
Abstract
Background
The clinical significance of SPINK1 intronic variants in chronic pancreatitis has been previously assessed by various approaches including a cell culture-based full-length gene assay. A close correlation between the results of this assay and in silico splicing prediction was apparent. However, until now, a clinical diagnostic pipeline specifically designed to classify SPINK1 intronic variants accurately and efficiently has been lacking. Herein, we present just such a pipeline and explore its efficacy and potential utility in potentiating the classification of newly described SPINK1 intronic variants.
Results
We confirm a close correlation between in silico splicing prediction and results from the cell culture-based full-length gene assay in the context of three recently reported pathogenic SPINK1 intronic variants. We then integrated in silico splicing prediction and the full-length gene assay into a stepwise approach and tested its utility in the classification of two novel datasets of SPINK1 intronic variants. The first dataset comprised 16 deep intronic variants identified in 52 genetically unexplained Chinese chronic pancreatitis patients by sequencing the entire intronic sequence of the SPINK1 gene. The second dataset comprised five novel rare proximal intronic variants identified through the routine analysis of the SPINK1 gene in French pancreatitis patients. Employing a minor allele frequency of > 5% as a population frequency filter, 6 of the 16 deep intronic variants were immediately classified as benign. In silico prediction of the remaining ten deep intronic variants and the five rare proximal intronic variants with respect to their likely impact on splice site selection suggested that only one proximal intronic variant, c.194 + 5G > A, was likely to be of functional significance. Employing the cell culture-based full-length gene assay, we functionally analyzed c.194 + 5G > A, together with seven predicted non-functional variants, thereby validating their predicted effects on splicing in all cases.
Conclusions
We demonstrated the accuracy and efficiency of in silico prediction in combination with the cell culture-based full-length gene assay for the classification of SPINK1 intronic variants. Based upon these findings, we propose an operational pipeline for classifying SPINK1 intronic variants in the clinical diagnostic setting.
Similar content being viewed by others

Background
Chronic pancreatitis has traditionally been defined as a chronic inflammatory process of the pancreas that leads to progressive and irreversible impairment of both exocrine and endocrine functions, with a focus on morphological changes. More recently, the disease has been redefined as a “pathologic fibro-inflammatory syndrome of the pancreas in individuals with genetic, environmental and/or other risk factors who develop persistent pathologic responses to parenchymal injury or stress”, with a focus on underlying pathogenic mechanisms [1]. In particular, genetic studies over the past two decades have underscored the importance of a trypsin-dependent pathway in the etiology of the disease [2,3,4,5]. One of the most extensively studied pancreatitis susceptibility genes, SPINK1 (encoding pancreatic secretory trypsin inhibitor; MIM# 167790), is characterized by a diverse range of reported variants from point mutations to whole gene deletions (for a complete list, see ref. [6]). Pathogenic SPINK1 variants predispose to pancreatitis by lowering the inhibitory capacity of prematurely activated trypsin within the pancreas. The clinical relevance of canonical splice site variants, nonsense mutations, or large-scale genomic deletions in the SPINK1 gene is generally clear. By contrast, the clinical relevance of SPINK1 promoter and enhancer variants [7,8,9], missense variants [10, 11], or intronic variants occurring outwith the canonical splice sites [12, 13] has often had to be ascertained by in vitro functional analysis.
In silico splicing prediction programs have been widely used to evaluate the functional effects of intronic variants in clinical genetics, either on their own or in combination with an in vitro splicing assay [14, 15]. In this regard, we have previously employed a cell culture-based full-length gene assay to systematically assess the functional impact of a series of SPINK1 intronic variants [12, 13] and, more recently, we have noted a close correlation between the results from this assay and in silico splicing predictions [16]. The full-length gene assay has at least two advantages over the commonly used minigene splicing assay. First, the full-length gene assay preserves better the natural genomic context of the studied variants, a point of key importance given the highly context-dependent nature of splicing regulation [17]. Second, the full-length gene splicing assay can be readily used to evaluate intronic variants located near the first exon or last exon of the gene; by contrast, special adaptation of the minigene would normally be required for such variants to be analyzed, as exemplified by a recent publication [18].
SPINK1 intronic variants continue to be reported in the literature [19,20,21,22] and additional SPINK1 intronic variants, including those located in deep intronic regions, are certainly likely to emerge with the application of high-throughput whole-genome sequencing [23]. To rise to this challenge, establishment of a clinical diagnostic pipeline for the classification of SPINK1 intronic variants is required. The aim of the present study was to develop such a pipeline and assess its efficacy and utility. To this end, we further explored the correlation of in silico splicing prediction and our cell-based full-length gene assay in the context of three recently reported pathogenic SPINK1 variants. Then we integrated both the in silico splicing prediction procedure and the full-length gene assay into a stepwise approach in order to classify a series of SPINK1 intronic variants newly discovered in Chinese and French pancreatitis patients.
Results and discussion
Further correlation of in silico splicing prediction and functional assay data in the context of three recently reported SPINK1 splice site variants
Before going into the detail of the current study, we would like to make two points. The first refers to the experimental evaluation of the functional effect of intronic variants. Ideally, the disease-affected tissue/cells or surrogate tissue/cells that also highly express the gene of interest from the patients should be analyzed whenever possible. SPINK1 mRNA is most abundantly expressed in the pancreas, with a median transcripts per kilobase million (TPM) of 4361 in accordance with the Genotype-Tissue Expression (GTEx) dataset [24]. Stomach ranks second for SPINK1 mRNA expression, although the corresponding TPM is only 285 [24]. Neither tissue, but particularly the pancreas, is accessible in practice in terms of biopsy samples. The next and most commonly used strategy is to perform a splicing assay in a transient expression system, in which human cell lines of pathophysiological relevance should be employed whenever possible owing to the tissue specificity of the splicing process in some instances (see [25] and references therein). Unfortunately, no human pancreatic acinar cell lines are currently available. In the present study, we used human embryonic kidney 293 T (HEK293T) cells for the splicing assay as previously described [12, 13, 16]. It is possible that splicing in HEK293T cells may not always reflect the in vivo situation, a general drawback of splicing assay that employ non-pathophysiologically relevant cells [26].
The second point refers to in silico prediction of the impact of intronic variants on splicing. Of particular relevance, we have previously observed a close correlation of results from our full-length gene assay with those from the in silico splicing predictions in the context of 24 SPINK1 intronic variants [16]. Findings pertaining to two variants merit especial attention. First, c.194 + 2T > C (or IVS3 + 2T > C) was shown to retain partial ability to generate wild-type transcripts by reverse transcription-polymerase chain reaction (RT-PCR) analysis of patient-derived stomach tissue [27] and also by our full-length gene assay [12]. It was predicted to be associated with only an ~ 12% decrease in the score for SpliceSiteFinder-like (wild-type score of 82.6 vs mutant score of 72.3) but invariably a score of 0 for MaxEntScan, NNSPLICE, and Human Splicing Finder [16]. Both predictions were correct, depending on one’s viewpoint. Thus, the prediction of SpliceSiteFinder-like was correct from the standpoint that c.194 + 2T > C was able to generate an appreciable level of wild-type transcript; the predictions of MaxEntScan, NNSPLICE, and Human Splicing Finder were correct from the standpoint that c.194 + 2T > C resulted in a significantly reduced level of normally spliced SPINK1 transcript as compared to that of the wild-type allele. The second case involved the c.194 + 13T > G variant, which was predicted by different programs to generate a new and viable donor splice site but resulted in the generation of a trace amount of aberrantly spliced transcripts that was only detectable using specially designed allele-specific primers [16]. With hindsight, these two particular cases might reflect a limitation in splicing prediction programs analogous to that seen in missense variant prediction programs: “The main issue lies within the binary output of most models, which predict whether or not a variant has an effect but not its magnitude” [28]. Nonetheless, at least in the context of the 24 SPINK1 intronic variants analyzed [16], the in silico prediction tools were collectively not found to yield false negative findings.
Bearing in mind the aforementioned considerations, we decided to further explore the cross-correlation of in silico predictions and our full-length gene splicing assay in the context of three recently reported SPINK1 splice site variants, c.55 + 1G > A [19], c.194 + 1G > A [20], and c.88-1G > A [22]. These three variants are of unequivocally clinical significance by virtue of their disruption of splice site consensus sequences (Additional file 1: Figure S1), as predicted by SpliceSiteFinder-like, MaxEntScan, NNSPLICE, and GeneSplicer, made available via the Alamut software suite, under default conditions [29].
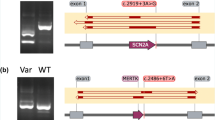
We then characterized the splicing defects associated with the three splice site variants by means of our previously established cell culture-based full-length gene assay [12, 13]. RT-PCR analysis of HEK293T cells transfected with the full-length SPINK1 gene construct harboring the c.55 + 1G > A variant showed two aberrant transcripts (Fig. 1a). Subsequent sequencing revealed that the shorter transcript resulted from the activation of a cryptic splice donor site within exon 1 (at position c.7_8), resulting in the deletion of the 3′ end of exon 1; the longer transcript resulted from activation of a cryptic splice acceptor site within intron 1 (at position c.55 + 141_55 + 142), resulting in the insertion of the 5′ end of intron 1 into the transcript (Fig. 2).

Results from the cell culture-based full-length gene assay. a, b RT-PCR analyses of HEK293T cells transfected with full-length SPINK1 gene expression constructs carrying respectively the wild-type and indicated intronic variants. Normal transcripts (confirmed by sequencing) are indicated by arrows. In b, the lower bands generated by c.194 + 2 T > C and c.194 + 5G > A were found to be identical by sequencing, with exon 3 being skipped. See Fig. 2 for the precise splicing outcomes of the three recently reported SPINK1 splice site variants

Splicing outcomes of the three recently reported SPINK1 splice site variants as determined by the cell culture-based full-length gene assay. Normal splicing in the context of the wild-type sequence and aberrant splicing in the context of the mutant sequence are illustrated for each of the three variants. The splice donor signal (GT) and splice acceptor signal (AG) are highlighted in blue. Variants occurred within the splice sites are highlighted in green. In c.55 + 1G > A, the two novel splice donor sites used for aberrant splicing are highlighted in red. In c.[88-1G > A; 88-7 T > A], the c.88-1G > A variant shifted the AG site by one base, resulting in the skipping of the first nucleotide of exon 3 (i.e., the G highlighted in red). S shorter, L longer, M mutant
The c.194 + 1G > A variant was found to result in the generation of a single aberrant transcript (Fig. 1a), in which exon 3 was skipped (Fig. 2).
The c.88-1G > A variant [22] was found to be in cis with c.88-7 T > A, located only five bases away (Additional file 1: Figure S2). Given that neither of these two variants is present in the Genome Aggregation Database (genomAD) [30], it is possible that they were generated simultaneously as a single mutational event [31, 32]. Irrespective of whether or not the two variants were generated simultaneously, they should be named c.[88-1G > A; 88-7 T > A] in accordance with the Human Genome Variation Society (HGVS) recommendations [33].
The c.[88-1G > A; 88-7 T > A] variant is very likely to affect splicing due to the c.88-1G > A component (Additional file 1: Figure S1). However, the splicing outcome of the c.88-1G > A variant may be modified by the juxtaposition of the c.88-7 T > A variant, even although the latter on its own was predicted not to significantly affect splice site selection (Additional file 1: Figure S3). To explore this possibility, we compared the splicing outcomes in vitro of c.88-1G > A alone, c.88-7 T > A alone, and c.[88-1G > A; 88-7 T > A]. RT-PCR analyses of HEK293T cells transfected with the corresponding full-length gene constructs invariably generated a single band of similar size to that of the wild-type (Fig. 1a). Subsequent sequencing revealed that c.88-1G > A alone and c.[88-1G > A; 88-7 T > A] alone resulted in identical skipping of the first nucleotide of exon 3 (i.e., the splice site was shifted by one nucleotide; Fig. 2) of the SPINK1 gene while c.88-7 T > A alone generated only wild-type transcripts. Consequently, it may be concluded that the functional effect of c.[88-1G > A; 88-7 T > A] was conferred solely by the c.88-1G > A variant.
Taken together, we have provided further evidence for a good correlation between in silico splicing predictions and our functional assay of SPINK1 intronic variants. Indeed, our cell culture-based full-length gene assay not only validated the predicted impact on splicing but also elucidated the precise mRNA splicing consequences of specific pathogenic variants. The latter is key to understanding the genotype-phenotype relationship since aberrantly spliced transcripts may not invariably lead to the synthesis of proteins characterized by complete loss-of-function.
Integration of splicing prediction and functional assay into a stepwise procedure for classifying newly found SPINK1 variants
Data directly comparing the incidences and clinical features of chronic pancreatitis between Chinese and European populations are lacking. By contrast, marked ethnic differences were noted between them in terms of genetic predisposition to the disease, exemplified by three recent findings: the CEL-HYB risk allele [34] was found to be absent in the Chinese population [35]; rare functional CPA1 variants [36] were not enriched in Chinese chronic pancreatitis patients [37]; and the common CTRB1-CTRB2 inversion allele [38] did not contribute to disease risk in the Chinese population due to near allele fixation [39]. Significant differences also exist between Chinese and European populations in terms of the spectrum and frequency of variants in each of the four firmly established pancreatitis susceptibility genes (i.e., SPINK1 [40], PRSS1 [41], CTRC [42, 43], and CFTR [44, 45]) [22].
A comprehensive analysis of the SPINK1, PRSS1, CTRC, and CFTR genes in 253 young French chronic pancreatitis revealed that ~ 52% of the studied patients remained genetically unexplained [46]. Remarkably, the proportion of Chinese patients that remained genetically unexplained after mutational analysis of the above four genes (i.e., ~ 50%) [22] is quite comparable to that in the French patients. As part of our attempt to identify the “missing heritability,” we performed targeted resequencing of the deep intronic sequence of the SPINK1 gene in 52 genetically unexplained Chinese chronic pancreatitis patients using previously described methods [13]. [Note that the proximal intronic regions of the SPINK1 gene had previously been analyzed [22].] This resulted in the identification of 16 deep SPINK1 intronic variants (Table 1). In addition, during the routine analysis of the SPINK1 gene (focusing on coding and proximal intronic sequences) in French pancreatitis patients, we identified five rare proximal SPINK1 intronic variants that had not previously been described in the literature (Table 2). In the five respective French carriers, no known disease-causing variants in the PRSS1 gene were found but other pancreatitis susceptibility genes remain to be analyzed. This does not affect the conclusion of the present study in any way.
In the following sections, we describe how we attempted to integrate in silico splicing predictions and our full-length gene assay into a stepwise approach to classify the above two datasets of SPINK1 intronic variants.
First step: population frequency filtering
Demonstrating the functionality of a given variant is a prerequisite for any claim of pathogenicity to be credible. A primary consideration when predicting whether a variant is likely to have a functional effect is its population frequency [47]; the rarer the variant, the more likely it is to exert a pathogenic effect. A minor allele frequency (MAF) of > 1% in the control population is the most frequently used threshold for defining “common” variants. Here, we employed a conservative threshold, a MAF of > 5%, for population frequency filtering, using data from genomAD [30] as a reference. Using allele frequency in the East Asian population as a filter would have resulted in 6 of the 16 deep intronic variants found in the Chinese patients being classified as benign (Table 1). Indeed, all six of these common variants have previously been described and annotated as benign in the Genetic Risk Factors in Chronic Pancreatitis Database [6]. It should be noted that in the case of four of the six common variants, it is the minor allele that is used as the reference sequence. We did not attempt to convert the corresponding major allele frequencies to the alternative MAFs in these cases (Table 1).
Second step: in silico prediction on splice site selection
None of the remaining ten deep SPINK1 intronic variants, all of which had an allele frequency of < 5% in the East Asian population (Table 1), have been previously described in the Genetic Risk Factors in Chronic Pancreatitis Database [6]. These variants, together with the five rare proximal variants found in the French patients (Table 2), were subjected to in silico splicing prediction (i.e., disruption of known splice sites or creation of novel splice sites were sought) by means of SpliceSiteFinder-like, MaxEntScan, NNSPLICE, and GeneSplicer made available via the Alamut software suite, under default conditions [29]. However, only the proximal c.194 + 5G > A variant was predicted to be of functional significance by virtue of it significantly reducing the splice site consensus scores (defined here as a reduction of ≥ 10% of the wild-type value across all four prediction programs) as compared to the wild-type allele (Fig. 3). By contrast, none of the ten rare deep intronic variants found in Chinese patients or the other four rare proximal variants found in French patients were predicted to significantly reduce the splice site consensus scores or generate a novel splice site (Additional file 1: Figures S4 and S5). Additionally, we performed the same predictions for the six common deep SPINK1 intronic variants (Table 1) but none were predicted to have a functional effect (Additional file 1: Figure S6). In short, of the 16 common and 5 rare SPINK1 intronic variants, the proximal c.194 + 5G > A variant was the only one predicted to be of functional significance (Tables 1 and 2).

Splicing effect of the proximal c.194 + 5G > A variant as predicted by the Alamut software suite
Third step: functional validation
We performed functional analysis of the predicted functionally significant c.194 + 5G > A variant by means of our cell culture-based full-length gene assay. We also included, as negative controls, seven variants predicted to be non-functional (including both deep and proximal variants; Tables 1 and 2), as a means to cross-correlate in silico prediction and the results of our functional assay. RT-PCR analyses of the respectively transfected HEK293T cells confirmed the splicing predictions in all cases. Thus, a single transcript of similar size to the wild-type was observed in all seven predicted non-functional variants (i.e., c.56-324 T > A, c.194 + 1278C > T, c.195-1414 T > A, c.195-862 T > C, and c.195-854C > T in Fig. 1a; and c.87 + 13 T > G and c.88-48C > A in Fig. 1b); subsequent sequencing confirmed that all these transcripts were identical to the wild-type sequence. By contrast, c.194 + 5G > A generated a splicing pattern that was very similar to that of the pathogenic c.194 + 2 T > C variant [12], comprising a normally spliced band and an aberrantly spliced band (exon 3 skipped). It should be noted that the c.194 + 5G > A variant retained fewer normally spliced transcripts as compared to the c.194 + 2 T > C variant (Fig. 1b), an observation that argues for it being a novel pathogenic variant.
The precise splicing outcomes of pathogenic SPINK1 intronic variants described to date are summarized in Table 3. All these pathogenic intronic variants are located either in the canonical splice sites or very close to the exon/intron junctions. This concurs with findings from many disease genes, probably for two reasons: splice-defining cis-acting sequence elements are predominantly located within proximal intronic regions [48] and the large size of the intronic regions renders routine screening impractical. In this regard, take two examples of recent large-scale analyses, one in the context of human cancer [49] and the other in the context of Stargardt disease [26]: none of the intronic variants under study were located within deep intronic regions. This notwithstanding, pathogenic variants do occur within deep intronic regions, and are often discovered by transcript analysis or whole-genome sequencing (e.g., [50,51,52]). In terms of their functional consequences, pathogenic variants in deep intronic regions often appear to be able to generate some wild-type transcripts (e.g., all three USH2A deep intronic variants reported in [50] were shown to do so in a minigene assay). In terms of their clinical consequences, pathogenic variants in deep intronic regions may be associated with a broad phenotypic spectrum, as exemplified by three CFTR deep intronic variants [52]. Pathogenic variants in the deep intronic regions of the SPINK1 gene may be identified in the future, when whole-genome sequencing is routinely used for clinical diagnosis.
Finally, it is pertinent to mention that any SPINK1 intronic variant that has been classified as benign may actually occur in cis with a functional variant located elsewhere in the coding sequence or regulatory regions of the gene. In order to explore this possibility, we searched all the currently studied SPINK1 intronic variants with a known rs number (Tables 1 and 2) in the GTEx dataset [24]. Only three SNPs, rs6580502, rs17703305, and rs4705202, all of which have a MAF of > 5 in the general population, were associated with a reduced SPINK1 expression; all the expression data were obtained from the lung tissue.
Conclusions
In the context of three recently reported SPINK1 splice site variants, we have provided further evidence for a close correlation between in silico splicing predictions and the results of our functional assay of SPINK1 intronic variants. In the context of two new datasets of SPINK1 intronic variants, we then demonstrated the accuracy and efficiency of in silico splicing prediction in combination with the cell culture-based full-length gene assay in variant classification. In so doing, we elucidated the precise splicing consequences of the three recently reported SPINK1 splice site variants and identified and functionally characterized a novel pathogenic variant, c.194 + 5G > A. Based on the findings of this study and previous studies, we propose the following clinical diagnostic pipeline for classifying SPINK1 intronic variants. The first step applies a population frequency filter using data in genomAD as a reference and employing a conservative MAF of ≥ 5% as a threshold. In the second step, the impact of the remaining rare variants on splice site selection is predicted. These two steps proved highly effective at classifying most of the detected SPINK1 intronic variants as benign. Thus, in practice, only a very small number of SPINK1 intronic variants (those predicted to affect splice site selection) actually needed to be functionally validated. We believe that the application of this procedure will greatly facilitate the classification of SPINK1 intronic variants in a clinical diagnostic setting. This notwithstanding, it should be noted that the number of in-parallel tested SPINK1 intronic variants is still relatively small. Consequently, we would recommend that functional analysis be employed once an intronic variant is suggested to be of functional significance by two or even one splicing prediction programs. Moreover, the threshold MAF for population frequency filtering (the reference population must be the same as the proband population) may be redefined as more data become available. Finally, it should be appreciated that an accurate determination of the pathogenic relevance of any SPINK1 intronic variant in chronic pancreatitis is not only important from a mechanistic viewpoint [1] but also provides potential therapeutic targets as shown in other genes (e.g., [51, 53]).
Methods
Identification of SPINK1 intronic variants in Chinese and French pancreatitis patients
Fifty-two Han Chinese chronic pancreatitis patients, whose age of disease onset was known to be ≤ 20 years or whose disease diagnosis was made at ≤ 20 years, participated this study. These patients, whose pancreatitis had remained genetically unexplained after mutational analysis of the entire coding regions and exon/intron boundaries of four pancreatitis susceptibility genes (i.e., SPINK1 [40], PRSS1 [41], CTRC [42, 43], and CFTR [44, 45]) [22], were searched for possible pancreatitis-predisposing variants occurring within deep SPINK1 intronic regions in accordance with previously described procedures [13]. Proximal SPINK1 intronic variants were identified through routine mutational screening of the entire coding region and exon/intron boundaries of the SPINK1 gene in French pancreatitis patients by means of high-resolution DNA melting (HRM) analysis [54]. All SPINK1 intronic variants were subjected to independent PCR amplification and Sanger sequencing. Informed consent was obtained from each participant. This study was approved by the respective Ethics Committees of Changhai Hospital in Shanghai and the University Hospital in Brest.
Nomenclature of SPINK1 intronic variants
Nomenclature for the description of SPINK1 intronic sequence variants followed HGVS recommendations [33]. It should however be noted that whereas the SPINK1 gene comprises five exons, in accordance with mRNA reference sequence accession NM_003122.3, the gene expressed in the exocrine pancreas comprises only four exons [55, 56]. It is the latter gene structure that is used by both pancreatitis genetics researchers [6, 9, 13, 40, 57] and Ensembl (refer to ENSG00000164266) [58]. The traditional IVS (InterVening Sequence; i.e., an intron) nomenclature for describing SPINK1 intronic variants corresponds to the four-exon gene structure. In this study, in accordance with convention, we used the four-exon gene structure of pathophysiological relevance to define SPINK1 intron numbers. Thus, in the current work, the SPINK1 gene is regarded as harboring three introns. Since the first exon in accordance with mRNA reference sequence accession NM_003122.3 is non-coding, the variant nomenclature following HGVS recommendations is unaffected.
Allele frequency reference
Data in genomAD [30] were used as a reference for population frequency filtering.
In silico prediction of impact on splice site selection
In silico prediction of the impact of specific variants on splice site choice was performed using Alamut® Visual v.2.11 rev. 0 that included four prediction algorithms viz. SpliceSiteFinder-like, MaxEntScan, NNSPLICE and GeneSplicer under default conditions [29].
Cell culture-based full-length gene assay
The wild-type expression vector harboring the full-length genomic SPINK1 gene has been previously described [59]. It was used to generate the full-length expression constructs harboring respectively the selected SPINK1 intronic variants by means of the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). In vitro mutagenesis, HEK293T cell culture, transfection, RT-PCR, and real-time quantitative RT-PCR analyses were performed essentially as previously described [12, 16].
Abbreviations
- genomAD:
-
The Genome Aggregation Database
- GTEx:
-
Genotype-tissue expression
- MAF:
-
Minor allele frequency
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- TPM:
-
Transcripts per kilobase million
References
Whitcomb DC, Frulloni L, Garg P, Greer JB, Schneider A, Yadav D, et al. Chronic pancreatitis: an international draft consensus proposal for a new mechanistic definition. Pancreatology. 2016;16:218–24.
Chen JM, Férec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2009;10:63–87.
Whitcomb DC. Genetic aspects of pancreatitis. Annu Rev Med. 2010;61:413–24.
Hegyi E, Sahin-Tóth M. Genetic risk in chronic pancreatitis: the trypsin-dependent pathway. Dig Dis Sci. 2017;62:1692–701.
Kleeff J, Whitcomb DC, Shimosegawa T, Esposito I, Lerch MM, Gress T, et al. Chronic pancreatitis. Nat Rev Dis Primers. 2017;3:17060.
Sahin-Tóth M, Nemeth B. Genetic risk factors in chronic pancreatitis http://www.pancreasgenetics.org/index.php (accessed 31 Oct 2018).
Boulling A, Witt H, Chandak GR, Masson E, Paliwal S, Bhaskar S, et al. Assessing the pathological relevance of SPINK1 promoter variants. Eur J Hum Genet. 2011;19:1066–73.
Derikx MH, Geisz A, Kereszturi E, Sahin-Tóth M. Functional significance of SPINK1 promoter variants in chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2015;308:G779–84.
Boulling A, Masson E, Zou WB, Paliwal S, Wu H, Issarapu P, et al. Identification of a functional enhancer variant within the chronic pancreatitis-associated SPINK1 c.101A>G (p.Asn34Ser)-containing haplotype. Hum Mutat. 2017;38:1014–24.
Boulling A, Le Maréchal C, Trouvé P, Raguénès O, Chen JM, Férec C. Functional analysis of pancreatitis-associated missense mutations in the pancreatic secretory trypsin inhibitor (SPINK1) gene. Eur J Hum Genet. 2007;15:936–42.
Király O, Wartmann T, Sahin-Tóth M. Missense mutations in pancreatic secretory trypsin inhibitor (SPINK1) cause intracellular retention and degradation. Gut. 2007;56:1433–8.
Zou WB, Boulling A, Masson E, Cooper DN, Liao Z, Li ZS, et al. Clarifying the clinical relevance of SPINK1 intronic variants in chronic pancreatitis. Gut. 2016;65:884–6.
Zou WB, Masson E, Boulling A, Cooper DN, Li ZS, Liao Z, et al. Digging deeper into the intronic sequences of the SPINK1 gene. Gut. 2016;65:1055–6.
Leman R, Gaildrat P, Gac GL, Ka C, Fichou Y, Audrezet MP, et al. Novel diagnostic tool for prediction of variant spliceogenicity derived from a set of 395 combined in silico/in vitro studies: an international collaborative effort. Nucleic Acids Res. 2018;46:7913–23.
Moles-Fernandez A, Duran-Lozano L, Montalban G, Bonache S, Lopez-Perolio I, Menendez M, et al. Computational tools for splicing defect prediction in breast/ovarian cancer genes: how efficient are they at predicting RNA alterations? Front Genet. 2018;9:366.
Zou WB, Wu H, Boulling A, Cooper DN, Li ZS, Liao Z, et al. In silico prioritization and further functional characterization of SPINK1 intronic variants. Hum Genomics. 2017;11:7.
Fu XD, Ares M Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15:689–701.
Chen Y, Huang L, Jiao X, Riazuddin S, Riazuddin SA, Fielding HJ. A novel LRAT mutation affecting splicing in a family with early onset retinitis pigmentosa. Hum Genomics. 2018;12:35.
Palermo JJ, Lin TK, Hornung L, Valencia CA, Mathur A, Jackson K, et al. Genophenotypic analysis of pediatric patients with acute recurrent and chronic pancreatitis. Pancreas. 2016;45:1347–52.
Cho SM, Shin S, Lee KA. PRSS1, SPINK1, CFTR, and CTRC pathogenic variants in Korean patients with idiopathic pancreatitis. Ann Lab Med. 2016;36:555–60.
Kereszturi E, Sahin-Toth M. Pancreatic cancer cell lines heterozygous for the SPINK1 p.N34S haplotype exhibit diminished expression of the variant allele. Pancreas. 2017;46:e54–e5.
Zou WB, Tang XY, Zhou DZ, Qian YY, Hu LH, Yu FF, et al. SPINK1, PRSS1, CTRC and CFTR genotypes influence disease onset and clinical outcomes in chronic pancreatitis. Clin Transl Gastroenterol. 2018;9:204.
Cooper DN. Functional intronic polymorphisms: buried treasure awaiting discovery within our genes. Hum Genomics. 2010;4:284–8.
The Genotype-Tissue Expression (GTEx) Dataset. https://gtexportal.org/home/. Accessed 05 January 2019.
Lin JH, Tang XY, Boulling A, Zou WB, Masson E, Fichou Y, et al. First estimation of the scale of canonical 5′ splice site GT>GC mutations generating wild-type transcripts and their medical genetic implications. bioRxiv 479493; doi: https://doiorg/10.101101/479493. Posted November 27, 2018.
Sangermano R, Khan M, Cornelis SS, Richelle V, Albert S, Garanto A, et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018;28:100–10.
Kume K, Masamune A, Kikuta K, Shimosegawa T. [−215G>A; IVS3+2T>C] mutation in the SPINK1 gene causes exon 3 skipping and loss of the trypsin binding site. Gut. 2006;55:1214.
Raraigh KS, Han ST, Davis E, Evans TA, Pellicore MJ, McCague AF, et al. Functional assays are essential for interpretation of missense variants associated with variable expressivity. Am J Hum Genet. 2018;102:1062–77.
Alamut. https://www.interactive-biosoftware.com/. Accessed 31 Oct 2018.
The Genome Aggregation Database (gnomAD). http://gnomad.broadinstitute.org/. Accessed 31 Oct 2018.
Chen JM, Férec C, Cooper DN. Closely spaced multiple mutations as potential signatures of transient hypermutability in human genes. Hum Mutat. 2009;30:1435–48.
Chen JM, Férec C, Cooper DN. Patterns and mutational signatures of tandem base substitutions causing human inherited disease. Hum Mutat. 2013;34:1119–30.
den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–9.
Fjeld K, Weiss FU, Lasher D, Rosendahl J, Chen JM, Johansson BB, et al. A recombined allele of the lipase gene CEL and its pseudogene CELP confers susceptibility to chronic pancreatitis. Nat Genet. 2015;47:518–22.
Zou WB, Boulling A, Masamune A, Issarapu P, Masson E, Wu H, et al. No association between CEL-HYB hybrid allele and chronic pancreatitis in Asian populations. Gastroenterology. 2016;150:1558–60.e5.
Witt H, Beer S, Rosendahl J, Chen JM, Chandak GR, Masamune A, et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet. 2013;45:1216–20.
Wu H, Zhou DZ, Berki D, Geisz A, Zou WB, Sun XT, et al. No significant enrichment of rare functionally defective CPA1 variants in a large Chinese idiopathic chronic pancreatitis cohort. Hum Mutat. 2017;38:959–63.
Rosendahl J, Kirsten H, Hegyi E, Kovacs P, Weiss FU, Laumen H, et al. Genome-wide association study identifies inversion in the CTRB1-CTRB2 locus to modify risk for alcoholic and non-alcoholic chronic pancreatitis. Gut. 2018;67:1855–63.
Tang XY, Zou WB, Masson E, Hu LH, Ferec C, Chen JM, et al. The CTRB1-CTRB2 risk allele for chronic pancreatitis discovered in European populations does not contribute to disease risk variation in the Chinese population due to near allele fixation. Gut. 2018;67:1368–9.
Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213–6.
Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141–5.
Rosendahl J, Witt H, Szmola R, Bhatia E, Ozsvari B, Landt O, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78–82.
Masson E, Chen JM, Scotet V, Le Maréchal C, Férec C. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum Genet. 2008;123:83–91.
Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998;339:653–8.
Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339:645–52.
Masson E, Chen JM, Audrézet MP, Cooper DN, Férec C. A conservative assessment of the major genetic causes of idiopathic chronic pancreatitis: data from a comprehensive analysis of PRSS1, SPINK1, CTRC and CFTR genes in 253 young French patients. PLoS One. 2013;8:e73522.
Kobayashi Y, Yang S, Nykamp K, Garcia J, Lincoln SE, Topper SE. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017;9:13.
Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17:19–32.
Shiraishi Y, Kataoka K, Chiba K, Okada A, Kogure Y, Tanaka H, et al. A comprehensive characterization of cis-acting splicing-associated variants in human cancer. Genome Res. 2018;28:1111–25.
Liquori A, Vache C, Baux D, Blanchet C, Hamel C, Malcolm S, et al. Whole USH2A gene sequencing identifies several new deep intronic mutations. Hum Mutat. 2016;37:184–93.
Rendu J, Montjean R, Coutton C, Suri M, Chicanne G, Petiot A, et al. Functional characterization and rescue of a deep intronic mutation in OCRL gene responsible for Lowe syndrome. Hum Mutat. 2017;38:152–9.
Bergougnoux A, Deletang K, Pommier A, Varilh J, Houriez F, Altieri JP, et al. Functional characterization and phenotypic spectrum of three recurrent disease-causing deep intronic variants of the CFTR gene. J Cyst Fibros; doi: 101016/jjcf201810012 [Epub ahead of print]. 2018.
Igreja S, Clarke LA, Botelho HM, Marques L, Amaral MD. Correction of a cystic fibrosis splicing mutation by antisense oligonucleotides. Hum Mutat. 2016;37:209–15.
Venet T, Masson E, Talbotec C, Billiemaz K, Touraine R, Gay C, et al. Severe infantile isolated exocrine pancreatic insufficiency caused by the complete functional loss of the SPINK1 gene. Hum Mutat. 2017;38:1660–5.
Yamamoto T, Nakamura Y, Nishide J, Emi M, Ogawa M, Mori T, et al. Molecular cloning and nucleotide sequence of human pancreatic secretory trypsin inhibitor (PSTI) cDNA. Biochem Biophys Res Commun. 1985;132:605–12.
Horii A, Kobayashi T, Tomita N, Yamamoto T, Fukushige S, Murotsu T, et al. Primary structure of human pancreatic secretory trypsin inhibitor (PSTI) gene. Biochem Biophys Res Commun. 1987;149:635–41.
Kereszturi E, Kiraly O, Sahin-Toth M. Minigene analysis of intronic variants in common SPINK1 haplotypes associated with chronic pancreatitis. Gut. 2009;58:545–9.
Ensembl. https://www.ensembl.org/. Accessed 31 Oct 2018.
Boulling A, JM Chen, I Callebaut, C Férec. Is the SPINK1 p.Asn34Ser missense mutation per se the true culprit within its associated haplotype? WebmedCentral GENETICS. 2012;3:WMC003084 (Available at: https://www.webmedcentral.com/article_view/3084). Accessed 14 Nov 2018.
Acknowledgements
Not applicable.
Funding
J.H.L., a joint PhD student between the Changhai Hospital and INSERM U1078, was in receipt of a 20-month scholarship from the China Scholarship Council (No. 201706580018). Support for this study came from the National Natural Science Foundation of China (81470884 and 81770636 (to Z.L.), 81873588 (to Z.S.L) and 81700565 (to W.B.Z.)), the Shanghai Pujiang Program (Grant No. 17PJD044 (to W.B.Z.)), the Chang Jiang Scholars Program of Ministry of Education (Q2015190 (to Z.L.)), and the Scientific Innovation Program of Shanghai Municipal Education Committee (to Z.L.), China; the Institut National de la Santé et de la Recherche Médicale (INSERM), the Association des Pancréatites Chroniques Héréditaires, and the Association de Transfusion Sanguine et de Biogénétique Gaetan Saleun, France. The funding bodies had no role in the study design, the collection, analysis, and interpretation of data, or the writing of the article and the decision to submit it for publication.
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its Supplementary information files).
Author information
Authors and Affiliations
Contributions
X.Y.T., J.H.L., W.B.Z., E.M., A.B., and S.J.D. performed the functional assay and/or mutational analysis. J.M.C., Z.L., Z.S.L., and C.F. designed the study. JMC drafted the paper. All authors analyzed the data, contributed to revision of the manuscript, and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethics approval for this study was obtained from the Ethics Committees of Changhai Hospital, Shanghai, China and the Université de Brest, Brest, France. Written informed consent was obtained from all participating subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Alamut-predicted impact on splice site selection of the three recently reported SPINK1 spice site variants. Figure S2. Presence of the c.88-1G > A (chr5:g.147207692C > T) in cis with a closely spaced variant, c.88-7 T > A (chr5:g.147207698A > T), in a Chinese patient with chronic pancreatitis. Figure S3. Alamut-predicted impact on splice site selection of the proximal c.88-7 T > A variant. Figure S4. Alamut-predicted impact on splice site selection of the 10 deep SPINK1 intronic variants with a minor allele frequency of < 5% in the East Asian population. Figure S5. Alamut-predicted impact on splice site selection of the other four proximal SPINK1 intronic variants found in the French pancreatitis patients. Figure S6. Alamut-predicted impact on splice site selection of the six deep SPINK1 intronic variants with a minor allele frequency of ≥5% in the East Asian population. (PDF 2391 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tang, XY., Lin, JH., Zou, WB. et al. Toward a clinical diagnostic pipeline for SPINK1 intronic variants. Hum Genomics 13, 8 (2019). https://doi.org/10.1186/s40246-019-0193-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-019-0193-7