan A, Armstrong T, Bettcher D, Branca F, Chisholm D, Ezzati M, Garfield R, MacLean D, Mathers C, Mendis S, Poznyak V, Riley L, Tang KC, Wild C. WHO Global Status Report on Noncommunicable Diseases 2010. Geneva: World Health Organization; 2011.
Neupane D, McLachlan CS, Sharma R, Gyawali B, Khanal V, Mishra SR, et al. Prevalence of hypertension in member countries of South Asian Association for Regional Cooperation (SAARC): systematic review and meta-analysis. Medicine (Baltimore). 2014;93(13):e74.
Tailakh A, Evangelista LS, Mentes JC, Pike NA, Phillips LR, Morisky DE. Hypertension prevalence, awareness, and control in Arab countries: a systematic review. Nurs Health Sci. 2014;16:126–30.
Ataklte F, Erqou S, Kaptoge S, Taye B, Echouffo-Tcheugui JB, Kengne AP. Burden of undiagnosed hypertension in sub-Saharan Africa: a systematic review and meta-analysis. Hypertension. 2015;65(2):291–8.
Anwar MA, Saleh AI, Al Olabi R, Al Shehabi TS, Eid AH. Glucocorticoid-induced fetal origins of adult hypertension: Association with epigenetic events. Vascul Pharmacol. 2016;82:41–50.
Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–62.
Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118(6):1021–40.
Cuspidi C, Rescaldani M, Sala C. Prevalence of echocardiographic left-atrial enlargement in hypertension: a systematic review of recent clinical studies. Am J Hypertens. 2013;26(4):456–64.
Triantafyllou A, Anyfanti P, Pyrpasopoulou A, Triantafyllou G, Aslanidis S, Douma S. Capillary rarefaction as an index for the microvascular assessment of hypertensive patients. Curr Hypertens Rep. 2015;17(5):33.
Tsioufis C, Dimitriadis K, Katsiki N, Tousoulis D. Microcirculation in Hypertension: An Update on Clinical Significance and Therapy. Curr Vasc Pharmacol. 2015;13(3):413–7.
Yamamoto S, Kita S, Iyoda T, Yamada T, Iwamoto T. New molecular mechanisms for cardiovascular disease: cardiac hypertrophy and cell-volume regulation. J Pharmacol Sci. 2011;116(4):343–9.
Goldhaber JI, Philipson KD. Cardiac sodium-calcium exchange and efficient excitation-contraction coupling: implications for heart disease. Adv Exp Med Biol. 2013;961:355–64.
Abriel H, Syam N, Sottas V, Amarouch MY, Rougier JS. TRPM4 channels in the cardiovascular system: physiology, pathophysiology, and pharmacology. Biochem Pharmacol. 2012;84(7):873–81.
Vennekens R. Emerging concepts for the role of TRP channels in the cardiovascular system. J Physiol. 2011;589(Pt 7):1527–34.
Morrow JP, Marx SO. Novel approaches to examine the regulation of voltage-gated calcium channels in the heart. Curr Mol Pharmacol. 2015;8(1):61–8.
Huo R, Sheng Y, Guo WT, Dong DL. The potential role of Kv4.3 K+ channel in heart hypertrophy. Channels (Austin). 2014;8(3):203–9.
Wang Y, Tandan S, Hill JA. Calcineurin-dependent ion channel regulation in heart. Trends Cardiovasc Med. 2014;24(1):14–22.
Tsutsui M, Tanimoto A, Tamura M, Mukae H, Yanagihara N, Shimokawa H, Otsuji Y. Significance of nitric oxide synthases: Lessons from triple nitric oxide synthases null mice. J Pharmacol Sci. 2015;127(1):42–52.
Rubattu S, Sciarretta S, Valenti V, Stanzione R, Volpe M. Natriuretic peptides: an update on bioactivity, potential therapeutic use, and implication in cardiovascular diseases. Am J Hypertens. 2008;21(7):733–41.
Silver MA. The natriuretic peptide system: kidney and cardiovascular effects. Curr Opin Nephrol Hypertens. 2006;15(1):14–21.
Mergia E, Stegbauer J. Role of Phosphodiesterase 5 and Cyclic GMP in Hypertension. Curr Hypertens Rep. 2016;18(5):39.
Niu X, Watts VL, Cingolani OH, Sivakumaran V, Leyton-Mange JS, Ellis CL, Miller KL, Vandegaer K, Bedja D, Gabrielson KL, Paolocci N, Kass DA, Barouch LA. Cardioprotective effect of beta-3 adrenergic receptor agonism: role of neuronal nitric oxide synthase. J Am Coll Cardiol. 2012;59(22):1979–87.
Westermeier F, Bustamante M, Pavez M, García L, Chiong M, Ocaranza MP, Lavandero S. Novel players in cardioprotection: Insulin like growth factor-1, angiotensin-(1–7) and angiotensin-(1–9). Pharmacol Res. 2015;101:41–55.
Sumners C, de Kloet AD, Krause EG, Unger T, Steckelings UM. Angiotensin type 2 receptors: blood pressure regulation and end organ damage. Curr Opin Pharmacol. 2015;21:115–21.
Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/Angiotensin 1–7 Axis of the Renin-Angiotensin System in Heart Failure. Circ Res. 2016;118(8):1313–26.
Kim M, Tian R. Targeting AMPK for cardiac protection: opportunities and challenges. J Mol Cell Cardiol. 2011;51(4):548–53.
Zhang YH, Casadei B. Sub-cellular targeting of constitutive NOS in health and disease. J Mol Cell Cardiol. 2012;52(2):341–50.
Zhang YH, Jin CZ, Jang JH, Wang Y. Molecular mechanisms of neuronal nitric oxide synthase in cardiac function and pathophysiology. J Physiol. 2014;592(15):3189–200.
Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel JL, Heymes C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004;363(9418):1365–7.
Damy T, Ratajczak P, Robidel E, Bendall JK, Oliviéro P, Boczkowski J, Ebrahimian T, Marotte F, Samuel JL, Heymes C. Up-regulation of cardiac nitric oxide synthase 1-derived nitric oxide after myocardial infarction in senescent rats. FASEB J. 2003;17(13):1934–6.
Bendall JK, Damy T, Ratajczak P, Loyer X, Monceau V, Marty I, Milliez P, Robidel E, Marotte F, Samuel JL, Heymes C. Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in beta-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation. 2004;110(16):2368–75.
Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodelling from chronic pressure load. J Clin Invest. 2005;115:1221–31.
Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric oxide synthase. J Biol Chem. 2003;278:22546–54.
Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in argininedepleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci U S A. 1996;93:6770–4.
Ichinose F, Bloch KD, Wu JC, Hataishi R, Aretz HT, Picard MH, Scherrer-Crosbie M. Pressure overloadinduced hypertrophy and dysfunction in mice are exacerbated by congenital NOS3 deficiency. Am J Physiol Heart Circ Physiol. 2004;286:H1070–5.
Buys ES, Raher MJ, Blake SL, Neilan TG, Graveline AR, Passeri JJ, Llano M, Perez-Sanz TM, Ichinose F, Janssens S, Zapol WM, Picard MH, Bloch KD, Scherrer-Crosbie M. Cardiomyocyte restricted restoration of nitric oxide synthase 3 attenuates left ventricular remodelling after chronic pressure overload. Am J Physiol Heart Circ Physiol. 2007;293:H620–7.
Ozaki M, Kawashima S, Yamashita T, Hirase T, Ohashi Y, Inoue N, Hirata K, Yokoyama M. Overexpression of endothelial nitric oxide synthase attenuates cardiac hypertrophy induced by chronic isoproterenol infusion. Circ J. 2002;66:851–6.
Janssens S, Pokreisz P, Schoonjans L, Pellens M, Vermeersch P, Tjwa M, Jans P, Scherrer-Crosbie M, Picard MH, Szelid Z, Gillijns H, Van de Werf F, Collen D, Bloch KD. Cardiomyocyte-specific overexpression of nitric oxide synthase 3 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circ Res. 2004;94:1256–62.
Massion PB, Balligand JL. Relevance of nitric oxide for myocardial remodelling. Curr Heart Fail Rep. 2007;4:18–25.
Massion PB, Feron O, Dessy C, Balligand JL. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res. 2003;93:388–98.
Watts VL, Sepulveda FM, Cingolani OH, Ho AS, Niu X, Kim R, Miller KL, Vandegaer K, Bedja D, Gabrielson KL, Rameau G, O'Rourke B, Kass DA, Barouch LA. Anti-hypertrophic and anti-oxidant effect of beta3-adrenergic stimulation in myocytes requires differential neuronal NOS phosphorylation. J Mol Cell Cardiol. 2013;62:8–17.
Umar S, Hessel M, Steendijk P, Bax W, Schutte C, Schalij M, van der Wall E, Atsma D, van der Laarse A. Activation of signalling molecules and matrix metalloproteinases in right ventricular myocardium of rats with pulmonary hypertension. Pathol Res Pract. 2007;203:863–72.
Idigo WO, Reilly S, Zhang MH, Zhang YH, Jayaram R, Carnicer R, Crabtree MJ, Balligand JL, Casadei B. Regulation of endothelial nitric-oxide synthase (NOS) S-glutathionylation by neuronal NOS: evidence of a functional interaction between myocardial constitutive NOS isoforms. J Biol Chem. 2012;287(52):43665–73.
Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, Li D, Berkowitz DE, Hare JM. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101(45):15944–8.
Jin CZ, Jang JH, Wang Y, Kim JG, Bae YM, Shi J, Che CR, Kim SJ, Zhang YH. Neuronal nitric oxide synthase is up-regulated by angiotensin II and attenuates NADPH oxidase activity and facilitates relaxation in murine left ventricular myocytes. J Mol Cell Cardiol. 2012;52(6):1274–81.
Zhang YH, Dingle L, Hall R, Casadei B. The role of nitric oxide and reactive oxygen species in the positive inotropic response to mechanical stretch in the mammalian myocardium. Biochim Biophys Acta. 2009;1787(7):811–7.
Burkard N, Williams T, Czolbe M, Blömer N, Panther F, Link M, Fraccarollo D, Widder JD, Hu K, Han H, Hofmann U, Frantz S, Nordbeck P, Bulla J, Schuh K, Ritter O. Conditional overexpression of neuronal nitric oxide synthase is cardioprotective in ischemia/reperfusion. Circulation. 2010;122(16):1588–603.
Ward ME, Toporsian M, Scott JA, Teoh H, Govindaraju V, Quan A, Wener AD, Wang G, Bevan SC, Newton DC, Marsden PA. Hypoxia induces a functionally significant and translationally efficient neuronal NO synthase mRNA variant. J Clin Invest. 2005;115(11):3128–39.
Jang JH, Chun JN, Godo S, Wu G, Shimokawa H, Jin CZ, Jeon JH, Kim SJ, Jin ZH, Zhang YH. ROS and endothelial nitric oxide synthase (eNOS)-dependent trafficking of angiotensin II type 2 receptor begets neuronal NOS in cardiac myocytes. Basic Res Cardiol. 2015;110(3):21.
Jin CZ, Jang JH, Kim HJ, Wang Y, Hwang IC, Sadayappan S, Park BM, Kim SH, Jin ZH, Seo EY, Kim KH, Kim YJ, Kim SJ, Zhang YH. Myofilament Ca2+ desensitization mediates positive lusitropic effect of neuronal nitric oxide synthase in left ventricular myocytes from murine hypertensive heart. J Mol Cell Cardiol. 2013;60:107–15.
Tambascia RC, Fonseca PM, Corat PD, Moreno Jr H, Saad MJ, Franchini KG. Expression and distribution of NOS1 and NOS3 in the myocardium of angiotensin II-infused rats. Hypertension. 2001;37:1423–8.
Castro-Chaves P, Fontes-Carvalho R, Pintalhao M, Pimentel-Nunes P, Leite-Moreira AF. Angiotensin II-induced increase in myocardial distensibility and its modulation by the endocardial endothelium in the rabbit heart. Exp Physiol. 2009;94:665–74.
Gao J, Zucker IH, Gao L. Activation of central angiotensin type 2 receptors by compound 21 improves arterial baroreflex sensitivity in rats with heart failure. Am J Hypertens. 2014;27:1248–56.
Gao J, Zhang H, Le KD, Chao J, Gao L. Activation of central angiotensin type 2 receptors suppresses norepinephrine excretion and blood pressure in conscious rats. Am J Hypertens. 2011;24:724–30.
Chow BS, Kocan M, Bosnyak S, Sarwar M, Wigg B, Jones ES, Widdop RE, Summers RJ, Bathgate RA, Hewitson TD, Samuel CS. Relaxin requires the angiotensin II type 2 receptor to abrogate renal interstitial fibrosis. Kidney Int. 2014;86:75–85.
Brede M, Roell W, Ritter O, Wiesmann F, Jahns R, Haase A, Fleischmann BK, Hein L. Cardiac hypertrophy is associated with decreased eNOS expression in angiotensin AT2 receptor-deficient mice. Hypertension. 2003;42:1177–82.
Murphy E, Kohr M, Menazza S, Nguyen T, Evangelista A, Sun J, Steenbergen C. Signaling by S-nitrosylation in the heart. J Mol Cell Cardiol. 2014;73:18–25.
Fernhoff NB, Derbyshire ER, Marletta MA. A nitric oxide/cysteine interaction mediates the activation of soluble guanylate cyclase. Proc Natl Acad Sci U S A. 2009;106(51):21602–7.
Zhang YH, Zhang MH, Sears CE, Emanuel K, Redwood C, El-Armouche A, Kranias EG, Casadei B. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ Res. 2008;102(2):242–9.
Wang L, Li D, Dawson TA, Paterson DJ. Long-term effect of neuronal nitric oxide synthase over-expression on cardiac neurotransmission mediated by a lentiviral vector. J Physiol. 2009;587:3629–37.
Burkard N, Rokita AG, Kaufmann SG, Hallhuber M, Wu R, Hu K, Hofmann U, Bonz A, Frantz S, Cartwright EJ, Neyses L, Maier LS, Maier SK, Renné T, Schuh K, Ritter O. Conditional neuronal nitric oxide synthase overexpression impairs myocardial contractility. Circ Res. 2007;100(3):e32–44.
Mohamed TM, Oceandy D, Prehar S, Alatwi N, Hegab Z, Baudoin FM, Pickard A, Zaki AO, Nadif R, Cartwright EJ, Neyses L. Specific role of neuronal nitric-oxide synthase when tethered to the plasma membrane calcium pump in regulating the beta-adrenergic signal in the myocardium. J Biol Chem. 2009;284(18):12091–8.
Khairallah M, Khairallah RJ, Young ME, Allen BG, Gillis MA, Danialou G, Deschepper CF, Petrof BJ, Des Rosiers C. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc Natl Acad Sci U S A. 2008;105(19):7028–33.
Tsai EJ, Liu Y, Koitabashi N, Bedja D, Danner T, Jasmin JF, Lisanti MP, Friebe A, Takimoto E, Kass DA. Pressure-Overload-Induced Subcellular Relocalization/Oxidation of Soluble Guanylate Cyclase in the Heart Modulates Enzyme Stimulation. Circ Res. 2012;110(2):295–303.
Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res. 2003;92(5):e52–9.
Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res. 2006;98(3):403–11.
Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105(27):9355–60.
Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7(4):381–91.
Acknowledgement
Not applicable.
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2013068067); by the Brain Korea 21 Graduate Programme of the Korean Ministry of Education, Science and Technology, Seoul National University Hospital, the Korean Society of Hypertension (2013), SK Telecom Research Fund (no. 3420130290).
Availability of data and materials
Not applicable.
Author’s contributions
YHZ designed and wrote the manuscript.
Competing interests
The author declares that he/she has no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, Y.H. Neuronal nitric oxide synthase in hypertension – an update. Clin Hypertens 22, 20 (2016). https://doi.org/10.1186/s40885-016-0055-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40885-016-0055-8