
Abstract
Background
Candida susceptibility commonly occurs in breast cancer patients. Of which, Candida albicans is considered as a common pathogen causing candidiasis. Martinella iquitosensis (Bignoniaceae) is one of the species belonged to Martinella, distributed widely in Amazon basin. Its root extract yielded two complex substituted tetrahydroquinolines, Martinelline and Martinellic acid which were the first natural non-peptide bradykinin receptor antagonists identified.
Findings
In this study, a novel martinelline type analogue, named 2,3,3a,4,5,9b-hexahydro-8-phenoxy-4-(pyridin-2-yl)furo[3,2-c]quinoline, was synthesized and its preliminary anticancer activity and antifungal potential were investigated. This compound showed potential anticancer activity against MDAMB-231 breast cancer cells. Meanwhile it could enhance the fungistatic activity of miconazole against Candida albicans.
Conclusions
These findings provide an implication for the continue investigation and development of martinelline type analogues as therapeutic agents in the future.
Similar content being viewed by others


Background
Breast cancer patients are commonly susceptible to candidiasis. Candida albicans (C. albicans) is one of the opportunistic fungi especially observed in immunocompromised patients (Calderone and Fonzi 2001), such as cancer patients. Previous studies have investigated the prevalence of candidiasis in cancer patients. Among 845 women with multiple lymph node positive or metastatic breast carcinoma receiving high-dose chemotherapy and autologous bone marrow transplantation at Duke University Medical Center during 1992–1997, 29 of them (3.4 %) developed candidemia. Of which, 23 % of them were found to be infected with C. albicans. The mortality was highest for the women who were infected by C. albicans (71 %) (Gottfredsson et al. 2003). In 2005, 400 adult patients with the hematological malignancy, head neck or solid tumor (including breast cancer) were recruited randomly into the study on admission to the regional cancer center of the Norfolk and Norwich University Hospital. There were 56.8 % (227 of 400) of all cancer patients and 18.9 % (43 of 227) of those who had clinical and microbiological evidence of oral candidiasis. Among 269 yeast isolates recovered from 227 patients, C. albicans was the most common yeast (74 %) causing colonization and infection (Schelenz et al. 2011).
Martinella can be used as indigenous medicine for eye disease caused by bacteria in various ethnolinguistic groups of some South American countries (Witherup et al. 1995). Martinella iquitosensis (Bignoniaceae), one of the species belonged to Martinella, is a tropical plant with dark purple corolla in South American and distributed widely in Amazon basin (Zuntini and Lohmann 2014). Its root extract yielded two complex substituted tetrahydroquinolines, Martinelline and Martinellic acid (Fig. 1a) which were the first natural non-peptide bradykinin receptor antagonists identified by Witherup et al. (1995). Afterwards, the studies on the synthesis of new compounds with this interesting core have been increased. For instance, hexahydro-2H-pyrano[3,2-c]quinolines were found to be a potent agent against pathogenic gram-negative bacteria and Magesh et al. discovered that one of the synthesized compounds exhibits good bacteriolytic activity against Virbio vulnificus and Vibrio parahaemolticus (Magesh et al. 2004). Another research group (Kantevari et al. 2011) also synthesized a series of hexahydro-2H-pyrano[3,2-c]quinolines and studied their activity against the Mycobacrtium tuberculosis H37Rv. Three of the synthesized compounds showed a comparable activity as ethambutol. In addition, it was reported that hexahydro-2H-pyrano[3,2-c]quinolines could be used as selective σ 1 receptor ligand for the treatment of pain (Diaz et al. 2013).

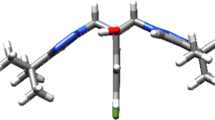
Structure of a Martinellic acid and Martinelline; b compound 1; c MOLECULAR structure of compound 1
Heterocyclic compounds are important candidates in the development of new class of structural entities for medicinal applications. Quinoline is a heterocyclic aromatic nitrogen containing compound characterized by a double-ring structure that has a benzene ring fused to pyridine at two adjacent carbon atoms (Keri and Patil 2014). Among these structures, tetrahydroquinoline derivatives demonstrated extensive biological activities. They included anticancer (Subramanian et al. 2011), antioxidant (Dorey et al. 2000) and antifungal (Vargas Méndez et al. 2010) activity. The construction and stereochemistry of the tricyclic ring system have also been recently reported (Calleja et al. 2014). In the previous years, we have prepared and studied some simple 2-subsituted terahydroquinoline alkaloid analogues as antitumor agents possessing notable cytotoxicity towards human Hep3B heptocellular carcinoma cells (Lam et al. 2013). In addition, we have reported the antimicrobial activity of some novel synthesized quinoline analogues (Lam et al. 2014; Chung et al. 2015). Inspired by unique core moiety of Martinelline and the reported promising bioactivity of tetrahydroquinolines and their related natural product analogues, we have designed compound 1 (Fig. 1b) and its potential biological activity was tested on MDAMB-231 breast cancer cells and C. albicans.
Strategically, we aimed to develop a simple and mild reaction pathway for the synthesis of compound 1 using one-pot multicomponent reaction with substituted aniline, aldehyde and alkene. Various catalysts, such as NbCl5 (da Silva et al. 2014), Fe2(SO4)·xH2O (Khan et al. 2011; Das et al. 2014), In(OTf)3 (Priestley et al. 2013) and BiCl3 (Kouznetsov et al. 2011) were reported to be effective for this reaction. Particularly, bismuth and its compounds are recognized as safe and green Lewis acid catalysts (Leonard et al. 2002; Mohan 2010). With increasing concern on the advancement of “green reaction” in last decade, the application of bismuth(III) compounds and their important roles in organic synthesis have been clearly addressed in recent researches (Gaspard-Iloughmane and Le Roux 2004; Bothwell et al. 2011; Ollevier 2013). Herein, we report a bismuth(III)-catalyzed synthesis of substituted tetrahydroquionlines.
Methods
General procedure for the synthesis of 2,3,3a,4,5,9b-hexahydro-8-phenoxy-4-(pyridin-2-yl)furo[3,2-c]quinoline (compound 1)
All reagents were purchased from Sigma-Aldrich. All the synthesized compounds were characterized by 1H NMR, 13C NMR, mass spectrometry and X-ray crystallography. NMR spectra were recorded on a Bruker DPX400 Fourier transform spectrometer using CDCl3 as solvent unless otherwise specified. X-ray crystallographic analysis was performed by Bruker D8_VENTURE PHOTON 100. To a mixture of 2-Pyridinecarboxaldehyde (1.0 mol equiv) and 4-Phenoxyaniline (1.2 mol equiv) was added in solvent (2 mL) and stirred. Catalyst (0.2 mol equiv) and 2,3-Dihydrofuran (1.2 mol equiv) were then added. The reaction mixture was stirred at room temperature for 1.5 h. Solvent was then removed and the residue was extracted with dichloromethane. The organic layer was washed with 10 % Na2CO3 solution and water. Dried over anhydrous Na2SO4 and removed the solvent by rotary evaporator. The crude product was purified by silica gel column chromatography.
Synthesis and characterization of compound 1
1H NMR (400 MHz, CDCl3): δ 1.55–1.57 (m, 1H), 2.03–2.09 (m, 1H), 3.07–3.14 (m, 1H), 3.76–3.80 (m, 2H), 4.53 (s, 1H), 4.83 (d, 1H, J = 2.8 Hz), 5.30 (d, 1H, J = 8.0 Hz), 6.70 (d, 1H, J = 8.8 Hz), 6.84–6.98 (m, 1H), 7.00–7.06 (m, 3H), 7.11 (d, 1H, J = 2.4 Hz), 7.25–7.27 (m, 1H), 7.31–7.33 (m, 2H), 7.50 (d, 1H, J = 8.0 Hz), 7.75–7.77 (m, 1H), 8.64 (d, 1H, J = 4.4 Hz); 13C NMR (100 MHz, CDCl3): δ 24.40, 43.33, 57.67, 66.62, 75.91, 116.17, 117.60, 120.43, 120.56, 120.98, 122.18, 122.42, 123.85, 129.53, 136.79, 140.93, 149.10, 149.16, 158.66, 160.13; HRMS (ESI): Calcd. for C22H21N2O2 [M + H]+, 345.1598. found 345.1597. Melting point = 132.4–134.2 °C. Optimized yield = 26.8 %.
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (MTS) assay and cellular morphology
MDAMB-231 cells were obtained from American Type of Culture Collection. Changes in the cellular viability of compound 1-treated cells were monitored using the MTS activity assay (20). Briefly, MDAMB-231 breast carcinoma cells were seeded at day 0. After 24 h, complete medium was changed and compound 1 was added at different concentrations (starting with 50 µM containing 0.1 % dimethyl sulfoxide (DMSO) as vehicle). Doxorubicin at 8 µM was used as a positive control. After 48 h of incubation, incubation medium was removed and fresh medium with MTS (Promega)/phenazine methosulfate as electron coupling agent mixed solution was added. Lastly, optical absorbance was determined at 490 nm using a microplate reader (Perkin Elmer Victor V) according to the user manual. Additionally, any morphological changes associated with compound 1 and doxorubicin treated breast cancer cells were recorded under an inverted microscope after fixing the cells with trichloroacetic acid and cellular protein was stained with sulforhodamine B after 24 h (Kok et al. 2007; Lam et al. 2015a, b).
Determination of minimum inhibitory concentration (MIC) and sensitization assay
Candida albicans was obtained from American Type of Culture Collection. The MIC values of synthesized compound 1 and miconazole nitrate were determined by the broth dilution method. Various concentrations of compound 1 and miconazole were loaded from a starting concentration of 50 µM containing 0.1 % dimethyl sulfoxide (DMSO) as vehicle and they were diluted serially. DMSO (0.1 %) was used as a vehicle control. The fungal samples were then incubated at 37 °C for 48 h. The minimum concentrations of compound 1 and miconazole that induced a complete growth inhibition would be determined as their MIC values. For sensitization study, with compound 1 at 50 µM, miconazole was added at a starting concentration of 1.56, 0.78 and 0.39 µM respectively. After incubation, the fungal samples were treated with MTS/PMS as above (Lam et al. 2015a).
Results and discussion
Synthesis of compound 1
We first screened different metal salts as catalyst for this reaction (Table 1). It was found that only metal(III) salts (Table 1, Entry 5–7) gave our desired compound 1 and, in particular, bismuth(III) nitrate pentahydrate provided the highest yield among all the selected catalysts. With such preliminary screening, we used Bi(NO3)3·5H2O as catalyst for further optimization of the reaction under various conditions aiming to enhance the product yield (Table 2). It is noted that addition of catalyst in 0.2 mol equivalent and the reaction carried in ethanol (Table 2, Entry 7) provided the best yield. With the present work, further investigation is ongoing to develop a greener and more effective reaction for the synthesis of compound 1.


Anticancer activity of compound 1
Compound 1 at 50 µM (~17 µg/ml) could readily induce cell death on MDAMB-231 cells with significant cellular morphological changes when compared with the untreated control (Fig. 2a) such as cell rounding and shrinkage (Fig. 2b) which were similar to those from the positive reference, doxorubicin (Fig. 2c) at 8 µM after 24 h. As shown in Fig. 2d, a dose dependent cytotoxicity of compound 1 on MDAMB-231 breast cancer cells was observed after a 48 h of incubation.

a–c Cellular morphology of MDAMB-231 cells after 24 h: a Untreated control; b compound 1 at 50 µM; c Doxorubicin at 8 µM as positive reference; and d MTS activity assay to determine the effect of compound 1 on MDAMB-231 cells after 48 h. Reported results represent the mean ± SD from triplicate tests. This figure shows a representative experiment taken from three independent experiments giving similar results
MIC of compound 1
The MIC value of miconazole on C. albicans was found to be 3 µM. Compound 1, however, did not exhibit antifungal activity on C. albicans of up to 50 µM. However, we found that compound 1 could enhance the antifungal activity of miconazole on C. albicans. In the subsequent tests, compound 1 was added simultaneously with different concentrations of miconazole. As shown in Fig. 3, compound 1 could significantly potentiate the antifungal action of miconazole. Recently, we have shown that corilagin could sensitize Hep3B hepatoma cells to cisplatin and doxorubicin (Gambari et al. 2014). Here we suggest that compound 1 at 50 µM could significantly improve the fungistatic property of miconazole against C. albicans.

MTS activity assay to determine the effect of miconazole alone and micronazole with compound 1 (50 µM) on C. albicans. Reported results represent the mean ± SD from triplicate tests. This figure shows a representative experiment taken from three independent experiments giving similar results
Conclusions
In this work, the synthesis and preliminary in vitro biological application of a novel martinelline type analogue, named 2,3,3a,4,5,9b-hexahydro-8-phenoxy-4-(pyridin-2-yl)furo[3,2-c]quinoline, were described. Interestingly, this compound showed potential anticancer activity against MDAMB-231 breast cancer cells and it could simultaneously potentiate the fungistatic activity of miconazole against a common human pathogenic fungus, C. albicans. As the obtained compound 1 consists of three chiral centres, there should be eight stereoisomers exist. Further work will be carried out to isolate each isomer and investigate their individual potential biological activity in order to elucidate if chirality is important in this group of compounds from the pharmaceutical point of view.
Abbreviations
- C. albicans :
-
Candida albicans
- DMSO:
-
dimethyl sulfoxide
- MIC:
-
minimum inhibitory concentration
- MTS:
-
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]
References
Bothwell JM, Krabbe SW, Mohan RS (2011) Applications of bismuth(III) compounds in organic synthesis. Chem Soc Rev 40(9):4649–4707
Calderone RA, Fonzi WA (2001) Virulence factors of Candida albicans. Trends Microbiol 9:327–335
Calleja J, González-Pérez AB, de Lera ÁR, Álvarez R, Fañanás FJ, Rodríguez F (2014) Enantioselective synthesis of hexahydrofuro[3,2-c] quinolines through a multicatalytic and multicomponent process. A new “aromatic sandwich” model for BINOL-phosphoric acid catalyzed reactions. Chem Sci 5:996–1007
Chung PY, Gambari R, Chen YX, Cheng CH, Bian ZX, Chan ASC, Tang JCO, Leung PHM, Chui CH, Lam KH (2015) Development of 8-benzyloxy-substituted quinoline ethers and evaluation of their antimicrobial activities. Med Chem Res 24:1568–1577
da Silva BHST, Marana NL, Mafud AC, da Silva-Filho LC (2014) A theoretical and experimental study to unequivocal structural assignment of tetrahydroquinoline derivatives. Struct Chem 25:327–337
Das DK, Sarkar S, Khan AT, Saravanan P, Patra S (2014) Synthesis of fused tetrahydropyrido[2,3-c]coumarin derivatives as potential inhibitors for dopamine d3 receptors, catalyzed by hydrated ferric sulfate. RSC Adv 4:3581–3590
Diaz JL, Christmann U, Fernandez A, Luengo M, Bordas M, Enrech R, Carro M, Pascual R, Burgueno J, Merlos M, Benet-Buchholz J, Ceron-Bertran J, Ramirez J, Reinoso RF, Fernandez de Henestrosa AR, Vela JM, Almansa C (2013) Synthesis and biological evaluation of a new series of hexahydro-2H-pyrano[3,2-c]quinolines as novel selective sigma1 receptor ligands. J Med Chem 56:3656–3665
Dorey G, Lockhart B, Lestage P, Casara P (2000) New quinolinic derivatives as centrally active antioxidants. Bioorg Med Chem Lett 10:935–939
Gambari R, Hau DKP, Wong WY, Chui CH (2014) Sensitization of Hep3B hepatoma cells to cisplatin and doxorubicin by corilagin. Phytother Res 28:781–783
Gaspard-Iloughmane H, Le Roux C (2004) Bismuth(III) triflate in organic synthesis. Eur J Org Chem 2004(12):2517–2532
Gottfredsson M, Vredenburgh JJ, Xu J, Schell WA, Perfect JR (2003) Candidemia in women with breast carcinoma treated with high-dose chemotherapy and autologous bone marrow transplantation. Cancer 98:24–30
Kantevari S, Yempala T, Surineni G, Sridhar B, Yogeeswari P, Sriram D (2011) Synthesis and antitubercular evaluation of novel dibenzo[b, d]furan and 9-methyl-9H-carbazole derived hexahydro-2H-pyrano[3,2-c]quinolines via Povarov reaction. Eur J Med Chem 46:4827–4833
Keri RS, Patil SA (2014) Quinoline: a promising antitubercular target. Biomed Pharmacother 68:1161–1175
Khan AT, Das DK, Khan MM (2011) Ferric sulfate [Fe2(SO4)3·xH2O]: an efficient heterogeneous catalyst for the synthesis of tetrahydroquinoline derivatives using Povarov reaction. Tetrahedron Lett 52:4539–4542
Kok SHL, Gambari R, Chui CH, Lau FY, Cheng GYM, Lai PBS, Lam WS, Chan ASC, Cheng CH, Teo ITN, Yu MWY, Tang JCO, Cheung F, Wong RSM (2007) Paradoxical proliferative potential of iron(II) sulphate on cancer cells after the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. Int J Mol Med 19:971–975
Kouznetsov VV, Gomez CM, Parada LK, Bermudez JH, Mendez LY, Acevedo AM (2011) Efficient synthesis and free-radical scavenging capacity of new 2,4-substituted tetrahydroquinolines prepared via BiCl(3)-catalyzed three-component Povarov reaction, using N-vinylamides. Mol Divers 15:1007–1016
Lam KH, Lee KKH, Gambari R, Wong RSM, Cheng GYM, Tong SW, Chan KW, Lau FY, Lai PBS, Wong WY, Chan ASC, Kok SHL, Tang JCO, Chui CH (2013) Preparation of Galipea officinalis Hancock type tetrahydroquinoline alkaloid analogues as anti-tumour agents. Phytomedicine 20:166–171
Lam KH, Gambari R, Lee KKH, Chen YX, Kok SHL, Wong RSM, Lau FY, Cheng CH, Wong WY, Bian ZX, Chan ASC, Tang JCO, Chui CH (2014) Preparation of 8-hydroxyquinoline derivatives as potential antibiotics against Staphylococcus aureus. Bioorg Med Chem Lett 24:367–370
Lam PL, Gambari R, Kok SHL, Lam KH, Tang JCO, Bian ZX, Lee KKH, Chui CH (2015a) Non-toxic agarose/gelatin-based microencapsulation system containing gallic acid for antifungal application. Int J Mol Med 35:503–510
Lam PL, Kok SHL, Gambari R, Kok TW, Leung HY, Choi KL, Wong CS, Hau DKP, Wong WY, Lam KH, Bian ZX, Lee KKH, Chui CH (2015b) Evaluation of berberine/bovine serum albumin nanoparticles for liver fibrosis therapy. Green Chem 17:1640–1646
Leonard NMW, Laura C, Mohan Ram S (2002) Applications of bismuth(III) compounds in organic synthesis. Tetrahedron 58(42):8373–8397
Magesh CJ, Magesh SV, Perumal PT (2004) Highly diastereoselective inverse electron demand (IED) Diels–Alder reaction mediated by chiral salen-AlCl complex: the first, target-oriented synthesis of pyranoquinolines as potential antibacterial agents. Bioorg Med Chem Lett 14:2035–2040
Mohan R (2010) Green bismuth. Nat Chem 2(4):336
Ollevier T (2013) New trends in bismuth-catalyzed synthetic transformations. Org Biomol Chem 11(17):2740–2755
Priestley ES, De Lucca I, Zhou J, Zhou J, Saiah E, Stanton R, Robinson L, Luettgen JM, Wei A, Wen X, Knabb RM, Wong PC, Wexler RR (2013) Discovery and gram-scale synthesis of BMS-593214, a potent, selective FVIIa inhibitor. Bioorg Med Chem Lett 23:2432–2435
Schelenz S, Abdallah S, Gray G, Stubbings H, Gow I, Baker P, Hunter PR (2011) Epidemiology of oral yeast colonization and infection in patients with hematological malignancies, head neck and solid tumors. J Oral Pathol Med 40:83–89
Subramanian S, Kim NS, Thanigaimalai P, Sharma VK, Lee KC, Kang JS, Kim HM, Jung SH (2011) Structure–activity relationship studies of novel arylsulfonylimidazolidinones for their anticancer activity. Eur J Med Chem 46:3258–3264
Vargas Méndez LY, Zacchino SA, Kouznetsov VV (2010) Synthesis of new 4-methyl-2-(4-pyridyl)-1,2,3,4-tetrahydroquinolines as potent antifungal compounds. J Braz Chem Soc 21:105–111
Witherup KMR, Richard W, Graham AC, Bernard AM, Salvatore MJ, Lumma WC, Anderson PS, Pitzenberger SM, Varga SL (1995) Martinelline and martinellic acid, novel G-protein linked receptor antagonists from the tropical plant Martinella iquitosensis (bignoniaceae). J Am Chem Soc 117:6682–6684
Zuntini AR, Lohmann LG (2014) Synopsis of Martinella Baill. (Bignonieae, Bignoniaceae), with the description of a new species from the Atlantic Forest of Brazil. PhytoKeys 37:15–24
Authors’ contributions
PYC carried out the chemistry experiments and drafted the manuscript. JCO participated in the chemistry experiments. CHC participated in the antifungal test. ZXB participated in the anticancer study. WYW participated in the chemistry experiments. KHL conceived of the study, participated in the design of the study and carried out the chemistry experiments. CHC conceived of the study, and participated in its design of the study, carried out the anticancer and antifungal studies and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This study has been supported by a grant from the Innovation Technology Commission to Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University and the research grants from Research Office of Hong Kong Baptist University (FRG1/14-15/004, FRG1/14-15/021, FRG1/14-15/078, 38-40-116 and 03-16-176). We would like to thank the support from Prof. Zhou Zhongyuan for the X-ray crystallography.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chung, PY., Tang, J.CO., Cheng, CH. et al. Synthesis of hexahydrofuro[3,2-c]quinoline, a martinelline type analogue and investigation of its biological activity. SpringerPlus 5, 271 (2016). https://doi.org/10.1186/s40064-016-1890-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-1890-5