
Abstract
Background
The Great Plains of the United States includes a large number of hybrid and contact zones between bird species. The amount of gene flow between sister species in these zones ranges from very rare hybridization events to widespread and prevalent introgression. Some of these avian systems have been studied extensively, while others have been indeterminate of whether hybridization exists in areas of sympatry. Using genomic-level approaches allows investigation of genomic patterns of hybridization and gene flow between species—or lack thereof.
Methods
We investigated a narrow zone of sympatry in Nebraska, USA between pewee species (Contopus sordidulus and C. virens), for which no hybridization has been confirmed. We used thousands of single nucleotide polymorphisms to identify potential hybridization and investigate genomic patterns of differentiation between these two species.
Results
We found evidence of multiple hybrid individuals in the contact zone. Little genomic variation was fixed between species, but a large proportion had differentiated allele frequencies between species. There was a positive relationship between genetic differentiation and chromosome size.
Conclusions
We provided the first conclusive evidence of hybridization between C. sordidulus and C. virens, in a region where secondary contact likely occurred due to human disturbance and habitat modification. The genomic patterns of differentiation affirm that these species split in the relatively recent past. Finally, the relationship of chromosome size and genetic differentiation may have resulted from differential rates of chromosomal recombination in songbirds and genetic differentiation between species largely due to genetic drift (possibly in concert with selection).
Similar content being viewed by others


Background
An enduring emphasis of evolutionary biology is investigating the mechanisms responsible for preventing gene flow between closely related species (Mayr 1942). The “natural laboratories” to study these questions are contact zones between closely related species (Hewitt 1988), where species may—or may not—hybridize. A classic clustering of contact and hybrid zones is located in the Great Plains of south-central Canada and the central United States (Rising 1983; Swenson and Howard 2005) where more than a dozen closely-related pairs of bird species meet in partial sympatry, including genera of owls (Megascops), woodpeckers (Colaptes, Melanerpes), and songbirds (Contopus, Cyanocitta, Poecile, Baeolophus, Sturnella, Icterus, Pheucticus, Passerina, and Pipilo; Rising 1983). Hybridization in these closely-related species ranges from only a few reports (e.g., Cyanocitta cristata and C. stelleri) to widespread and frequent hybridization [e.g., Colaptes auratus (red-shafted and yellow-shafted phenotypic variants); Grudzien et al. 1987]. The nature of several of these hybrid zones, including Passerina buntings, Icterus orioles, and Pheucticus grosbeaks have been investigated extensively using both morphological and genetic analysis techniques (Carling and Brumfield 2008; Carling et al. 2011; Mettler and Spellman 2009).
One of the contact zones, where the Western Wood-pewee (Contopus sordidulus) and Eastern Wood-pewee (C. virens) come into contact, has not been investigated thoroughly, and it is unknown whether hybridization occurs. The scarcity of sympatry between these species in breeding distributions and almost complete overlap in plumage morphology and morphometric characters (Rising and Schueler 1980; Rising 1983; Pyle 1997) has thus far precluded positive identification of hybridization. In Kansas and Montana, two investigations using museum voucher specimens and morphological measurements failed to conclusively identify hybrids (Rising 1965; Rising and Schueler 1980)—albeit with small sample sizes. The authors in both of these studies suggest that hybridization is possible, if not probable, between the two Contopus species, but lack data to document the phenomenon.
Because the species are generally identified via geography or song (Rising and Schueler 1980) and have a great deal of plumage overlap, investigation of possible hybridization may be best facilitated with genetic data. With the recent increase in genomic techniques for phylogeography and systematics [e.g., restriction-site associated DNA sequencing (RAD-seq); Miller et al. 2007], investigations into hybridization may now take a genomic approach. Because songbirds have strong interchromosomal synteny (Kawakami et al. 2014) and genomic resources (Estrildidae: Taeniopygia guttata annotated genome; Warren et al. 2010), these resources may be combined with thousands of genetic loci to identify genomic regions with reduced introgression via hybridization or increased levels of fixed differences in parental populations (e.g., the Z chromosome).
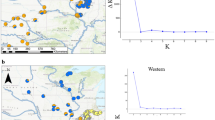
Using 20 Contopus individuals across a narrow zone of sympatry in Nebraska, USA (Fig. 1a) and 11 pure parental individuals (away from zone of sympatry), we obtained thousands of loci to investigate potential hybridization between C. sordidulus and C. virens, and ask the following questions:

Sampling and genetic structure of all individuals. a Localities sampled in this study in Nebraska and Missouri, USA. Colors correspond to pure C. sordidulus (dark red), pure C. virens (blue), or mixed (light purple). b STRUCTURE and NewHybrids results for the 50 and 75 % coverage matrices (CM). Each column represents the posterior probability of clustering to genetic groups (STRUCTURE) or parental/hybrid classes (NewHybrids). Localities with an “S” subscript had individuals with intermediate (i.e., possible hybrid) song that were unable to be collected (see “Results” and “Discussion”)
-
1.
Is there hybridization between Contopus species?
-
H0: There is a lack of evidence of hybridization between species.
-
HA: There is evidence of infrequent hybridization.
-
-
2.
If hybridization is detected, is gene flow between species biased to certain genomic regions?
-
H0: Gene flow is consistent across the genome.
-
HA: The Z chromosome has less gene flow between species.
-
Methods
Sampling
Fresh tissue samples of 31 C. sordidulus and C. virens were obtained from Nebraska and Missouri, USA (Fig. 1a; Table 1) during summer 2014. For some of these individuals, song was recorded (Table 1). Two sites consisted of relatively large Ponderosa Pine (Pinus ponderosa) plantations that were initially hand planted in 1902: (1) Steer Creek campground (Locality 2 of Fig. 1a; Table 1), Samuel McKelvie Nebraska National Forest, north-central Cherry County, Nebraska; (2) Bessey District of the Nebraska National Forest, Thomas County (Locality 6 of Fig. 1a; Table 1). Prior to planting both areas consisted of Sandhill prairie and thus would not have provided Contopus breeding habitat. Today, the mature plantations continue to be surrounded by grasslands (USDA Forest Service 2015). Five other sites were sampled in close proximity to these two hand-planted forests (Localities 3, 4, 5, 7, 8 of Fig. 1a; Table 1). An additional three sites were assessed with presumed pure populations of each taxon taken far removed from the contact zone in northwest Nebraska (sordidulus; Locality 1 of Fig. 1a; Table 1) and eastern Nebraska and northwest Missouri (virens; Localities 9 and 10 of Fig. 1a; Table 1). One sample of C. pertinax was included as an outgroup to confirm that ingroup samples were more closely related and no mistakes were made handling tissues. We used a QIAGEN DNeasy blood and tissue extraction kit to extract genomic DNA for each individual.
Laboratory procedures and SNP dataset creation
To obtain single nucleotide polymorphism (SNP) data from all individuals, we performed a modified RAD-seq (Miller et al. 2007) protocol identical to that used by Manthey and Moyle (2015). Briefly, we digested samples with the restriction enzyme NdeI, multiplexed with one barcode per individual, and size selected fragments between 500 and 600 bp using a Pippin Prep electrophoresis cassette (Sage Science). DNA quality and quantity was tested using quantitative polymerase chain reaction and the Agilent Tapestation, followed by sequencing of 100 bp single-end reads on a partial lane of an Illumina HiSeq2500 performed at the University of Kansas Genome Sequencing Core Facility.
We used the STACKS (Catchen et al. 2013) pipeline to assemble loci de novo from the Illumina sequencing run data files. Sequences were screened for quality, including removal of sequences lacking the restriction site or containing possible adapter contamination. We used the default settings of the ustacks, cstacks, and sstacks modules in STACKS. Finally, we used the populations module of STACKS to create SNP datasets, with the following restrictions: a minimum allele frequency of 0.05, a minimum stack depth of five, and observed heterozygosity less than 0.5 (to reduce paralogous loci included). With these restrictions, we created two datasets, where loci needed to be represented in 50 or 75 % of individuals to be included (i.e., 50 and 75 % coverage matrices, respectively). To assess robustness of minimum stack depth, we reran the last step of STACKS with different values of minimum stack depth (m = 1, 5, 10, 20). Based on these different values, genetic differentiation (F ST) among localities did not change (R > 0.96 all comparisons); we therefore continued all subsequent analyses with the original settings.
Genetic structure and identification of hybrids
We used the program STRUCTURE (Pritchard et al. 2000) to investigate genetic structure and potential admixture between species. We initially inferred lambda with a fixed number of genetic clusters (k = 1). In subsequent runs, we used the inferred lambda with a fixed number of genetic clusters (k = 2) and using the admixture model. Five replicates were run for each dataset, using 50,000 steps as burn-in, followed by 100,000 sampled iterations. To explicitly identify hybrids, we used the program NewHybrids (Anderson and Thompson 2002), which calculates the posterior probability of an individual being a parental, F1, F2, or backcross. Because the program would not work with the large number of SNPs in our datasets, we limited this analysis to the 75 % coverage matrix (CM) inclusive of only SNPs with a minor allele frequency greater than 0.3 (similar to the reduction technique used by Bell et al. 2015). We ran this program with 100,000 burn-in steps with 100,000 iterations subsequently sampled.
BLAST+ analyses
To investigate differential genetic structure between species per chromosome, we used the BLAST+ utility (Camacho et al. 2009) to match RAD-seq loci with Zebra Finch (Taeniopygia guttata) chromosomes. The high levels of interchromosomal synteny in songbirds (Kawakami et al. 2014) allow matching of loci to chromosome, but frequent intrachromosomal recombination precludes inference of chromosomal position of each locus. We used all loci in the 50 % coverage matrix, which is inclusive of all loci from the less restrictive matrix. Here, to be considered a match to Zebra Finch chromosomes, the sequence needed 70 % sequence identity and a maximum e value of 0.01. Multiple e values (0.01, 0.001, 0.0001) were tested to ensure robustness of results; all results with different e values showed highly related number of loci per chromosome (R 2 > 0.99). Thus, results with an e value of 0.01 were hereafter used.
For all individuals that were genetically pure based on STRUCTURE analyses (see “Results”, Fig. 1), we estimated the F ST between the two Contopus species. We did this in order to assess whether sex chromosomes showed increased differentiation between species (e.g., Passerina buntings, Carling and Brumfield 2009), or whether there was a pattern related with chromosome size and F ST (e.g., Certhia treecreepers, Manthey et al. 2015, 2016). All values of F ST were estimated using STACKS.
Results
Genetic data
Illumina sequencing of 31 Contopus sp. individuals resulted in a total of ~42 million sequencing reads (Table 1). The number of reads was highly variable among individuals (mean ~1.3 million reads, SD ~980 thousand reads). This resulted in a total of ~3.9 billion quality-trimmed sequenced base pairs. The coverage was generally high (mean ~77 reads per included SNP locus; Table 1) but also variable across individuals (SD ~56 reads per SNP locus). In the total dataset, there were 419285 RAD-tags; when limited to the 50 and 75 % coverage matrices, this resulted in datasets with 5538 loci (18838 SNPs) and 2064 loci (7499 SNPs), respectively.
Of the ten localities sampled, eight were genetically pure for C. sordidulus or C. virens (Fig. 1b) based on STRUCTURE results. Two localities had individuals with possible mixed ancestry (i.e., potential hybrids, Localities 4 and 5 in Fig. 1b). This was reinforced with results of the NewHybrids analysis, which identified strong probability for a backcross C. sordidulus and an F2 hybrid in the same localities (Fig. 1b).
Among polymorphic SNPs, little was fixed between pure C. sordidulus and C. virens (using non-hybrid individuals, Fig. 2a). The majority of fixed differences (~50 %) were on the largest chromosome (Chr. 2), with others spread across chromosomes (Chr. 1, 4, 8, 10). While a large proportion of polymorphisms were private to either sordidulus or virens, about one-third of genetic variation was shared between species (Fig. 2a).

Patterns of genetic diversity and differentiation. a The proportion of fixed, shared, and private polymorphisms from the 50 and 75 % coverage matrices (CM). b The proportion of non-private polymorphisms that are at differential allele frequencies for each dataset
Given the inherent lack of fixed differences between species and apparent strong genetic structure (Fig. 1b), we investigated differential allele frequencies between species. We found large numbers of segregating polymorphisms (Fig. 2b), with ~10 % of non-private genetic variation with allele frequency differences at a 90–10 % ratio (i.e., 90 % major allele in one lineage and less than 10 % in the other lineage). The high number of loci with strong allele frequency differences between species likely lead to the strong genetic structure patterns observed in STRUCTURE analyses (Fig. 1b).
Between the two species the Z chromosome had one of the highest F ST values (0.176) across all chromosomes, but it did not appear to be an outlier based on chromosome size (Fig. 3). Overall, across well-sampled chromosomes (≥10 loci per chromosome) there was a positive relationship between chromosome size and genetic differentiation (R 2 = 0.34, p = 0.006) assuming interchromosomal synteny in songbirds (Kawakami et al. 2014).

Relationship of chromosome size and genetic differentiation. The arrow points to the Z chromosome
Song recordings
For some specimens, in addition to genetic information, song recordings were also collected. Because the two species have distinctive primary songs (Rising and Schueler 1980), we investigated recordings in a qualitative fashion. We direct readers to Xeno-Canto (xeno-canto.org) and the Cornell Lab of Ornithology’s Macaulay Library (macaulaylibrary.org) where typical song types of both species may be simultaneously examined aurally and spectrographically. Here, we report on individuals as having typical or aberrant call types (accession information of recordings with specimens in Table 1).
In 2011, Robbins recorded individuals from Steer Creek campground (Locality 2 of Fig. 1a) and identified two presumed males in a minimum of seven pairs that may have involved hybrids (song accessions: ML 172380, 172385, 172387). These initial observations prompted collection of more recordings and genetic samples in 2014. Song recordings of individuals at the same location on 19 June 2014 (Locality 2 of Fig. 1a) produced only sordidulus males (at least 12 territorial males; ebird checklist: S18839266; ML 515869–72; 515874–79; six specimens with genetic data; Table 1). Further west (Localities 3 and 4 of Fig. 1a), all recorded individuals gave typical sordidulus call (ML 515893, 515907, 515909), even an individual identified as a backcross sordidulus (Table 1; ML 515908). At the Niobrara National Wildlife Refuge along the Niobrara River (Locality 5 of Fig. 1a), one genetically virens bird gave a typical virens call (ML 515906, 201685), while an F2 individual gave a somewhat aberrant virens call (ML 515903). To the south, at the Bessey District of Nebraska National Forest, both song types were recorded (Locality 6 of Fig. 1a); here, two virens-like individuals were audio recorded but not collected. One of these repeatedly gave a song that appeared to have characteristics of both Contopus. It gave a virens-like slurred whistle, but it had the burry quality of sordidulus (ML 515859). The other virens-like bird, also not collected, appeared to have a more typical virens-like song (ML 515863). Five other males recorded at this site—one that was collected (KU 123160)—were sordidulus (ML 515864–7). Lastly, one bird in western Keya Paha County (42.832, −100.154; ~1.5 km from Locality 7 of Fig. 1a), gave intermediate song (ML 515920), but was not collected.
Discussion
Distributional changes resulting in secondary contact and hybridization
Prior to European settlement most of the Great Plains was much less forested due to bison grazing and regular fires (Roe 1970; Brown 1993; Stewart 2002); therefore, Contopus contact during the breeding season, if there was any, would have been very limited and likely would have been restricted to narrow riparian corridors west of the 100th meridian west (Rising and Schueler 1980; Sharpe et al. 2001). With the elimination of bison, suppression of fire, and anthropogenic planting of trees during the past ca. 150 years much of the Great Plains has become forested, especially along river corridors; this has facilitated recent contact among a number of avian species, including Contopus (Rising 1983).
Within Nebraska, the earliest historical information indicates that the two Contopus species were not in contact in the early part of the 20th century, as it is believed that C. sordidulus was restricted to west of −100° longitude and C. virens had not yet expanded west to that meridian. Swenk and Dawson (1921) remarked that the two “do not anywhere meet”. During that period, C. sordidulus reached as far east as Thomas County along the Dismal River (Bruner et al. 1904). During subsequent decades both species likely expanded breeding distributions within the state, sordidulus eastward, virens westward. Short (1961) believed that limited contact might occur along the Niobrara River Valley between Valentine and the Pine Ridge region and he noted to the south that virens was found as far west as the Colorado border along the South Platte River.
In the region that we sampled, our genetic (STRUCTURE and NewHybrids results, Fig. 1b) and vocal data indicate a very narrow contact zone between these two Contopus species in north-central Nebraska (Fig. 1a). In 2011, Robbins noted multiple pairs (ebird checklist: S8389263) of Contopus breeding at the relatively large Ponderosa Pine (Pinus ponderosa) plantation at the Steer Creek campground (Locality 2 of Fig. 1a; Table 1); while initial recordings (from 2011) suggested possible hybrid individuals, sampling in 2014 indicated only sordidulus individuals (see “Results”). From the 2014 specimens with genetic data, two had a small probability of being sordidulus backcrosses (Fig. 1b). This may suggest that areas of sympatry vary through time. As both species are migratory, this may simply be a case of which individuals set up territories earliest in areas where pine and riparian habitats coincide.
In Central Nebraska, at the Bessey District of Nebraska National Forest (Locality 6 of Fig. 1a), one individual with genetic data was pure sordidulus. However, both species were present in this area based on song recordings, with one appearing to have characteristics of both Contopus (ML 515859; see “Results”). All individuals were in Ponderosa Pine-dominated upland forest. None were found along the narrow riparian strip, <2 km in length, of the Middle Loup River through this national forest. This area deserves further investigation.
The other area of contact that we identified was centered along the Niobrara River in the vicinity of Valentine. Just to the southwest of Valentine, where Ponderosa Pine was on the slopes and riparian vegetation was along the river, birds were audio recorded and collected (Locality 4 of Fig. 1a; Table 1). Here, one of four individuals with genetic samples was identified as a backcross sordidulus (Fig. 1b). A few kilometers to the east of Valentine, at Niobrara National Wildlife Refuge along the Niobrara River (Locality 5 of Fig. 1a; Table 1) sordidulus was in pines upslope from the lower riparian-inhabiting virens. At this location, one individual was strongly identified as an F2 hybrid, with another individual potentially being a backcross virens (Fig. 1b).
Patterns of genomic differentiation
Because of the presumed lack of hybridization until recently, the evolution of these sister species likely occurred in allopatry throughout much of the recent past. Cicero and Johnson (2002) found only 1.7 % sequence divergence in the mitochondrial cytochrome B gene between C. sordidulus and C. virens, less than intraspecific differences in other bird species with east–west splits in North America (e.g., Certhia americana, Manthey et al. 2011; Sitta carolinensis, Spellman and Klicka 2007), and suggestive that the split between these two Contopus species occurred relatively recently. It was thus not surprising that little of the nuclear genome was fixed (~0.3 %) between these species near a contact zone (using non-hybrid individuals, Fig. 2a).
Because of increased genetic differentiation and reduced gene flow on the Z chromosome in many hybridizing bird species, including Ficedula flycatchers (Ellegren et al. 2012), Luscinia nightingales (Storchova et al. 2010), Passer sparrows (Elgvin et al. 2011) and Passerina buntings (Carling and Brumfield 2008, 2009), we investigated differential patterns of genetic differentiation among chromosomes. The Z chromosome had one of the highest F ST values (0.176) between species across all chromosomes, but it did not appear to be an outlier based on chromosome size (Fig. 3). Across well-sampled chromosomes (≥10 loci per chromosome) there was a positive relationship between chromosome size and genetic differentiation (R 2 = 0.34, p = 0.006) assuming interchromosomal synteny in songbirds (Kawakami et al. 2014). This relationship has been found in one other North American songbird species (Certhia americana, Manthey et al. 2015, 2016), although the relationship observed here in pewees is not as strong (R 2 > 0.8 in Certhia).
In Certhia, this pattern was hypothesized to be due to genetic drift across chromosomes, with differential recombination frequencies among chromosomes due to negative scaling of recombination rates with chromosome size due to meiotic recombination requirements (Lynch 2007). This hypothesis was largely owing to an assumed lack of hybridization between Certhia lineages, leading to no strong patterns of similar selective pressures across lineages (i.e., only independent selective pressures) and the genomic signal sampled being due to genetic drift through time in allopatry. Many similarities exist between the Certhia and Contopus systems: (1) A presumed lack of widespread hybridization, at least until recently, (2) habitat differences between lineages/species which could lead to non-random gene flow and subsequent genetic differentiation (Edelaar and Bolnick 2012), and (3) dialect or song differences that could act as a pre-mating isolation mechanism. Because of a potential selective mechanism (e.g., song recognition signal or other factors) there appears to be no strong selection against hybrids allowing the signal of genetic drift to be the main force observed in our data, although our data does not preclude the possibility of selection contributing to this pattern. These similarities suggest that pre-mating isolation mechanisms may result in a positive relationship between chromosome size and genetic differentiation—at least in oscine passerines songbirds—due to the high variance in chromosome size (and relative recombination rates) and a steady rate of genome-wide genetic differentiation—easily observable in RAD-seq datasets compared to specific loci under selection—due to genetic drift.
Conclusions
We provide the first conclusive evidence of hybridization between C. sordidulus and C. virens in a narrow zone of sympatry in central Nebraska, USA based on thousands of single nucleotide polymorphisms across the genome. Contact is a result of contemporary human disturbance and did not likely occur in the past in Nebraska. The two species have little fixed differences (~0.3 % of genetic variation), although a large proportion of polymorphisms have highly differentiated allele frequencies between species. Additionally, we found a positive relationship between genetic differentiation and chromosome size, likely caused by minimal hybridization and a large proportion of observed genetic differentiation due to genetic drift, potentially in concert with selection.
References
Anderson EC, Thompson EA. A model-based method for identifying species hybrids using multilocus genetic data. Genetics. 2002;160:1217–29.
Bell RC, Drewes RC, Zamudio KR. Reed frog diversification in the Gulf of Guinea: overseas dispersal, the progression rule, and in situ speciation. Evolution. 2015;69:904–15.
Brown DA. Early nineteenth-century grasslands of the midcontinent plains. Ann Assoc Am Geogr. 1993;83:589–612.
Bruner L, Wolcott RH, Swenk MH. A preliminary review of the birds of Nebraska, with synopses. Omaha: Klopp & Bartlett Co.; 1904.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform. 2009;10:421.
Carling MD, Brumfield RT. Haldane’s rule in an avian system: using cline theory and divergence population genetics to test for differential introgression of mitochondrial, autosomal, and sex-linked loci across the Passerina bunting hybrid zone. Evolution. 2008;62:2600–15.
Carling MD, Brumfield RT. Speciation in Passerina buntings: introgression patterns of sex-linked loci identify a candidate gene region for reproductive isolation. Mol Ecol. 2009;18:834–47.
Carling MD, Serene LG, Lovette IJ. Using historical DNA to characterize hybridization between Baltimore Orioles (Icterus galbula) and Bullock’s Orioles (I. bullockii). Auk. 2011;128:61–8.
Catchen JM, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool for population genomics. Mol Ecol. 2013;22:3124–40.
Cicero C, Johnson NK. Phylogeny and character evolution in the Empidonax group of tyrant flycatchers (Aves: Tyrannidae): a test of WE Lanyon’s hypothesis using mtDNA sequences. Mol Phylogenet Evol. 2002;22:289–302.
Edelaar R, Bolnick DI. Non-random gene flow: an underappreciated force in evolution and ecology. Trends Ecol Evol. 2012;27:659–65.
Ellegren H, Smeds L, Burri R, Olason PI, Backström N, Kawakami T, Künstner A, Mäkinen H, Nadachowska-Brzyska K, Qvarnström A, Uebbing S, Wolf JBW. The genomic landscape of species divergence in Ficedula flycatchers. Nature. 2012;491:756–60.
Elgvin TO, Hermansen JS, Fuarczyk A, Bonnet T, Borge T, Sæther SA, Voje KL, Sætre GP. Hybrid speciation in sparrows II: a role for sex chromosomes? Mol Ecol. 2011;20:3823–937.
Grudzien TA, Moore WS, Cook JR, Tagle D. Genic population structure and gene flow in the Northern Flicker (Colaptes auratus) hybrid zone. Auk. 1987;104:654–64.
Hewitt GM. Hybrid zones-natural laboratories for evolutionary studies. Trends Ecol Evol. 1988;3:158–67.
Kawakami T, Smeds L, Backström N, Husby A, Qvarnström A, Mugal CF, Olason P, Ellegren H. A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol Ecol. 2014;23:4035–58.
Lynch M. The origins of genome architecture. Sunderland: Sinauer Associates; 2007.
Manthey JD, Moyle RG. Isolation by environment in White-breasted Nuthatches (Sitta carolinensis) of the Madrean Archipelago sky islands: a landscape genomics approach. Mol Ecol. 2015;24:3628–38.
Manthey JD, Klicka J, Spellman GM. Cryptic diversity in a widespread North American songbird: phylogeography of the Brown Creeper (Certhia americana). Mol Phylogenet Evol. 2011;58:502–12.
Manthey JD, Klicka J, Spellman GM. Chromosomal patterns of diversity and differentiation in creepers: a next-gen phylogeographic investigation of Certhia americana. Heredity. 2015;115:165–72.
Manthey JD, Robbins MB, Moyle RG. A genomic investigation of the putative contact zone between divergent Brown Creeper (Certhia americana) lineages: chromosomal patterns of genetic differentiation. Genome. 2016;59:115–25. doi:10.1139/gen-2015-0093.
Mayr E. Systematics and the origin of species, from the viewpoint of a zoologist. Cambridge: Harvard University Press; 1942.
Mettler RD, Spellman GM. A hybrid zone revisited: molecular and morphological analysis of the maintenance, movement, and evolution of a Great Plains avian (Cardinalidae: Pheucticus) hybrid zone. Mol Ecol. 2009;18:3256–67.
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007;17:240–8.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Pyle P. A further examination of wing and tail formulae in Contopus and Empidonax flycatchers. In: Dickman RW, editor. The era of Allan Phillips: a Festschrift. NM: Albuquerque; 1997. p. 147–54.
Rising JD. Summer birds from Cherokee County, Kansas. Kansas Orn Soc Bull. 1965;16:9–16.
Rising JD. The great plains hybrid zones. In: Johnston R, editor. Current ornithology. New York: Plenum Press; 1983.
Rising JD, Schueler FW. Identification and status of wood pewees (Contopus) from the Great Plains: what are sibling species? Condor. 1980;82:301–8.
Roe FG. The North American Buffalo. Toronto: University of Toronto Press; 1970.
Sharpe RS, Silcock WR, Jorgensen JG. Birds of Nebraska. Their distribution and temporal occurrence. Lincoln: University of Nebraska Press; 2001.
Short LL. Notes on bird distribution in the central Plains. Nebraska Bird Rev. 1961;29:2–22.
Spellman GM, Klicka J. Phylogeography of the white-breasted nuthatch (Sitta carolinensis): diversification in North American pine and oak woodlands. Mol Ecol. 2007;16:1729–40.
Stewart O. Forgotten fires, Native Americans and the transient wilderness. Norman: University of Oklahoma Press; 2002.
Storchova R, Reif J, Nachman MW. Female heterogamety and speciation: reduced introgression of the Z chromosome between two species of nightingales. Evolution. 2010;64:456–71.
Swenk MH, Dawson RW. Notes on the distribution and migration of Nebraska birds. 1. Tyrant flycatchers (Tyrannidae). Wilson Bull. 1921;33:132–41.
Swenson NG, Howard DJ. Clustering of contact zones, hybrid zones, and phylogeographic breaks in North America. Am Nat. 2005;166:581–91.
USDA Forest Service (2015). Nebraska national forest history. www.fs.usda.gov/main/nebraska/learning/history-culture. Accessed 1 June 2015.
Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Künstner A, Searle S, White S, Vilella AJ, Fairley S, Heger A, Kong L, Ponting CP, Jarvis ED, Mello CV, Minx P, Lovell P, Velho TAF, Ferris M, Balakrishnan CN, Sinha S, Blatti C, London SE, Li Y, Lin Y-C, George J, Sweedler J, Southey B, Gunaratne P, Watson M, Nam K, Backström N, Smeds L, Nabholz B, Itoh Y, Whitney O, Pfenning AR, Howard J, Völker M, Skinner BM, Griffin DK, Ye L, McLaren WM, Flicek P, Quesada V, Velasco G, Lopez-Otin C, Puente XS, Olender T, Lancet D, Smit AFA, Hubley R, Konkel MK, Walker JA, Batzer MA, Gu W, Pollock DD, Chen L, Cheng Z, Eichler EE, Stapley J, Slate J, Ekblom R, Birkhead T, Burke T, Burt D, Scharff C, Adam I, Richard H, Sultan M, Soldatov A, Lehrach H, Edwards SV, Yang S-P, Li XC, Graves T, Fulton L, Nelson J, Chinwalla A, Hou S, Mardis ER, Wilson RK. The genome of a songbird. Nature. 2010;464:757–62.
Authors’ contributions
MBR performed fieldwork, collecting specimens and song data. JDM collected and analyzed genomic data. MBR and JDM designed the research and worked on completion of the manuscript. Both authors read and approved the final manuscript.
Authors’ information
JDM was a graduate student at The University of Kansas working at the Biodiversity Institute and is currently a postdoctoral researcher at New York University in Abu Dhabi. He studies the evolutionary history of birds using ecological, geographic, and genomic landscapes. MBR is the ornithology collections manager at the University of Kansas Biodiversity Institute. His work has ranged from the study of North American avian contact zones, manakin social organization, systematics, wren intra-breeding season movements to the use of avian song rates and the implications for estimating populations.
Acknowledgements
We would like to thank the U.S. Fish and Wildlife Service, Fort Niobrara National Wildlife Refuge, and Nebraska Game and Parks Commission for pertinent permits. Specimen collection was performed following review of the Institutional Animal Care and Use Committee (IACUC) at University of Kansas. We thank John Kelly and Patrick Monnahan for lab work advice and materials. This work was funded through an NSF Doctoral Dissertation Improvement Grant (DEB-1406989). Thomas Labedz helped with logistics and identifying potential contact zone areas. We thank the use of facilities at the COBRE Genome Sequencing Core Laboratory at the University of Kansas, funded through NIH award number P20GM103638.
Competing interests
The authors declare that they have no competing interests.
Data accessibility All raw sequence data (FASTQ files) are available at the NCBI Sequence Read Archive accessioned under BioProject ID PRJNA315945. All song files are available at Macaulay Library, Cornell Laboratory of Ornithology (macaulaylibrary.org; see Table 1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Manthey, J.D., Robbins, M.B. Genomic insights into hybridization in a localized region of sympatry between pewee sister species (Contopus sordidulus × C. virens) and their chromosomal patterns of differentiation. Avian Res 7, 6 (2016). https://doi.org/10.1186/s40657-016-0040-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40657-016-0040-7