
Abstract
Fanconi anemia (FA) is a recessively inherited disease manifesting developmental abnormalities, bone marrow failure, and increased risk of malignancies. Whereas FA has been studied for nearly 90 years, only in the last 20 years have increasing numbers of genes been implicated in the pathogenesis associated with this genetic disease. To date, 19 genes have been identified that encode Fanconi anemia complementation group proteins, all of which are named or aliased, using the root symbol “FANC.” Fanconi anemia subtype (FANC) proteins function in a common DNA repair pathway called “the FA pathway,” which is essential for maintaining genomic integrity. The various FANC mutant proteins contribute to distinct steps associated with FA pathogenesis. Herein, we provide a review update of the 19 human FANC and their mouse orthologs, an evolutionary perspective on the FANC genes, and the functional significance of the FA DNA repair pathway in association with clinical disorders. This is an example of a set of genes––known to exist in vertebrates, invertebrates, plants, and yeast––that are grouped together on the basis of shared biochemical and physiological functions, rather than evolutionary phylogeny, and have been named on this basis by the HUGO Gene Nomenclature Committee (HGNC).
Similar content being viewed by others

Introduction
Fanconi anemia (FA) is an autosomal recessive disorder having an incidence of ~1/200,000–400,000 in most populations; the incidence is higher in Ashkenazi Jews (~1/30,000) and Afrikaners (~1/22,000) [1–4]. Most FA patients exhibit developmental abnormalities, developing early bone marrow failure and acute myelogenous leukemia (AML). Later, these patients display a higher risk of developing carcinomas of the head, neck, and anogenital region [5].
In recent decades, 19 human genes have been implicated in the causation of FA. These genes code for a group of proteins, viz. the Fanconi anemia subtype (FANC) proteins, which function cooperatively in a DNA damage recognition - and - repair pathway [6]. The FA pathway plays a crucial role in maintaining hematological homeostasis––particularly blood cell development and differentiation. FA is a congenital disease resulting from a somatic mutation in both alleles of the specific FANC gene; a notable exception is FANCB in which the mutation occurs on the X chromosome [7]. The primary causal factor in FA is generally thought to involve chromosomal instability in hematopoietic stem cells, a result of defective DNA repair caused by mutant FANC proteins [8, 9]. However, precise molecular mechanisms underlying the roles of specific FANC proteins (as well as their interactions with non-FANC proteins) in the FA pathway or other DNA damage repair pathways remain to be defined.
Genetics of FA: human FANC genes
FA was first reported in 1927 by Swiss pediatrician Guido Fanconi. After finding three siblings who suffered from complex physical defects relating to aplastic anemia, Dr. Fanconi described the intrinsic nature of the abnormalities as “panmyelopathy” and proposed that multiple genes were likely causing the underlying complexity of FA symptoms [10]. It was later recognized that a high degree of underlying genetic heterogeneity existed in this disease [11]. Sixty-five years after the initial FA case was reported, the first FANC gene (FANCC) was identified, following development of a complementation-cloning method [12]. This approach was subsequently used in identification of an additional four FANC genes [13–16]. Through the use of complementation cloning, positional cloning, protein association, and candidate-gene and whole-genome sequencing, 19 FANC genes have now been identified as being involved in the etiology of FA (Table 1). The FANC genes are phylogenetically unrelated and have been grouped together and named or aliased with the FANC root symbol, based on association of mutations in the encoded proteins having a FA-like disease phenotype in combination with cellular hypersensitivity to DNA cross-linking agents.
Information about most published FANC gene mutations is available on the public Fanconi Anemia Mutation Database (http://www.rockefeller.edu/fanconi/). Genetic studies have revealed that mutations in the FANCA, FANCC, and FANCG genes are most common and account for ~85 % of FA cases [5, 17]. The FANCA gene was the second FANC gene identified [13]. FANCA mutations account for nearly ~65 % of FA cases [18]. About 200 different mutated FANCA alleles, comprising almost all the known mutation types, have been reported to date. Large intragenic deletions appear to be the major form of mutations [18].
FANCC mutations account for ~14 % of FA cases [5]. Among all known mutants, 322delG and IVS4+4A>T occurring in exon 1 and intron 4, respectively, are the most commonly observed FANCC mutations [19, 20]. Deletion of a single G at base 322 (322delG) produces a truncated protein of 44 amino acids. The IVS4+4A>T mutation results in either deletion of the entire exon 4 or a 40-bp deletion leading to a frame-shift.
FANCG was found to be identical to the X-ray repair cross-complementing protein 9 (XRCC9) gene; the gene is named as FANCG, with XRCC9 as an alias for the DNA repair protein. FANCG mutations account for ~10 % of FA cases [5] and have been implicated as founder mutations in different populations. For instance, a deletion mutation at c.637_643delTACCGCC was found to be associated with 82 % of FA cases in black populations of Southern Africa [21]. Screening of 45 FA families in Japan showed that nine of the families carried a splice mutation of IVS3+1G>C, with three of these nine families also carrying a 1066C>T mutation. Haplotype analysis revealed IVS3+1G>C and 1066C>T to be associated with Japanese and Korean ethnicities [22], respectively.
Mutations in the FANCB, FANCD1, FANCD2, FANCE, and FANCF genes, combined, account for ~13 % of reported FA cases [5]. The FANCB gene is the only FANC gene not to be autosomal, but rather X-linked. Point mutations, small insertions, and large deletions in the FANCB gene have been reported. For instance, a frame-shift mutation in exon 8 and a 3314-bp deletion in exon 1 (that includes some of the promoter region of FANCB) were reported in cell lines derived from FA patients [7]. Most FANCB mutations result in truncation of the encoded protein [23].
FANCD1 is identical to the breast cancer susceptibility gene BRCA2; because the latter gene symbol was so extremely well-established, this gene is officially named BRCA2 by the HGNC but has the alias of FANCD1. Studies reveal that FA-D1 patients have biallelic mutations in the BRCA2 gene and express a truncated protein [24]. Heterozygotes in FA families (e.g., parents of FA patients) display increased risk of early-onset breast and ovarian cancers [25]. These findings connect FA and breast cancer, which share common mechanisms of defective DNA repair.
FANCD2 encodes a protein that plays a central role in the FA pathway of DNA repair. FANCD2 mutations involving large deletions and/or single-base changes have been reported [26, 27]. In addition, specific FANCD2 mutations (for example, c.458T>C and c.2715+1G>A) have been associated with T cell acute lymphoblastic leukemia (T-ALL) and testicular seminoma [26, 27].
Mutations in the FANCE and FANCF genes account for 5–8 % of reported FA cases [5, 17]. Multiple types of mutation have been reported for FANCE, including a 355C>T transition in exon 2 in the EUFA410 FA cell line and the IVS5-8G>A splice mutation in EUFA622 cells [16, 28]. Disease-associated mutations have been reported throughout the single coding exon of the FANCF gene. The most commonly seen FANCF mutations are short deletions, resulting in frame-shifts and premature termination of the protein [29].
Mutations in the remaining FA genes, FANCI, BRIP1 (FANCJ), FANCL, FANCM, PALB2 (FANCN), RAD51C (FANCO), SLX4 (FANCP), ERCC4 (FANCQ), BRCA1 (FANCS), and the recently reported RAD51(FANCR) and UBE2T (FANCT), contribute to <5 % of FA cases, combined [5, 17]. Documented mutations in these eleven genes––in FA patients and/or FA subtype cell lines––include point mutations, nucleotide insertions, splice-site mutations, and mutations resulting in protein truncation. For example, biallelic mutations [30] and a 30-residue C-terminal protein truncation [31] were reported for FANCI.
BRIP1 (BRCA1-interacting protein C-terminal helicase 1) is the official gene symbol for FANCJ. Amino-acid substitutions in BRIP1/FANCJ, such as R251C, Q255H, and A349P, were found to be disease-causing [32, 33].
A dinucleotide insertion (c.755-756insAT) in FANCL [34], c.5569G>A transition in FANCM [35], and 3549C>G transition and 3549C>A transversion mutations in PALB2/FANCN [36] were reported in FA families. PALB2 (partner and localizer of BRCA2) is the official gene symbol for FANCN. However, it should be noted that Lim et al. found individuals in the Finnish population who were homozygous for FANCM loss-of-function mutations but appeared to have no reported FA phenotype [37], casting doubt over the pathogenicity of this gene in FA.
RAD51C (RAD51 paralog C) is the official gene symbol for the FANCO gene. Meindl et al. reported 14 RAD51C/FANCO mutations––which included single-base-pair insertions, splice-site mutations, and sequence alterations [38]. SLX4 (SLX4 structure-specific endonuclease subunit) and ERCC4 (excision-repair cross-complementation group 4) are the official gene symbols for FANCP and FANCQ, respectively. Biallelic mutations in the SLX4/FANCP and ERCC4/FANCQ genes have been documented to cause FA subtypes [39–41].
BRCA1 (breast cancer 1, early onset) has recently been identified as the 17th FANC gene and assigned the FANCS synonym [42]. Despite knowing that the BRCA1 protein cooperates in the FA pathway through forming a complex with several FANC proteins at DNA repair loci [43], BRCA1 was previously precluded from being assigned as a FA gene due to the lack of reported biallelic BRCA1 mutations in patients. However, recently, two cases of individuals harboring biallelic deleterious BRCA1 mutations were reported [42, 44]. Detailed phenotypic and cellular characterization of one patient provided lines of evidence supporting the hypothesis that biallelic BRCA1 (FANCS) mutations cause a new Fanconi anemia subtype associated with increased breast and ovarian cancer susceptibility [42].
Most recently, researchers have identified two new genes, mutations of which cause FA-like symptoms. The RAD51 gene encodes a key recombinase essential for homologous recombination of DNA during double strand break repair [45]. RAD51 is a mammalian homologue of the bacterial DNA repair protein RecA. A novel heterozygous mutation (T131P) in RAD51 was identified in a FA-like patient [46]. Expression of this dominant-negative mutant RAD51 in the patient’s cell line disrupted interstrand cross-link (ICL) repair but spared homologous recombination [46]. The authors proposed that RAD51 plays an ICL-repair-specific function through protection of nascent DNA strands from excessive processing at the ICL sites; this function may be regulated by BRCA2/FANCD1 [46]. The UBE2T gene encodes an ubiquitin-conjugating enzyme E2T that was already known to act in the FA pathway [47, 48]. Two unrelated individuals were found with biallelic UBE2T missense mutations that rendered the UBE2T protein unable to interact with FANCL [48]. The RAD51 and UBE2T genes have now been given the aliases FANCR and FANCT, respectively.
Evolution of FANC genes
It was originally thought that the FA DNA repair pathway was restricted to vertebrates. However, counterparts of several FA proteins have been discovered in lower eukaryotes, including sea squirt (C. intestinalis) [49], fly [50-52], worm [53-55], and yeast [56, 57], as well as in plants [58]. Nevertheless, it appears that the majority of lower eukaryotes possess a simplified FA pathway (Fig. 1) consisting of FANCL, FANCM, UBE2T, FANCD2, FANCI, BRCA2, BRIP1, RAD51C, SLX4, ERCC4, and RAD51. These FA proteins represent two components of the FA core complex, the D2/I complex, and most of the FA downstream effector proteins (vide infra).

Overview of FA pathway genes identified in eukaryotic lineages. Representative species include mammals (Homo sapiens, Mus musculus, and Gallus gallus), amphibian (African clawed toad, Xenopus laevis), fish (zebrafish, Danio rerio), sea squirt (Ciona intestinalis), insect (Drosophila melanogaster), worm (Caenorhabditis elegans), yeast (Saccharomyces cerevisiae), and plant (Arabidopsis thaliana). FANC genes are grouped into three classes. Group I includes nine genes that encode proteins that form the FA core complex; group II encodes FANCD2 and FANCI that form the D2/I complex; group III comprises eight genes that encode FA effector proteins that function downstream of D2/I complex. Lower eukaryotes tend to be missing orthologues of the FA core complex genes. A = FANCA, B = FANCB, C = FANCC, D2 = FANCD2, E = FANCE, F = FANCF, G = FANCG, I = FANCI, L = FANCL, M = FANCM, D1 = BRCA2/FANCD1, J = BRIP1/FANCJ, N = PALB2/FANCN, O = RAD51C/FANCO, P = SLX4/FANCP, Q = ERCC4/FANCQ/XPF, R = RAD51/FANCR, S = BRCA1/FANCS, T = UBE2T/FANCT. If we extend this gene family update to include prokaryotes, it might be noted that, whereas no orthologs of any of the 19 eukaryotic FANC genes exist in prokaryote genomes, RAD51 (as a nineteenth FANC member in living organisms) qualifies as a homologue of bacterial RecA
What appear to have evolved later in the ancestral animal kingdom are most of the FA core complex proteins (Fig. 1). In addition to the 19 human FANC genes, two pseudogenes of the FANCD2 gene have been annotated in the human genome, FANCD2P1 (gene ID 100421239) and FANCD2P2 (gene ID 101929530).
Comparative genomic analysis revealed genes of the FA pathway to be conserved among vertebrate genomes [49, 59]. A single ortholog of each of the 19 human FANC genes has been identified in other vertebrates––including rodents, chicken, Xenopus, and zebrafish (Fig. 1). However, the zebrafish genome has no BRCA1 orthologue, suggesting that the entire vertebrate FA gene network had not been completely formed after the land-animal/sea-animal split.
In the mouse genome, 19 Fanc genes have been mapped to 13 chromosomes (Table 2). Mouse FANC proteins share 50–85 % sequence identity with their corresponding human orthologs. Transgenic knockout mouse lines have been generated for many Fanc genes [60-68]. In general, cells derived from Fanc(−/−) knockout mice recapitulate the phenotypes of human FA patient cells, whereas these mice only partially reproduce the clinical features of FA patients.
FANC complementation group proteins that cause the FA disorder
FA is characterized primarily by progressive bone marrow dysfunction and sensitivity to DNA cross-linking agents [5, 17]. This congenital disease results from loss-of-function of any of the 19 FANC genes, thereby revealing the essential roles of FANC proteins in maintaining chromosomal stability of hematopoietic stem cells. As noted above, more than 95 % of FA cases are attributed to mutations in known FANC genes [5]. In a few FA cases, genetic contributions remain unclassified. The unique clinical phenotype associated with FANC gene mutations implies that proteins encoded by these genes function in a common cellular pathway. This pathway, known as the FA/BRCA DNA repair pathway, functions pivotally in preserving genomic homeostasis in response to specific types of DNA damage [6].
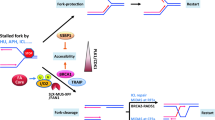
In a proposed model of the FA/BRCA DNA repair pathway (Fig. 2) [5, 17], eight FANC proteins––FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM––plus three FA-associated proteins, FAAP20, FAAP24, and FAAP100, form the nuclear FA core complex. Fanconi anemia core complex associated proteins (FAAP) are functionally equivalent to FANC proteins, except that, thus far, no FA patients have been found whose disease phenotype could be ascribed to mutations in a FAAP-encoding gene. The FA core complex functions to receive upstream signals in response to DNA damage, through phosphorylation of multiple FA core units. The activated FA core complex binds the enzyme UBE2T (FANCT) via the putative plant homeodomain (PHD) zinc finger of the FANCL subunit, and then mono-ubiquitinates the FANCD2/I complex. This then translocates to DNA damage sites and recruits eight downstream FA effector proteins, i.e., BRCA1, BRCA2, BRIP1, PALB2, RAD51C, SLX4, ERCC4, RAD51, plus other DNA repair molecules, to DNA damage sites.

FA/BRCA pathway and crosstalk between FA and other DNA repair pathways. In response to upstream DNA damage signaling (such as phosphorylation by ATR/ATM), in the FA/BRCA pathway [1], the FA core complex forms––comprising FANCA (A), FANCB (B), FANCC (C), FANCE (E), FANCF (F), FANCG (G), FANCM (M), and FANCL (L) proteins, plus FAAP20, FAAP24, and FAAP100 (FAAP). This core complex binds UBE2T(T) via FANCL, which then activates FANCD2/I dimers through mono-ubiquitination. The activated FANCD2/I (D2/I) complex then translocates to DNA - damage sites and recruits downstream FA effector proteins––including BRCA1 (S), BRCA2 (D1), RAD51 (R), BRIP1 (J), PALB2 (N), RAD51C (O), SLX4 (P), and ERCC4 (Q), plus other DNA repair molecules (such as FAN1) to the lesion site to repair damage. In the FANCM/BS pathway [2], the FA core complex binds to the BS complex by way of interactions between FANCM-RMI1 and TopoIIIα of the BS complex and translocates to the lesion site. In the FANCD2/ATM pathway [3], in response to ionizing radiation or ICL-inducing agents, FANCD2 is phosphorylated by ATM and co-localizes with the NMR complex to repair DNA damage or cause S-phase arrest. ICL interstrand cross-links, ATR ataxia-telangiectasia-Ser/Thr-protein kinase, ATM ataxia telangiectasia mutated kinase, FAAP FA-associated proteins, FAN1 FA-associated nuclease-1, BS Bloom’s syndrome protein, RMI1/2 RecQ-mediated genomic instability protein 1/2, TopoIIIα topoisomerase IIIα, NMR the NBS1/MRE11/RAD50 complex
Distinct roles of FA proteins in the FA core complex and FANCD2/I complex have been suggested. FANCE contributes to core complex integrity––by promoting nuclear accumulation of FANCC protein and FANCA—FANCC complex formation and directly targeting FANCD2 for ubiquitination [69]. Phe-522, at the highly conserved carboxyl terminus of FANCE, was found to be a critical residue for mediating mono-ubiquitination of the FANCD2/I complex [70]. FANCL is an E3 ubiquitin ligase containing three WD40 repeats and a PHD zinc finger motif. These account for protein-protein interactions and recruitment of UBE2T (FANCT), respectively [71], both of which function importantly in FANCD2 mono-ubiquitination. FANCM in the FA core complex was reported to possess endonuclease and DNA helicase domains and is believed to function in translocation of the FA core complex along DNA [51]. FANCD2 mono-ubiquitination is essential for FA pathway-mediated DNA repair. However, the specific role of FANCD2 in the repair process remains unknown. A higher frequency of telomere dysfunction-induced foci (TIFs) and telomere sister-chromatid exchanges (T-SCE) was observed in primary cells derived from FA patients carrying a mutated FANCD2 gene [72], suggesting its involvement in telomere regulation.
Of the 19 FANC proteins, some have been previously identified as part of different DNA damage repair pathways that may interact with the FA pathway [73, 74]. For example, FANCA has been reported to interact with DNA repair-associated proteins, such as the endonuclease ERCC4 and the ATP-dependent helicase SMARCA4 [75, 76]. In addition, FANCA mutant cells (derived from FA patients) exhibit defective mitochondrial respiration and impaired ATP production [77]. In human embryonic kidney (HEK 293T) cells, FANCA was found to influence centrosome integrity by way of its capacity to interact with NIMA-related kinase 2 (NEK2) [78].
FA pathway combats genomic instability
One of the hallmarks of FA is hypersensitivity of cells from FA patients to the clastogenic and cytostatic effects of DNA cross-linking agents such as diepoxybutane (DEB) and mitomycin C (MMC) [5, 17]. In particular, DNA ICLs are highly cytotoxic and difficult to repair because these affect both strands of the DNA helix [79]. The FA/BRCA DNA damage-response pathway is a complex network that functions to remove ICLs through the coordinated actions of FANC proteins––plus other non-FANC proteins such as FAAPs and FANCD2/I-associated nuclease-1 (FAN1) [5, 17].
From an evolutionary point of view, the complete FA pathway is conserved in higher eukaryotes as an efficient pathway to manage ICLs [59]. In this model (Fig. 2), the FA core complex, comprising eight FANC proteins and three FAAPs, forms in response to DNA - damage signaling. The assembled FA core complex binds the ubiquitin-conjugating enzyme UBE2T via the FANCL subunit, which then activates the FANCD2/I complex via mono-ubiquitination at the K561 residue of FANCD2. The ubiquitinated FANCD2/I complex translocates to sites of DNA damage following binding with BRCA1/2 and RAD51, which ultimately triggers DNA repair.
Extensive crosstalk occurs between the FA/BRCA pathway and other DNA repair pathways. FANCD2-deficient cells are hypersensitive to ionizing radiation [80]. In these cells, the FANCD2 mutant cannot be efficiently phosphorylated by the Ser/Thr kinase ATM, resulting in a defect in the ionizing radiation-inducible S-phase checkpoint. FANCD2 was found to initiate S-phase arrest, in response to ionizing radiation, by interacting with ATM-phosphorylated nibrin of the MRE11–RAD50-NBN/NBS1 (MRN) complex [81, 82]. In response to ICL agents such as mitomycin C (MMC), phosphorylated FANCD2 co-localizes with the MRN complex at the DNA damage sites. This FANCD2/ATM pathway operates independently of the FANCD2/BRCA pathway because FANCD2 mono-ubiquitination-mediated formation of nuclear foci is not affected in ATM-deficient (ATM(−/−)) cells. In addition, a non-ubiquitinated mutant K561R of FANCD2 has no effect on FANCD2 phosphorylation, following ionizing radiation. FANCM was reported to interact with the Bloom syndrome (BS) complex using its highly conserved protein-protein interaction motifs, MM1 and MM2 [83]. The BS complex binds to the FA core complex by means of MM1-mediated FANCM-FANCF interaction and MM2-mediated interaction between FANCM, RecQ-mediated genomic instability 1 (RMI1), and topoisomerase IIIα (TOP3A) of the BS complex. This FANCM-mediated FA-BS crosstalk is required for MMC resistance of cells.
FA and cancer
A high risk of carcinogenesis, particularly in hematopoietic and squamous cells, is another characteristic phenotype in FA patients, due to loss-of-function of FA proteins. FA-related malignancies vary between organs and cell types and in ages of onset and frequencies. Statistical analysis of 1300 identified FA cases revealed the highest frequency to be leukemia (9 %), followed by myelodysplastic syndrome (MDS) (7 %), solid tumors (5 %), and liver tumors (3 %) [84].
Using the International Fanconi Anemia Registry (IFAR) database to analyze 397 FA patients carrying FANCC mutations (specifically the IVS4 splice mutation in intron 4, 322delG or Q13X in exon 1 and R548X or L554P in exon 14) revealed that IVS4 and exon-14 mutations were highly correlated with severe congenital malformations and early onset of hematologic disorders at a median age of 2.7 and 2.1 years, respectively. Patients with exon-1 322delG or Q13X mutations exhibited mild congenital malformations and later onset of hematologic disease (median age 7.6 years) [85]. The IVS4 mutation caused distinct phenotypes in patients of Ashkenazi Jewish ancestry; interestingly, Japanese carriers display no significant clinical abnormality [86]. Patients carrying biallelic mutations in FANCD1 demonstrate an early-onset leukemia (median age 2.2 years), compared with 13.4 years in other FA patients [87]. Increased risk of esophageal squamous cell carcinoma (ESCC) was reported to be associated with mutations in FANCD2, FANCE, FANCL, and FANCA in patients from an Iranian population [88]. Screening of germline DNA in 421 pancreatic cancer cases showed that, besides homozygote-associated cancer formation, heterozygous FANCC mutations were associated with increased susceptibility to pancreatic cancer [89]. A BRIP1 mutation (c.2040_2041insTT), found at an allelic frequency of 0.41 % in an Icelandic cohort of 323,000 samples, was found to confer an elevated risk of ovarian cancer and is associated with an overall decrease of 3.6 years in lifespan––due to all cancers [90].
Summary
Many of the currently identified 19 FA proteins have already been characterized with respect to their relative roles in FA etiology and FA-associated neoplasm. However, our understanding of the full range of molecular actions of these important and intriguing proteins remains to be clarified. For example, the specific role of individual FA protein members and their interaction with non-FA pathways in response to DNA damage are not fully understood. Reported cases of certain heterozygous FANC mutations, resulting in altered FA proteins, that predispose some individuals to cancer might involve non-FA mechanisms in specific organ or cell types. The observation that a specific FANC mutation can induce a different phenotype, depending on each patient’s specific genetic background, suggests that non-FA factor(s), or unidentified FA proteins, might also be participating in cancer risk. Further investigation of FA proteins should provide valuable insights into understanding molecular mechanisms involved in maintaining genomic integrity.
Finally, it should be mentioned that almost all previous Human Genomics “Nomenclature Update” reviews have emphasized gene nomenclature based on evolutionary divergence from an original ancestral gene. In the case of the FANC gene group––or “family”––we show here that the HGNC-approved classification of these 19 genes is based on a common phenotype resulting from a shared biochemical or physiological functions, rather than evolutionary divergence and, as such, this is not a classical “gene family.”
References
Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006;25(43):5875–84.
Barankin B, Metelitsa AI, Schloss EH, Wasel NR. Skin disorders in Ashkenazi Jews: a review. Int J Dermatol. 2005;44(8):630–5.
Tipping AJ, Pearson T, Morgan NV, Gibson RA, Kuyt LP, Havenga C, et al. Molecular and genealogical evidence for a founder effect in Fanconi anemia families of the Afrikaner population of South Africa. Proc Natl Acad Sci U S A. 2001;98(10):5734–9.
Whitney MA, Jakobs P, Kaback M, Moses RE, Grompe M. The Ashkenazi Jewish Fanconi anemia mutation: incidence among patients and carrier frequency in the at-risk population. Hum Mutat. 1994;3(4):339–41.
Alter BP, and Kupfer G (1993–2015) Fanconi Anemia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. Available From http://www.ncbi.nlm.nih.gov/books/NBK1401/ Access date February 7, 2013.
Wang LC, Gautier J. The Fanconi anemia pathway and ICL repair: implications for cancer therapy. Crit Rev Biochem Mol Biol. 2010;45(5):424–39.
Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004;36(11):1219–24.
Yung SK, Tilgner K, Ledran MH, Habibollah S, Neganova I, Singhapol C, et al. Brief report: human pluripotent stem cell models of Fanconi anemia deficiency reveal an important role for Fanconi anemia proteins in cellular reprogramming and survival of hematopoietic progenitors. Stem Cells. 2013;31(5):1022–9.
Alter BP. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol. 2014;27(3–4):214–21.
Lobitz S, Velleuer E. Guido Fanconi (1892–1979): a jack of all trades. Nat Rev Cancer. 2006;6(11):893–8.
Rogers KJ, Fu W, Akey JM, Monnat Jr RJ. Global and disease-associated genetic variation in the human Fanconi anemia gene family. Hum Mol Genet. 2014;23(25):6815–25.
Strathdee CA, Gavish H, Shannon WR, Buchwald M. Cloning of cDNAs for Fanconi’s anaemia by functional complementation. Nature. 1992;356(6372):763–7.
Lo Ten Foe JR, Rooimans MA, Bosnoyan-Collins L, Alon N, Wijker M, Parker L, et al. Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nat Genet. 1996;14(3):320–3.
de Winter JP, Waisfisz Q, Rooimans MA, van Berkel CG, Bosnoyan-Collins L, Alon N, et al. The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nat Genet. 1998;20(3):281–3.
de Winter JP, Rooimans MA, van Der WL, van Berkel CG, Alon N, Bosnoyan-Collins L, et al. The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nat Genet. 2000;24(1):15–6.
de Winter JP, Leveille F, van Berkel CG, Rooimans MA, van Der WL, Steltenpool J, et al. Isolation of a cDNA representing the Fanconi anemia complementation group E gene. Am J Hum Genet. 2000;67(5):1306–8.
Schneider M, Chandler K, Tischkowitz M, Meyer S. Fanconi anaemia: genetics, molecular biology, and cancer––implications for clinical management in children and adults. Clin Genet. 2014;88(1):13–24.
Solomon PJ, Margaret P, Rajendran R, Ramalingam R, Menezes GA, Shirley AS, et al. A case report and literature review of Fanconi Anemia (FA) diagnosed by genetic testing. Ital J Pediatr. 2015;41:38.
Pang Q, Christianson TA, Keeble W, Diaz J, Faulkner GR, Reifsteck C, et al. The Fanconi anemia complementation group C gene product: structural evidence of multifunctionality. Blood. 2001;98(5):1392–401.
Kutler DI, Auerbach AD. Fanconi anemia in Ashkenazi Jews. Fam Cancer. 2004;3(3–4):241–8.
Yagasaki H, Oda T, Adachi D, Nakajima T, Nakahata T, Asano S, et al. Two common founder mutations of the Fanconi anemia group G gene FANCG/XRCC9 in the Japanese population. Hum Mutat. 2003;21(5):555.
Park J, Chung NG, Chae H, Kim M, Lee S, Kim Y, et al. FANCA and FANCG are the major Fanconi anemia genes in the Korean population. Clin Genet. 2013;84(3):271–5.
Moldovan GL, D’Andrea AD. How the Fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49.
Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297(5581):606–9.
Petrucelli N, Daly MB, Feldman GL. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet Med. 2010;12(5):245–59.
Smetsers S, Muter J, Bristow C, Patel L, Chandler K, Bonney D, et al. Heterozygote FANCD2 mutations associated with childhood T Cell ALL and testicular seminoma. Fam Cancer. 2012;11(4):661–5.
Flynn EK, Kamat A, Lach FP, Donovan FX, Kimble DC, Narisu N, et al. Comprehensive analysis of pathogenic deletion variants in Fanconi anemia genes. Hum Mutat. 2014;35(11):1342–53.
Waisfisz Q, Saar K, Morgan NV, Altay C, Leegwater PA, de Winter JP, et al. The Fanconi anemia group E gene, FANCE, maps to chromosome 6p. Am J Hum Genet. 1999;64(5):1400–5.
Gille JJ, Floor K, Kerkhoven L, Ameziane N, Joenje H, de Winter JP. Diagnosis of Fanconi anemia: mutation analysis by multiplex ligation-dependent probe amplification and PCR-based Sanger sequencing. Anemia. 2012;2012:603253.
Sims AE, Spiteri E, Sims III RJ, Arita AG, Lach FP, Landers T, et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol. 2007;14(6):564–7.
Colnaghi L, Jones MJ, Cotto-Rios XM, Schindler D, Hanenberg H, Huang TT. Patient-derived C-terminal mutation of FANCI causes protein mislocalization and reveals putative EDGE motif function in DNA repair. Blood. 2011;117(7):2247–56.
Wu Y, Sommers JA, Suhasini AN, Leonard T, Deakyne JS, Mazin AV, et al. Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood. 2010;116(19):3780–91.
Guo M, Vidhyasagar V, Ding H, Wu Y. Insight into the roles of helicase motif Ia by characterizing Fanconi anemia group J protein (FANCJ) patient mutations. J Biol Chem. 2014;289(15):10551–65.
Ameziane N, Sie D, Dentro S, Ariyurek Y, Kerkhoven L, Joenje H, et al. Diagnosis of Fanconi anemia: mutation analysis by next-generation sequencing. Anemia. 2012;2012:132856.
Garcia MJ, Fernandez V, Osorio A, Barroso A, Fernandez F, Urioste M, et al. Mutational analysis of FANCL, FANCM and the recently identified FANCI suggests that among the 13 known Fanconi anemia genes, only FANCD1/BRCA2 plays a major role in high-risk breast cancer predisposition. Carcinogenesis. 2009;30(11):1898–902.
Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39(2):162–4.
Lim ET, Wurtz P, Havulinna AS, Palta P, Tukiainen T, Rehnstrom K, et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10(7):e1004494.
Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42(5):410–4.
Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat Genet. 2011;43(2):142–6.
de Garibay GR, Diaz A, Gavina B, Romero A, Garre P, Vega A, et al. Low prevalence of SLX4 loss-of-function mutations in non-BRCA1/2 breast and/or ovarian cancer families. Eur J Hum Genet. 2013;21(8):883–6.
Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92(5):800–6.
Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, Majewski J, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5(2):135–42.
Stecklein SR, Jensen RA. Identifying and exploiting defects in the Fanconi anemia/BRCA pathway in oncology. Transl Res. 2012;160(3):178–97.
Domchek SM, Tang J, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013;3(4):399–405.
Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013;5(11):a012740.
Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, et al. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol Cell. 2015;59(3):478–90.
Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D’Andrea AD, et al. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell. 2006;23(4):589–96.
Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet. 2015;96(6):1001–7.
Titus TA, Selvig DR, Qin B, Wilson C, Starks AM, Roe BA, et al. The Fanconi anemia gene network is conserved from zebrafish to human. Gene. 2006;371(2):211–23.
Marek LR, Bale AE. Drosophila homologs of FANCD2 and FANCL function in DNA repair. DNA Repair (Amst). 2006;5(11):1317–26.
Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 2005;37(9):958–63.
Lo T, Pellegrini L, Venkitaraman AR, Blundell TL. Sequence fingerprints in BRCA2 and RAD51: implications for DNA repair and cancer. DNA Repair (Amst). 2003;2(9):1015–28.
Petalcorin MI, Sandall J, Wigley DB, Boulton SJ. CeBRC-2 stimulates D-loop formation by RAD-51 and promotes DNA single-strand annealing. J Mol Biol. 2006;361(2):231–42.
Collis SJ, Barber LJ, Ward JD, Martin JS, Boulton SJ. C. elegans FANCD2 responds to replication stress and functions in interstrand cross-link repair. DNA Repair (Amst). 2006;5(11):1398–406.
Youds JL, Barber LJ, Ward JD, Collis SJ, O’Neil NJ, Boulton SJ, et al. DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol Cell Biol. 2008;28(5):1470–9.
Daee DL, Myung K. Fanconi-like crosslink repair in yeast. Genome Integr. 2012;3(1):7.
Ward TA, Dudasova Z, Sarkar S, Bhide MR, Vlasakova D, Chovanec M, et al. Components of a Fanconi-like pathway control Pso2-independent DNA interstrand crosslink repair in yeast. PLoS Genet. 2012;8(8):e1002884.
Knoll A, Higgins JD, Seeliger K, Reha SJ, Dangel NJ, Bauknecht M, et al. The Fanconi anemia ortholog FANCM ensures ordered homologous recombination in both somatic and meiotic cells in Arabidopsis. Plant Cell. 2012;24(4):1448–64.
Zhang XY, Langenick J, Traynor D, Babu MM, Kay RR, Patel KJ. Xpf and not the Fanconi anaemia proteins or Rev3 accounts for the extreme resistance to cisplatin in Dictyostelium discoideum. PLoS Genet. 2009;5(9):e1000645.
Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, et al. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003;17(16):2021–35.
Whitney MA, Royle G, Low MJ, Kelly MA, Axthelm MK, Reifsteck C, et al. Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88(1):49–58.
Hakem R, de la Pompa JL, Mak TW. Developmental studies of Brca1 and Brca2 knock-out mice. J Mammary Gland Biol Neoplasia. 1998;3(4):431–45.
Cheng NC, van de Vrugt HJ, van der Valk MA, Oostra AB, Krimpenfort P, de VY, et al. Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum Mol Genet. 2000;9:1805–11.
Yang Y, Kuang Y, De Montes OR, Hays T, Moreau L, Lu N, et al. Targeted disruption of the murine Fanconi anemia gene, Fancg/Xrcc9. Blood. 2001;98(12):3435–40.
Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 2003;12(16):2063–76.
Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, et al. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum Mol Genet. 2009;18(18):3484–95.
Bouwman P, Drost R, Klijn C, Pieterse M, van der Gulden H, Song JY, et al. Loss of p53 partially rescues embryonic development of Palb2 knockout mice but does not foster haploinsufficiency of Palb2 in tumour suppression. J Pathol. 2011;224(1):10–21.
Bakker ST, van de Vrugt HJ, Visser JA, Delzenne-Goette E, van der Wal A, Berns MA, et al. Fancf-deficient mice are prone to develop ovarian tumours. J Pathol. 2012;226(1):28–39.
Taniguchi T, D’Andrea AD. The Fanconi anemia protein, FANCE, promotes the nuclear accumulation of FANCC. Blood. 2002;100(7):2457–62.
Polito D, Cukras S, Wang X, Spence P, Moreau L, D’Andrea AD, et al. The carboxyl terminus of FANCE recruits FANCD2 to the Fanconi Anemia (FA) E3 ligase complex to promote the FA DNA repair pathway. J Biol Chem. 2014;289(10):7003–10.
Gurtan AM, Stuckert P, D’Andrea AD. The WD40 repeats of FANCL are required for Fanconi anemia core complex assembly. J Biol Chem. 2006;281(16):10896–905.
Joksic I, Vujic D, Guc-Scekic M, Leskovac A, Petrovic S, Ojani M, et al. Dysfunctional telomeres in primary cells from Fanconi anemia FANCD2 patients. Genome Integr. 2012;3(1):6.
Garaycoechea JI, Patel KJ. Why does the bone marrow fail in Fanconi anemia? Blood. 2014;123(1):26–34.
Howard SM, Yanez DA, Stark JM. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015;11(1):e1004943.
Otsuki T, Furukawa Y, Ikeda K, Endo H, Yamashita T, Shinohara A, et al. Fanconi anemia protein, FANCA, associates with BRG1, a component of the human SWI/SNF complex. Hum Mol Genet. 2001;10(23):2651–60.
Sridharan D, Brown M, Lambert WC, McMahon LW, Lambert MW. Nonerythroid alpha II spectrin is required for recruitment of FANCA and XPF to nuclear foci induced by DNA interstrand cross-links. J Cell Sci. 2003;116(Pt 5):823–35.
Ravera S, Vaccaro D, Cuccarolo P, Columbaro M, Capanni C, Bartolucci M, et al. Mitochondrial respiratory chain Complex I defects in Fanconi anemia complementation group A. Biochimie. 2013;95(10):1828–37.
Kim S, Hwang SK, Lee M, Kwak H, Son K, Yang J, et al. Fanconi anemia complementation group A (FANCA) localizes to centrosomes and functions in the maintenance of centrosome integrity. Int J Biochem Cell Biol. 2013;45(9):1953–61.
Scharer OD. DNA interstrand crosslinks: natural and drug-induced DNA adducts that induce unique cellular responses. Chembiochem. 2005;6(1):27–32.
Castillo P, Bogliolo M, Surralles J. Coordinated action of the Fanconi anemia and ataxia telangiectasia pathways in response to oxidative damage. DNA Repair (Amst). 2011;10(5):518–25.
Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, et al. Convergence of theFanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109(4):459–72.
Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, et al. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol. 2002;4(12):913–20.
Deans AJ, West SC. FANCM connects the genome instability disorders Bloom’s syndrome and Fanconi anemia. Mol Cell. 2009;36(6):943–53.
Alter BP. Cancer in Fanconi anemia, 1927–2001. Cancer. 2003;97(2):425–40.
Gillio AP, Verlander PC, Batish SD, Giampietro PF, Auerbach AD. Phenotypic consequences of mutations in the Fanconi anemia FANCC gene: an International Fanconi Anemia Registry study. Blood. 1997;90(1):105–10.
Futaki M, Yamashita T, Yagasaki H, Toda T, Yabe M, Kato S, et al. The IVS4 + 4 A to T mutation of the Fanconi anemia gene FANCC is not associated with a severe phenotype in Japanese patients. Blood. 2000;95(4):1493–8.
Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, Satagopan J, et al. Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood. 2004;103(8):3226–9.
Akbari MR, Malekzadeh R, Lepage P, Roquis D, Sadjadi AR, Aghcheli K, et al. Mutations in Fanconi anemia genes and the risk of esophageal cancer. Hum Genet. 2011;129(5):573–82.
Couch FJ, Johnson MR, Rabe K, Boardman L, McWilliams R, de AM, et al. Germ line Fanconi anemia complementation group C mutations and pancreatic cancer. Cancer Res. 2005;65(2):383–6.
Rafnar T, Gudbjartsson DF, Sulem P, Jonasdottir A, Sigurdsson A, Jonasdottir A, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43(11):1104–7.
Acknowledgements
We thank our colleagues for valuable discussions during the course of writing this review and careful readings of this manuscript. This work was supported in part by NIH grants AA022057 and AA021724 (V.V.), U41HG003345 (E.A.B.) and P30 ES006096 (D.W.N.) and Wellcome Trust grant 099129/Z/12/Z (E.A.B).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VV and DWN proposed and oversaw the analyses; HD and VV drafted the manuscript; EAB drafted the gene nomenclature sections; EAB, DCT, HJ, DWN and VV edited the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dong, H., Nebert, D.W., Bruford, E.A. et al. Update of the human and mouse Fanconi anemia genes. Hum Genomics 9, 32 (2015). https://doi.org/10.1186/s40246-015-0054-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-015-0054-y