
Abstract
In this study, a β-agarase gene, agaB-4, was isolated for the first time from the agar-degrading bacterium Paenibacillus agarexedens BCRC 17346 by using next-generation sequencing. agaB-4 consists of 2652 bp and encodes an 883-amino acid protein with an 18-amino acid signal peptide. agaB-4 without the signal peptide DNA was cloned and expressed in Escherichia coli BL21(DE3). His-tagged recombinant AgaB-4 (rAgaB-4) was purified from the soluble fraction of E. coli cell lysate through immobilized metal ion affinity chromatography. The optimal temperature and pH of rAgaB-4 were 55 °C and 6.0, respectively. The results of a substrate specificity test showed that rAgaB-4 could degrade agar, high-melting point agarose, and low-melting point agarose. The Vmax and Km of rAgaB-4 for low-melting point agarose were 183.45 U/mg and 3.60 mg/mL versus 874.61 U/mg and 9.29 mg/mL for high-melting point agarose, respectively. The main products of agar and agarose hydrolysis by rAgaB-4 were confirmed to be neoagarotetraose. Purified rAgaB-4 can be used in the recovery of DNA from agarose gels and has potential application in agar degradation for the production of neoagarotetraose.
Similar content being viewed by others


Introduction
Agar is a hydrophilic colloid extracted from the cell walls of red algae (Rhodophyceae), such as Gelidium spp., Gracilaria spp., and Porphyra spp. It is a heterogeneous polysaccharide which consists of agarose and porphyran (Chi et al. 2012). Agarose is a neutral polysaccharide that forms a gel; its molecular weight is approximately 120 kDa; and it consists of alternating β-d-galactose and 3,6-anhydro-α-l-galactopyranose linked by α-1,3 and β-1,4 glycosidic bonds (Armisén 1991; Yun et al. 2017). Porphyran, the non-gelling fraction, is a linear sulfated galactan; its composition is similar to that of agarose, except that some 3,6-anhydro-α-l-galactose are replaced with α-L-galactose-6-sulfate (Knutsen et al. 1994; Chi et al. 2012).
Agarases are enzymes that catalyze the hydrolysis of agar into oligosaccharides; these enzymes cleave glycosidic bonds at different positions. Thus, agarases can be classified into α-agarases (EC 3.2.1.158), β-agarases (EC 3.2.1.81), and β-porphyranases (EC 3.2.1.178) according to the cleavage pattern (Chi et al. 2012). α-Agarases act on the α-1,3 glycosidic bonds of agarose, producing agaro-oligosaccharides with a 3,6-anhydro-α-l-galactose residue at the reducing end. β-agarases, on the other hand, act on the β-1,4 glycosidic bonds of agarose, producing neoagaro-oligosaccharides with a d-galactose residue at the reducing end (Fu and Kim 2010). β-porphyranases act on the β-1,4 glycosidic bonds of porphyran, producing oligosaccharides with a d-galactose residue at the reducing end.
Various microbes from seawater, marine sediments, seaweed, marine mollusks, soil, solar salt, city drain water, and hot spring produce agarases. Seawater isolates, including Alteromonas agarlyticus GJ1B (Potin et al. 1993) and Thalassomonas sp. JAMB-A33 (Ohta et al. 2005), produce α-agarases. Based on the amino acid sequence similarity, known α-agarases belong to the glycoside hydrolase (GH) family GH96. Compared with the source of α-agarases, more bacterial strains produce β-agarases, such as Vibrio sp. JT0107 (Sugano et al. 1993) and Catenovulum sp. X3 (Xie et al. 2013) from seawater; Vibrio sp. PO-303 (Dong et al. 2007) and Agarivorans sp. HZ105 (Hu et al. 2009) from marine sediments; Vibrio sp. AP-2 (Aoki et al. 1990) and Pseudoalteromonas antarctica N-1 (Vera et al. 1998) from seaweed; Agarivorans albus YKW-34 (Fu et al. 2008) from marine mollusks; Paenibacillus sp. SSG-1 (Song et al. 2014) and Alteromonas sp. E-1 (Kirimura et al. 1999) from soil; Halococcus sp. 197A (Minegishi et al. 2013) from solar salt; Alcaligenes sp. Yen (Sie et al. 2009) from city drain water; and Bacillus sp. BI-3 (Li et al. 2014) from hot spring. Based on the amino acid sequence similarity, known β-agarases are classified into the four GH families of GH16, GH50, GH86, and GH118 (Lombard et al. 2014). Unlike the source of β-agarases, only two bacterial strains, including Zobellia galactanivorans DSM 12802 from the red alga and Bacteroides plebeius DSM 17135 from Japanese individuals (Hehemann et al. 2010, 2012), produce β-porphyranases. Based on the amino acid sequence similarity, known β-porphyranases belong to the GH families of GH16 and GH86.
Previous studies reported that α-agarases and β-agarases have various applications, for example, those in the recovery of DNA from agarose gel (Finkelstein and Rownd 1978), preparation of seaweed protoplasts (Araki et al. 1998), and production of agar-derived oligosaccharides (Fu and Kim 2010). Studies have shown that the oligosaccharides generated by the hydrolysis of agar or seaweed polysaccharide crude extracts by agarases have numerous biological activities, such as antioxidative activity (Wu and Pan 2004), hepatoprotective potential (Chen et al. 2006), immunostimulatory activity (Lee et al. 2017), antiobesity effect (Hong et al. 2017a), whitening effect on melanoma cells (Jang et al. 2009), moisturizing effect on skin (Kobayashi et al. 1997), and prebiotic effect (Hu et al. 2006). Hence, agar-derived oligosaccharides can be used as new-generation, high-value functional oligosaccharides in cosmetic, health food, and pharmaceutical industries.
In the present study, the newly identified β-agarase gene agaB-4 from Paenibacillus agarexedens BCRC 17346 was cloned and expressed in the cytoplasm of Escherichia coli BL21(DE3). The characteristics and potential applications of the purified recombinant enzyme were also analyzed.
Materials and methods
Bacterial strains, plasmids, and culture condition
Paenibacillus agarexedens BCRC 17346 was purchased from BCRC (Bioresource Collection and Research Center, Hsinchu, Taiwan) and was used for the isolation of genomic DNA. Escherichia coli ECOS™ 9-5 (Yeastern, Taipei, Taiwan) was used for the propagation and manipulation of recombinant DNA. E. coli BL21(DE3) (Merck Millipore, Darmstadt, Germany) was used as the expression host. pJET1.2 (Fermentas, Maryland, USA) and pET-29a(+) (Merck Millipore) were used as cloning and expression vectors, respectively. P. agarexedens BCRC 17346 was cultured at 30 °C in nutrient broth medium supplemented with 0.1% (w/v) urea and 1% (w/v) glucose. E. coli was grown at 37 °C in Luria–Bertani (LB) medium (Difico, Detroit, USA) containing 30 μg/mL kanamycin, when required.
General DNA techniques
Bacterial genomic DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Plasmid DNA was isolated using the Plasmid Miniprep Purification Kit II (GMbiolab, Taichung, Taiwan) according to the manufacturer’s instructions. DNA fragments were amplified using GDP-HiFi DNA Polymerase (Genedirex, Las Vegas, USA) according to the manufacturer’s recommendations. All polymerase chain reactions (PCRs) were performed on a TProfessional TRIO thermocycler (Biometra GmbH, Göttingen, Germany). PCR products were purified using a PCR Clean-Up Kit (GMbiolab). Restriction enzyme digestions were performed according to the supplier’s recommendations (Thermo Fisher Scientific, Waltham, USA). DNA fragments were recovered from gels by using a Gel Elution Kit (GMbiolab). DNA ligation reactions were performed using a DNA Ligation Kit (Yeastern). The purified PCR product was cloned into pJET1.2 by using a CloneJET PCR Cloning Kit (Fermentas) according to the manufacturer’s recommendations. Plasmids were introduced into E. coli through heat shock transformation according to the manufacturer’s instructions.
Whole-genome sequencing of P. agarexedens
The genomic DNA of P. agarexedens was isolated and quantified using a Quant-iT dsDNA BR assay (Thermo Fisher Scientific). The quality of the extracted genomic DNA was verified on a 0.6% agarose gel. DNA libraries were constructed using an Illumina TruSeq DNA LT Sample Prep Kit. Mate-pair libraries were constructed using an Illumina Mate Pair Library Prep Kit v2. For the de novo assembly of the complete genome, we used Velvet (Zerbino and Birney 2008) to assemble paired-end and mate-pair reads to scaffolds. Genes on these assembled scaffolds were predicted using GeneMark.hmm (Besemer and Borodovsky 1999) and were annotated using a BLAST (blastp) search against the NCBI nr protein database, with an e-value cutoff of 0.00001. For the functional annotation of predicted genes, the accession numbers from BLAST hits were mapped to GO terms by querying the GO database (Ashburner et al. 2000). Enzyme code annotations were retrieved by mapping to GO terms, and enzyme codes were retrieved by querying the GO database.
Construction of expression vector
Based on the results of whole-genome sequencing of P. agarexedens, the forward primer PBAGA4F (5′-GATATAGGTACCGCCACGCCGTTCCCTACTC-3′, KpnI site underlined) and the reverse primer PBAGA4R (5′-CAATATCTCGAGTTAGTGGTGGTGGTGGTGGTGCTTTGAGATTAGCAGACGATCCATTA-3′; XhoI site underlined, stop codon in italics, and His tag DNA in bold) were designed and used to amplify the 2598-bp DNA fragment encoding the mature β-agarase AgaB-4 lacking the predicted signal peptide through PCR. This fragment was generated from the genomic DNA of P. agarexedens BCRC 17346. The PCR product was purified and then ligated to pJET1.2. The ligation mixture was transformed into E. coli ECOS™ 9-5 competent cells for the generation of recombinant plasmids. The recombinant plasmids were confirmed by DNA sequencing. The resulting plasmid containing the agarase DNA fragment was named pJET-AGAB-4. The agarase DNA fragment was excised from pJET1.2 by using KpnI and XhoI and was subsequently subcloned into pET-29a(+) at the corresponding restriction sites. The recombinant plasmid, designated pET-AgaB-4, was confirmed by DNA sequencing and was then transformed into E. coli BL21(DE3).
Expression and detection of rAgaB-4 in the soluble fraction
E. coli BL21(DE3)(pET-AgaB-4) cells were cultured in LB medium containing kanamycin (30 μg/mL), with shaking at 37 °C. On the next day, 0.1 mL of the overnight culture was inoculated into 10 mL of LB medium containing kanamycin (30 μg/mL) and was grown at various temperatures (37, 30, 25, 20, 16 °C), with shaking. When the optical density at 600 nm (OD600) of the cultures reached 0.4–0.6, isopropyl-β-d-thiogalactopyranoside (IPTG) at a final concentration of 0.1 mM was added. After 4 and 24-h incubation, cells were harvested by centrifugation at 10,000×g for 10 min at 4 °C and were then resuspended in lysis buffer (50 mM Tris–HCl and 500 mM NaCl, pH 8.0). Cells were lysed by sonication in an ice water bath. The suspensions (total cell lysate) were centrifuged at 10,000×g for 10 min at 4 °C. The clear supernatant (soluble fraction) was collected, and the remaining pellet (insoluble fraction) was resuspended in an equal volume of lysis buffer. Equal volumes of the total cell lysate, soluble fraction, and insoluble fraction were analyzed through 12.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Laemmli 1970), using a minigel apparatus (model AE-6450; ATTO, Tokyo, Japan).
Western blot analysis
Protein samples were separated through 12.5% SDS-PAGE. After electrophoresis, proteins were electrophoretically transferred onto methanol-activated polyvinylidene fluoride membrane (Merck Millipore). The membrane was blocked with 5% skim milk in phosphate-buffered saline and incubated with the Penta·His antibody (1:20,000) (Qiagen), followed by incubation with an alkaline phosphatase-conjugated anti-mouse antibody (1:20,000) (Bethyl, Montgomery, USA). Immunoreactive bands were visualized using BCIP/NBT substrate solution (PerkinElmer, Waltham, USA).
Purification of rAgaB-4
E. coli BL21(DE3)(pET-AgaB-4) cells were cultured in 1 L of LB broth containing kanamycin (30 μg/mL), with shaking at 37 °C. Cells were cultured to an OD600 of 0.4–0.6. Subsequently, IPTG (0.1 mM) was added to induce rAgaB-4 expression at 20 °C for 24 h. Cells were harvested by centrifugation at 8000×g for 30 min and were then resuspended in lysis buffer. The cell suspension was disrupted using Constant Cell Disruption Systems (Constant Systems Ltd, Warwick, UK). The cell lysate was centrifuged at 8000×g for 15 min at 4 °C, and the resulting supernatant was filtered through a 0.22-μm membrane and applied to a 5-mL HiTrap™ excel affinity chromatography column (GE Healthcare, Uppsala, Sweden) according to the manufacture’s instruction. The purity of the eluted fusion protein was analyzed through 12.5% SDS-PAGE, and the protein concentration was determined using a Protein Quantification Assay Kit (MACHEREY–NAGEL, Düren, Germany).
Enzyme activity measurements
Agarase activity was measured by determining the amount of reducing sugars generated from hydrolysis, according to the DNS method developed by Miller (1959), with minor modifications. Briefly, 50 μL of suitably diluted rAgaB-4 solution was mixed with 950 μL of phosphate buffer (50 mM, pH 6) containing 0.2% (w/v) low-melting point (LMP) agarose. After incubation at 40 °C for 10 min, the sample was mixed with 1.0 mL of 3,5-dinitrosalicylic acid reagent solution, heated in a boiling water bath for 10 min, and then cooled in an ice water bath. Absorbance (OD) readings at 540 nm were obtained on an Infinite 200 PRO microplate reader (Tecan Group Ltd, Männedorf, Switzerland). The amount of enzyme required to produce 1 μmol d-galactose per min under the assay conditions was defined as one unit (U) of agarase. d-galactose was used as a reference reducing sugar for preparing the standard curve.
Effects of pH and temperature on agarase activity and stability
The effect of pH on rAgaB-4 activity was assayed at 40 °C in 50 mM buffer solutions containing 0.2% LMP agarose and 1.51 μg of purified rAgaB-4 with a pH range of 3–10 (at 1.0 intervals). The buffer solutions used were citric acid/sodium citrate buffer (pH 3–6), phosphate buffer (pH 6–8), and glycine–NaOH buffer (pH 9–10). The effect of temperature on rAgaB-4 activity was determined by monitoring agarase activity at temperatures ranging from 20 to 80 °C in 50 mM phosphate buffer (pH 6) containing 0.2% LMP agarose and 1.51 μg of purified rAgaB-4 for 10 min. The thermostability of rAgaB-4 was determined by measuring the residual enzyme activity after incubation at temperatures ranging from 20 to 80 °C in 50 mM phosphate buffer (pH 6) containing 0.2% LMP agarose and 1.51 μg of purified rAgaB-4 for 1 h.
Effect of various metal ions and ethylenediaminetetraacetic acid (EDTA) on enzyme activity
The effects of various metal ions and EDTA on rAgaB-4 activity were assayed in 50 mM sodium phosphate buffer (pH 6) containing 0.2% LMP agarose and 1.91 μg of purified rAgaB-4 by adding metal ions or EDTA at a final concentration of 1 mM. Hydrolysis reactions were performed at 55 °C for 10 min. Relative activity was calculated as the enzyme activity of rAgaB-4 with added metal ion or EDTA/activity of rAgaB-4 × 100.
Substrate specificity of rAgaB-4
The substrate specificity of rAgaB-4 was measured using high-melting point (HMP) agarose, LMP agarose, agar, sodium alginate, carrageenan, soluble starch, and sodium carboxymethyl cellulose. Hydrolysis reactions were performed at 55 °C for 10 min in 50 mM sodium phosphate buffer (pH 6) containing 0.2% substrates and 1.91 μg of purified rAgaB-4. Relative activity was defied as the percentage of activity determined with the respect to the maximum agarase activity.
Determination of kinetic parameters
The kinetic parameters of purified rAgaB-4 (1.54 μg) were determined in 50 mM phosphate buffer (pH 6) containing LMP agarose and HMP agarose (molecular mass, 120 kDa), ranging in concentration from 2 to 30 mg/mL. The reaction mixture was incubated at 55 °C for 10 min. Km and Vmax for LMP agarose and HMP agarose were determined from Lineweaver–Burk plots using SigmaPlot 12 software (Systat Software, San Jose, USA). Subsequently, the Kcat (turnover number) and Kcat/Km (catalytic efficiency) values were calculated based on the Vmax, Km, and [E] (concentration of rAgaB-4) values.
Thin layer chromatography analysis of hydrolysis products
The products of LMP agarose, HMP agarose, and agar hydrolysis by rAgaB-4 were detected using thin layer chromatography (TLC) performed on silica gel 60 plates (Merck Millipore) as previously described (Li et al. 2014) with some modifications. Hydrolysis reactions were conducted at 40 °C for 24 h in 50 mM sodium phosphate buffer (pH 6) containing 1% (w/v) LMP agarose, HMP agarose and agar with 1.54 μg of purified rAgaB-4, respectively. The reaction mixtures were centrifuged at 20,630×g for 10 min at 4 °C to pellet the undigested agarose and agar. Subsequently, 2 μL of each supernatant was applied to a silica gel 60 plate and was developed using an n-butanol-acetic acid-water solution (2:2:1, by volume). The developed oligosaccharides were detected by spraying the plate with aniline phthalate solution (Merck Millipore) and by heating it on a hot plate at 180 °C.
Evaluation of rAgaB-4 ability for DNA recovery from gel
The pUC19 plasmid (2.5 μg) was embedded in 1% LMP agarose. The agarose containing the pUC19 plasmid was incubated at 70 °C for 10 min and was treated with 1 U rAgaB-4 at 40 °C for 1 h. The mixture was centrifuged at 20,630×g for 10 min at 4 °C to remove the undigested residue. The DNA in the supernatant was precipitated by adding 0.6 volumes of isopropanol in the presence of 2.5 M ammonium acetate and 1 μg/μL glycogen. The mixture was centrifuged at 20,630×g for 10 min at 4 °C to pellet the DNA. The precipitated DNA was washed twice with 70% ethanol, dried, and dissolved in sterile Tris–HCl buffer (pH 8.0). Equal amounts of recovered DNA and the original pUC19 plasmid were analyzed through agarose gel electrophoresis.
Nucleotide sequence accession number
The nucleotide sequence of agaB-4 reported in this study has been submitted to the GenBank database under the accession number MF998080.
Results
In silico analysis and cloning of the β-agarase gene agaB-4
The bioinformatics analysis of whole-genome sequencing data revealed that P. agarexedens may have four β-agarase genes (data not shown). According to the number of nucleotides in each gene, the genes were named in the descending order of size as agaB-1, agaB-2, agaB-3, and agaB-4. The present study focused on agaB-4. According to the bioinformatics analysis, the total length of agaB-4 is 2652 bp, and its start and stop codons are ATG and TAA, respectively; this gene encodes an 883-amino acid protein. Signal peptide prediction (SignalP 4.1 Server, http://www.cbs.dtu.dk/services/SignalP/) revealed that AgaB-4 may be a secretory protein with an N-terminal signal peptide (MILAIIAGLTGQPGAAAA) consisting of 18 amino acids, and the cleavage site of the signal peptidase is located between Ala18 and Ala19. The molecular weight of the mature protein without the signal peptide is 94,136 Da, and the predicted isoelectric point is 5.57.
Amino acid sequence similarities searches were performed using the BLASTP program in NCBI. The results showed that the sequence of AgaB-4 was the most similar to that of agarase AgaW (GenBank accession No. AKV62624) from the soil bacterium Cohnella sp. LGH (Li et al. 2015), which belongs to the GH50 family. The degree of identity and similarity between AgaB-4 and AgaW was 93.6 and 97.5%, respectively. Amino acid sequence alignment of AgaB-4 was performed with agarase AgaW, Aga50D (GenBank accession No. ABD81904) (Kim et al. 2010), AgWH50A (GenBank accession No. AFP32918) (Liu et al. 2014), and HZ2 (GenBank accession No. ADY17919) (Lin et al. 2012), which all belong to the GH50 family. The results revealed higher conservation of the C-terminal sequence in these GH50 family members (Additional file 1: Figure S1). According to the amino acid sequence alignment results, AgaB-4 was determined to belong to the GH50 family.
To investigate whether the protein encoded by agaB-4 exhibits the enzymatic activity of β-agarase, we conducted gene cloning, expression, and purification of rAgaB-4; and subsequently analyzed the enzyme properties. For gene cloning, specific primers were designed and used to amplify the DNA fragment encoding the mature AgaB-4 through PCR. The PCR product was then cloned into pJET1.2 to yield pJET-AGAB-4. Subsequently, the agarase DNA fragment was excised from pJET-AGAB-4 and inserted into pET-29a(+) to yield the agarase expression vector pET-AGAB-4.
Expression and purification of rAgaB-4
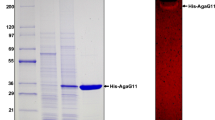
The agarase expression vector pET-AGAB-4 was transformed into E. coli BL21(DE3) cells, and IPTG was then added to induce protein expression. According to the results of SDS-PAGE and Western blot analyses, compared with E. coli BL21(DE3) transformants containing pET-29a(+), E. coli BL21(DE3) transformants containing the expression vector pET-AGAB-4 expressed the rAgaB-4 with the His-tag (attached to the C-terminal end); the molecular weight of the band was similar to the expected molecular weight of 97.32 kDa (Fig. 1). When protein expression was induced at 37 °C, rAgaB-4 mainly existed in the intracellular insoluble fraction. Protein expression induction at 20 °C and 16 °C significantly improved the solubility of rAgaB-4 (Additional file 1: Figure S2). This result suggests that lowering the induction temperature increases the solubility of rAgaB-4 expressed in E. coli. rAgaB-4 in intracellular soluble fraction from E. coli was purified using immobilized metal ion affinity chromatography. In SDS-PAGE, a single band with a molecular weight of approximately 100 kDa was observed, representing the homogenous composition of the purified protein (Fig. 2).

a SDS-PAGE and b Western blot analyses of rAgaB-4 expression in E. coli BL21(DE3). Lane M PageRuler™ Prestained Protein Ladder; lanes 1 and 2 total cell lysates of E. coli BL21(DE3) containing pET-29a(+) induced by IPTG at 37 °C for 4 and 24 h, respectively; lanes 3 and 4 total cell lysates of E. coli BL21(DE3) containing pET-AGAB-4 induced by IPTG at 37 °C for 4 and 24 h, respectively. Arrows indicate rAgaB-4 expression

SDS-PAGE analysis of purified rAgaB-4. Lane M PageRuler™ Prestained Protein Ladder; lane 1 purified rAgaB-4
Characteristics of rAgaB-4
The optimal pH of rAgaB-4 was 6; more than 90% of enzyme activity could be maintained over a pH range of 5–7 (Fig. 3a). The optimal temperature of the enzyme was 55 °C; when the temperature increased to 60 °C, enzyme activity decreased sharply. When the temperature increased to up to 70 °C, enzyme activity was almost undetectable (Fig. 3b). The effect of temperature on enzyme stability was also investigated. The results indicated that when the enzyme was incubated at a temperature less than 45 °C for 1 h, 94% of enzyme activity was maintained. However, when the enzyme was incubated at a temperature higher than 50 °C for 1 h, the enzyme became completely inactivated (Fig. 3b). Moreover, the results showed that several metal ions, including Cu2+, K+, Fe2+, Ba2+, Na+, Sr2+, Co2+, Mg2+, Mn2+, Ca2+, and Al3+, may enhance the activity of rAgaB-4. Among these ions, Mn2+ was the most effective in enhancing enzyme activity, which increased up to 95% with Mn2+ addition. On the other hand, no significant activation or inhibition was observed by Zn2+ and EDTA (Table 1). The results of a substrate specificity test showed that the activity of rAgaB-4 was the highest for HMP agarose hydrolysis. Moreover, the relative enzyme activity for LMP agarose and agar was 62 and 94%, respectively. However; rAgaB-4 could not hydrolyze sodium alginate, carrageenan, soluble starch, and sodium carboxymethyl cellulose (Table 2). According to the Lineweaver–Burk plots, Vmax and Km of rAgaB-4 for LMP agarose were 183.45 U/mg and 3.60 mg/mL, respectively, while those for HMP agarose were 874.61 U/mg and 9.29 mg/mL, respectively (Fig. 4). The calculated Kcat and Kcat/Km values were, respectively 2.98 × 102/s and 9.92 × 106/s/M for LMP agarose, and 1.42 × 103/s and 1.83 × 107/s/M for HMP agarose. The results of kinetic analysis revealed that rAgaB-4 had higher catalytic efficiency (Kcat/Km) toward HMP agarose than that toward LMP agarose. TLC analysis showed that neoagarotetraose was the main product formed from the hydrolysis of LMP agarose, HMP agarose and agar by rAgaB-4 (Fig. 5).

Effects of pH and temperature on rAgaB-4 activity. a Effects of pH on rAgaB-4 activity. The buffer solutions used for different pH were 50 mM citric acid/sodium citrate buffer (pH 3–6), phosphate buffer (pH 6–8), and glycine-NaOH buffer (pH 9–10). b Effects of temperature on rAgaB-4 activity and stability. Filled circle, optimal temperature; Filled square, thermostability. Error bars represent standard errors from triplicate experiments

Lineweaver–Burk plots for determining the kinetic parameters of rAgaB-4 acting on low-melting point agarose (open circle) and high-melting point agarose (closed circle). V velocity; S substrate concentration. All data are mean values from triplicate experiments

Thin layer chromatography analysis of the products of low-melting point agarose, high-melting point agarose, and agar hydrolysis by rAgaB-4. Lane 1 neoagarobiose (NA2); lane 2 neoagarotetraose (NA4); lane 3 neoagarohexaose (NA6); lane 4 hydrolysis products from low-melting point agarose; lane 5 hydrolysis products from high-melting point agarose; lane 6 hydrolysis products from agar
rAgaB-4 is used in the recovery of DNA from gels
The feasibility of using rAgaB-4 to recover DNA from gels was tested using pUC19 DNA as the model DNA. The results showed that rAgaB-4 could be used for the recovery of DNA from gels, and the recovery efficiency was more than 95% (Additional file 1: Figure S3).
Discussion
P. agarexedens is an agar-degrading bacterium that was isolated from meadow soil by Miehlmanni in 1972 (Uetanabaro et al. 2003). The culture conditions, physiological characteristics, and genetic characteristics of this bacterium have been studied. Agarase production by this bacterium has been proven by the appearance of depressions around bacterial colonies when cultured in solid culture medium containing agar. However, the type of agarase produced by this bacterium and the cellular localization, gene and enzyme characteristics, and applications of agarase have yet to be analyzed. In this study, the agarase gene of this bacterium was explored through whole-genome sequencing and bioinformatics analysis. Subsequently, cloning and expression of agaB-4 were performed to reveal the characteristics of the recombinant enzyme.
The sequence of AgaB-4 reported in this study was the most similar to that of agarase AgaW from Cohnella sp. LGH, which belongs to the GH50 family. In addition, the partial C-terminal sequence is conserved among various GH50 family members. Therefore, AgaB-4 was determined to belong to the GH50 family. To date, according to the CAZy database (http://www.cazy.org/Glycoside-Hydrolases.html), the GH50 family is composed of 476 types of β-agarases; the products of agarose hydrolysis by agarases belonging to the GH50 family are neoagarobiose, neoagarotetraose, or a mixture of the two compounds. The main hydrolysis products of AgaB-4 and AgaW from Cohnella sp. LGH are identical, namely neoagarotetraose. However, these enzymes still differ in terms of specific properties. For example, the optimal pH of AgaB-4 and AgaW is 6 and 7, respectively. When incubated at 50 °C for 1 h, AgaB-4 is completely inactivated. By contrast, AgaW can maintain 50% activity. This finding shows that the difference in amino acid sequence affects enzyme properties. Currently, of the agarases in the GH50 family, only the structure of Aga50D from Saccharophagus degradans has been studied (Pluvinage et al. 2013). Its possible catalytic residues are Glu-534 and Glu-695. Compared with these catalytic residues, the possible catalytic residues of AgaB-4 are Glu-455 and Glu-617, indicating a remarkable conservation of catalytic residues. Additional studies should evaluate the structure and function of AgaB-4 to understand its possible catalytic mechanism.
The production of agaro-oligosaccharides or neoagaro-oligosaccharides by using the enzymatic hydrolysis method has several advantages over the acid hydrolysis method (Chen et al. 2004, 2005; Yang et al. 2009). The advantages include (1) enzymes selectively cleave specific glycosidic bonds to produce oligosaccharides with a specific degree of polymerization; (2) the degradation conditions are easier to control; (3) the optimal temperature for enzyme activity is lower than the optimal temperature for acid hydrolysis, reducing energy consumption; (4) the operation process of the enzymatic hydrolysis method is simpler than that of the acid hydrolysis method, without the need for reactions between acid and alkali and desalination; (5) no acids are required for the enzymatic hydrolysis method; thus, this method is safer and less likely to pollute the environment; (6) and enzymatic hydrolysis can produce agaro-oligosaccharides or neoagaro-oligosaccharides (Fu and Kim 2010). Acid hydrolysis can only produce agaro-oligosaccharides and cannot produce products with a uniform degree of polymerization; the polymerization degree is approximately 2–22 (Chen et al. 2004). However, the agarase used in the industry must exhibit high activity and stability at a temperature higher than the agar gelling temperature (1.5% of agar solution solidifies at 32–43 °C). In the present study, rAgaB-4 exhibited high activity and stability at 45 °C. Therefore, it can be used to produce neoagarotetraose.
Studies have shown that neoagarotetraose exhibits various biological activities, and because of its nontoxic properties, neoagarotetraose can be used in cosmetic, health food, and pharmaceutical industries (Jang et al. 2009; Hong et al. 2017b). For example, Jang et al. (2009) pointed out that 0.1 μg/mL neoagarotetraose could reduce the melanin content in murine melanoma B16F10 cells and could inhibit tyrosinase activity in B16F10 cells and mushroom tyrosinase activity in vitro; thus, it is a potential skin-whitening agent. Zhang et al. (2017) reported that neoagarotetraose could protect mice from intense exercise-induced fatigue damage by regulating the composition and function of intestinal microbes, and that it could be used as an ingredient in health foods. Wang et al. (2017) demonstrated that neoagarotetraose is a potential anti-inflammatory agent because it inhibits anti-inflammatory reactions in LPS-induced macrophages. In the future, we will use rAgaB-4 to hydrolyze agar and further investigate whether neoagarotetraose exhibits other biological active properties, such as anticancer and immunomodulatory activities.
The present study focused on AgaB-4 from P. agarexedens BCRC 17346. The results showed that rAgaB-4 exhibits β-agarase activity and can be used for the recovery of DNA from agarose gel and for the production of neoagarotetraose. Future research should focus on the cloning and expression of agaB-1, agaB-2, and agaB-3 from P. agarexedens BCRC 17346 and should also evaluate whether recombinant proteins exhibit agarase activity.
Abbreviations
- rAgaB-4:
-
recombinant AgaB-4
- GH:
-
glycoside hydrolase
- BCRC:
-
Bioresource Collection and Research Center
- LB:
-
Luria–Bertani
- PCR:
-
polymerase chain reaction
- BLAST:
-
basic local alignment search tool
- OD600 :
-
optical density at 600 nm
- IPTG:
-
isopropyl-β-d-thiogalactopyranoside
- SDS-PAGE:
-
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- LMP:
-
low-melting point
- DNS:
-
3,5-Dinitrosalicylic acid
- EDTA:
-
ethylenediaminetetraacetic acid
- HMP:
-
high-melting point
- TLC:
-
thin layer chromatography
- CAZy:
-
carbohydrate-active enzymes
References
Aoki T, Araki T, Kitamikado M (1990) Purification and characterization of a novel β-agarase from Vibrio sp. AP-2. Eur J Biochem 187:461–465. https://doi.org/10.1111/j.1432-1033.1990.tb15326.x
Araki T, Lu Z, Morishita T (1998) Optimization of parameters for isolation of protoplasts from Gracilaria verrucosa (Rhodophyta). J Mar Biotechnol 6:193–197
Armisén R (1991) Agar and agarose biotechnological applications. Hydrobiologia 221:157–166. https://doi.org/10.1007/BF00028372
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29. https://doi.org/10.1038/75556
Besemer J, Borodovsky M (1999) Heuristic approach to deriving models for gene finding. Nucleic Acids Res 27:3911–3920. https://doi.org/10.1093/nar/27.19.3911
Chen HM, Zheng L, Lin W, Yan XJ (2004) Product monitoring and quantitation of oligosaccharides composition in agar hydrolysates by precolumn labeling HPLC. Talanta 64:773–777. https://doi.org/10.1016/j.talanta.2004.04.002
Chen HM, Zheng L, Yan XJ (2005) The preparation and bioactivity research of agaro-oligosaccharides. Food Technol Biotechnol 43:29–36
Chen HM, Yan XJ, Zhu P, Lin J (2006) Antioxidant activity and hepatoprotective potential of agaro-oligosaccharides in vitro and in vivo. Nutr J 5:31. https://doi.org/10.1186/1475-2891-5-31
Chi WJ, Chang YK, Hong SK (2012) Agar degradation by microorganisms and agar-degrading enzymes. Appl Microbiol Biotechnol 94:917–930. https://doi.org/10.1007/s00253-012-4023-2
Dong JH, Tamaru Y, Araki T (2007) Molecular cloning, expression, and characterization of a β-agarase gene, agaD, from a marine bacterium, Vibrio sp. strain PO-303. Biosci Biotechnol Biochem 71:38–46. https://doi.org/10.1271/bbb.60304
Finkelstein M, Rownd TH (1978) A rapid method for extracting DNA from agarose gels. Plasmid 1:557–562. https://doi.org/10.1016/0147-619X(78)90012-4
Fu XT, Kim SM (2010) Agarase: review of major sources, categories, purification method, enzyme characteristics and applications. Mar Drugs 8:200–218. https://doi.org/10.3390/md8010200
Fu XT, Lin H, Kim SM (2008) Purification and characterization of a novel β-agarase, AgaA34, from Agarivorans albus YKW-34. Appl Microbiol Biotechnol 78:265–273. https://doi.org/10.1007/s00253-007-1303-3
Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G (2010) Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464:908–912. https://doi.org/10.1038/nature08937
Hehemann JH, Correc G, Thomas F, Bernard T, Barbeyron T, Jam M, Helbert W, Michel G, Czjzek M (2012) Biochemical and structural characterization of the complex agarolytic enzyme system from the marine bacterium Zobellia galactanivorans. J Biol Chem 287:30571–30584. https://doi.org/10.1074/jbc.M112.377184
Hong SJ, Lee JH, Kim EJ, Yang HJ, Park JS, Hong SK (2017a) Anti-obesity and anti-diabetic effect of neoagarooligosaccharides on high-fat diet-induced obesity in mice. Mar Drugs 15:90. https://doi.org/10.3390/md15040090
Hong SJ, Lee JH, Kim EJ, Yang HJ, Park JS, Hong SK (2017b) Toxicological evaluation of neoagarooligosaccharides prepared by enzymatic hydrolysis of agar. Regul Toxicol Pharmacol 90:9–21. https://doi.org/10.1016/j.yrtph.2017.08.001
Hu B, Gong QH, Wang Y, Ma YM, Li JB, Yu WG (2006) Prebiotic effects of neoagaro-oligosaccharides prepared by enzymatic hydrolysis of agarose. Anaerobe 12:260–266. https://doi.org/10.1016/j.anaerobe.2006.07.005
Hu Z, Lin BK, Xu Y, Zhong MQ, Liu GM (2009) Production and purification of agarase from a marine agarolytic bacterium Agarivorans sp. HZ105. J Appl Microbiol 106:181–190. https://doi.org/10.1111/j.1365-2672.2008.03990.x
Jang MK, Lee DG, Kim NY, Yu KH, Jang HJ, Lee SW, Jang HJ, Lee YJ, Lee SH (2009) Purification and characterization of neoagarotetraose from hydrolyzed agar. J Microbiol Biotechnol 19:1197–1200. https://doi.org/10.4014/jmb.0906.06045
Kim HT, Lee SY, Lee DH, Kim HS, Bang WG, Kim KH, Choi IG (2010) Overexpression and molecular characterization of Aga50D from Saccharophagus degradans 2-40: an exo-type β-agarase producing neoagarobiose. Appl Microbiol Biotechnol 86:227–234. https://doi.org/10.1007/s00253-009-2256-5
Kirimura K, Masuda N, Iwasaki Y, Nakagawa H, Kobayashi R, Usami S (1999) Purification and characterization of a novel β-agarase from an alkalophilic bacterium, Alteromonas sp. E-1. J Biosci Bioeng 87:436–441. https://doi.org/10.1016/S1389-1723(99)80091-7
Knutsen SH, Myslabodski DE, Larsen B, Usov AI (1994) A modified system of nomenclature for red algal galactans. Bot Mar. 37:163–169. https://doi.org/10.1515/botm.1994.37.2.163
Kobayashi R, Takisada M, Suzuki T, Kirimura K, Usami S (1997) Neoagarobiose as a novel moisturizer with whitening effect. Biosci Biotechnol Biochem 61:162–163. https://doi.org/10.1271/bbb.61.162
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. https://doi.org/10.1038/227680a0
Lee MH, Jang JH, Yoon GY, Lee SJ, Lee MG, Kang TH, Han HD, Kim HS, Choi WS, Park WS, Park YM, Jung ID (2017) Neoagarohexaose-mediated activation of dendritic cells via Toll-like receptor 4 leads to stimulation of natural killer cells and enhancement of antitumor immunity. BMB Rep 50:263–268. https://doi.org/10.5483/bmbrep.2017.50.5.014
Li J, Sha YJ, Seswita-Zilda D, Hu QS, He PQ (2014) Purification and characterization of thermostable agarase from Bacillus sp. BI-3, a thermophilic bacterium isolated from hot spring. J Microbiol Biotechnol 24:19–25. https://doi.org/10.4014/jmb.1308.08055
Li G, Sun MM, Wu J, Ye M, Ge XC, Wei W, Li HX, Hu F (2015) Identification and biochemical characterization of a novel endo-type β-agarase AgaW from Cohnella sp. strain LGH. Appl Microbiol Biotechnol 99:10019–10029. https://doi.org/10.1007/s00253-015-6869-6
Lin BK, Lu GY, Zheng YD, Xie W, Li SK, Hu Z (2012) Gene cloning, expression and characterization of a neoagarotetraose-producing β-agarase from the marine bacterium Agarivorans sp. HZ105. World J Microbiol Biotechnol 28:1691–1697. https://doi.org/10.1007/s11274-011-0977-y
Liu N, Mao XZ, Du ZJ, Mu BZ, Wei DZ (2014) Cloning and characterisation of a novel neoagarotetraose-forming-β-agarase, AgWH50A from Agarivorans gilvus WH0801. Carbohydr Res 388:147–151. https://doi.org/10.1016/j.carres.2014.02.019
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. https://doi.org/10.1093/nar/gkt1178
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428. https://doi.org/10.1021/ac60147a030
Minegishi H, Shimane Y, Echigo A, Ohta Y, Hatada Y, Kamekura M, Maruyama T, Usami R (2013) Thermophilic and halophilic β-agarase from a halophilic archaeon Halococcus sp. 197A. Extremophiles 17:931–939. https://doi.org/10.1007/s00792-013-0575-z
Ohta Y, Hatada Y, Miyazaki M, Nogi Y, Ito S, Horikoshi K (2005) Purification and characterization of a novel α-agarase from a Thalassomonas sp. Curr Microbiol 50:212–216. https://doi.org/10.1007/s00284-004-4435-z
Pluvinage B, Hehemann JH, Boraston AB (2013) Substrate recognition and hydrolysis by a family 50 exo-β-agarase, Aga50D, from the marine bacterium Saccharophagus degradans. J Biol Chem 288:28078–28088. https://doi.org/10.1074/jbc.M113.491068
Potin P, Richard C, Rochas C, Kloareg B (1993) Purification and characterization of the α-agarase from Alteromonas agarlyticus (Cataldi) comb. nov., strain GJ1B. Eur J Biochem 214:599–607. https://doi.org/10.1111/j.1432-1033.1993.tb17959.x
Sie YF, Yang HC, Lee Y (2009) The discoveery of agarolytic bacterium with agrarse gene containing plasmid, and sone enzymology characteristics. Int J App Sci Eng 1:25–41
Song T, Cao Y, Xu H, Zhang WJ, Fei BJ, Qiao DR, Cao Y (2014) Purification and characterization of a novel β-agarase of Paenibacillus sp. SSG-1 isolated from soil. J Biosci Bioeng 118:125–129. https://doi.org/10.1016/j.jbiosc.2014.02.008
Sugano Y, Terada I, Arita M, Noma M, Matsumoto T (1993) Purification and characterization of a new agarase from a marine bacterium, Vibrio sp. strain JT0107. Appl Environ Microbiol 59:1549–1554
Uetanabaro AP, Wahrenburg C, Hunger W, Pukall R, Spröer C, Stackebrandt E, de Canhos VP, Claus D, Fritze D (2003) Paenibacillus agarexedens sp. nov., nom. rev., and Paenibacillus agaridevorans sp. nov. Int J Syst Evol Microbiol 53:1051–1057. https://doi.org/10.1099/ijs.0.02420-0
Vera J, Alvarez R, Murano E, Slebe JC, Leon O (1998) Identification of a marine agarolytic Pseudoalteromonas isolate and characterization of its extracellular agarase. Appl Environ Microbiol 64:4378–4383
Wang W, Liu P, Hao C, Wu LJ, Wan WJ, Mao XZ (2017) Neoagaro-oligosaccharide monomers inhibit inflammation in LPS-stimulated macrophages through suppression of MAPK and NF-κB pathways. Sci Rep 7:44252. https://doi.org/10.1038/srep44252
Wu SC, Pan CL (2004) Preparation of algal-oligosaccharide mixtures by bacterial agarases and their antioxidative properties. Fish Sci 70:1164–1173. https://doi.org/10.1111/j.1444-2906.2004.00919.x
Xie W, Lin BK, Zhou ZR, Lu GY, Lun JS, Xia CY, Li SK, Hu Z (2013) Characterization of a novel β-agarase from an agar-degrading bacterium Catenovulum sp. X3. Appl Microbiol Biotechnol 97:4907–4915. https://doi.org/10.1007/s00253-012-4385-5
Yang B, Yu GL, Zhao X, Jiao GL, Ren SM, Chai WG (2009) Mechanism of mild acid hydrolysis of galactan polysaccharides with highly ordered disaccharide repeats leading to a complete series of exclusively odd-numbered oligosaccharides. FEBS J 276:2125–2137. https://doi.org/10.1111/j.1742-4658.2009.06947.x
Yun EJ, Yu S, Kim KH (2017) Current knowledge on agarolytic enzymes and the industrial potential of agar-derived sugars. Appl Microbiol Biotechnol 101:5581–5589. https://doi.org/10.1007/s00253-017-8383-5
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Brujin graphs. Genome Res 18:821–829. https://doi.org/10.1101/gr.074492.107
Zhang N, Mao XZ, Li RW, Hou EL, Wang YM, Xue CH, Tang QJ (2017) Neoagarotetraose protects mice against intense exercise-induced fatigue damage by modulating gut microbial composition and function. Mol Nutr Food Res 61:1600585. https://doi.org/10.1002/mnfr.201600585
Authors’ contributions
ZWC and JPW conceived and designed the study. HJL and WCH performed experiments, analyzed and interpreted data, and prepared figures and tables. All authors participated in discussion of the experimental results. ZWC and JPW wrote the manuscript. SLH, JHL, and JPW reviewed and edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This manuscript was edited by Wallace Academic Editing.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional information files.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This study was funded by Council of Agriculture, Executive Yuan, Taiwan, ROC (grant number 106AS-12.2.1-ST-a1).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Figure S1.
Multiple amino acid sequence alignment of AgaB-4 with known β-agarases from the GH50 family, including AgaW from Cohnella sp. LGH, Aga50D from Saccharophagus degradans 2-40, AgWH50A from Agarivorans gilvus WH0801, and HZ2 from Agarivorans sp. HZ105. A partially conserved catalytic residue of GH50 family is underlined in blue. Filled triangles indicate the active sites of Aga50D. Figure S2. SDS-PAGE analysis of total cell lysates (T), soluble (S), and insoluble (I) protein fractions from E. coli BL21 (DE3)(pET-AgaB-4) expressing rAgaB-4 after induction for 4 and 24 h at a 37 °C, b 30 °C, c 25 °C, d 20 °C, and e 16 °C with 0.1 mM IPTG added to the culture. Lane M, PageRuler™ Prestained Protein Ladder. The arrow indicates the protein bands of rAgaB-4. Figure S3. Recovery of pUC19 from low-melting point agarose by rAgaB-4. Lane M, 1-kb DNA Ladder; lane 1, original pUC19; lane 2, recovered pUC19 from low-melting point agarose.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chen, ZW., Lin, HJ., Huang, WC. et al. Molecular cloning, expression, and functional characterization of the β-agarase AgaB-4 from Paenibacillus agarexedens. AMB Expr 8, 49 (2018). https://doi.org/10.1186/s13568-018-0581-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-018-0581-8