
Abstract
Background
The mitochondrial DNA (mtDNA) control region is extensively used in the phylogeography of species. We examined sequence variations in the mtDNA control region of sambar (Rusa unicolor) populations from the South, Central and North India.
Results
Most of the samples collected from the south India exhibited a 40 bp insertion in the mtDNA control region. This insertion was not observed in the North and Central Indian populations.
Conclusion
This study provided a potential marker for molecular screening and identification of sambar populations in the form of a distinct 40 bp insertion. Some populations in South India did not exhibit this insertion. It indicates that there could be an ecological barrier that might be preventing the expansion of insertion-positive sambar population.
Similar content being viewed by others


Background
The sambar (Rusa unicolor) is the largest cervid species in Southeast Asia. In India, it is widely distributed from the Himalayan foothills to the southern limits. Seven subspecies of sambar are recognised, with R. u. unicolor occurring in India and Sri Lanka [1]. Despite a large distribution range, very limited information is available on genetic variations for this species.
The mitochondrial DNA (mtDNA) control region has been used extensively in studying the population genetics of wild species, for example the tiger, Panthera tigris [2]; wild pig, Sus scrofa [3]; cervids [4]; sika deer, Cervus nippon [5]; roe deer, Capreolus capreolus [6]; white-tailed deer, Odocoileus virginianus [7]; Chinese water deer, Hydropotes inermis inermis [8] and black muntjac, Muntiacus crinifrons [9]. In the present study, we investigated genetic variations among sambar populations of selected zones of India. Sequences of the mtDNA hypervariable region I (HVR-I) were compared among the sambar populations of the South, Central and North India for evaluation of genetic variation.
Results
Overall, the mtDNA HVR-I region demonstrated high variability among sambar populations, and 26 distinct haplotypes were identified (Table 1). PCR amplifications that were 40 bp longer than the expected size were obtained with DNA extracted from 23 samples from South India (Fig. 1). Three samples from South India and all 38 samples from Central and North India yielded amplifications of the expected size. It indicated that the majority of the animals of the South Indian population have a unique feature in the mtDNA control region. We obtained 543–623 bp sequences of the control region using a cervid specific primer pair. After alignment of the sequences, 23 samples of South India exhibited a unique 40 bp insertion after nucleotide (nt) position 233 (Table 1). This insertion in a majority of the South Indian samples indicated that there is a significant genetic variation in this population. Four out of the 23 insertion-positive south Indian samples were collected from a confined population from a zoological park in Goa. The 40 bp insertion was absent in three out of eight samples collected from Kalakkad-Mundunthurai Tiger Reserve (KMTR), Tamil Nadu. The 40 bp insertion was not observed in the samples of central and north Indian populations. Therefore, the 40 bp deletion in the control region after nt position 233 appears to be a potential marker for genetic screening and identification of Central and North Indian populations. The haplotype (RUC1–RUC26) sequences were submitted to GenBank (accession numbers KF133981-99 and KF648589-95, Table 1). Haplotypes RUC1–RUC5, RUC8–RUC15, RUC20 and RUC21 were molecular signatures of the insertion-positive South Indian population. The most frequent haplotype was RUC17, which was observed in 24 individuals from Central and North India. Haplotypes RUC2 and RUC18 were observed in four individuals from the South and Central India and RUC19 in three individuals from Central India (Table 1). The pairwise distance and percentage similarity matrices showed wide ranges of 0.002–0.113 and 91.60–99.83, respectively (Table 2). We observed 62 polymorphic sites (S) among 26 haplotypes. The average number of nucleotide differences (k) and nucleotide diversity (π) were 24.00615 and 0.04479, respectively. Median-joining (MJ) network tree exhibited a distinct clustering of insertion-positive (green colour) and insertion-negative (red colour) populations (Fig. 2).

Gel image showing 40 bp longer PCR amplification from the samples of South India (lanes 1–4) and shorter amplification from the samples of Central and North India (lanes 5–7). Lane M is 100 bp DNA ladder

Median-joining (MJ) network tree demonstrating distinct clustering of 40 bp insertion positive (green) and negative (red) haplotypes. Haplotype numbers are given in the circle and described in Table 1
Discussion and conclusions
In several studies, insertion–deletion (INDEL) markers have been used in population genetics and forensics [10–12] as it is easy to use. In the present study, we used INDEL to describe a unique 40 bp insertion in the HVR-I (control) region that is found mostly in the sambar population from the South India. This unique molecular feature differentiated it from the other populations of sambar from India suggesting further investigation. It is apparent from genetic data that INDEL has a significant role in population structuring in sambar.
This study provided further insight into the genetic makeup of sambar. Analysis of samples collected from various parts of India revealed a high level of genetic variations among different populations. The percentage similarity and pairwise distance indicate that the insertion-positive haplotypes (RUC1–RUC5, RUC8–RUC15, RUC20 and RUC21) were significantly related and had a higher genetic distance from the other haplotypes (Table 2). We suspect that there could be a possible ecological barrier operating, which is separating majority of the South Indian population from the North and Central India. The 40 bp INDEL in the mtDNA region is a rapid marker for genetic screening and identification of these populations using a simple PCR and sequencing-based analysis.
Methods
Sampling and DNA extraction
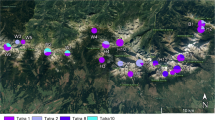
A total of 64 biological samples (54 tissue samples and ten fecal samples) were used in this study. Of these, 26 were from South India, and 38 were from Central and North India (Fig. 3). Out of the 64 samples, 36 had been seized from poachers and forwarded by enforcement agencies to us for forensic validation. The confiscated biological samples were validated for species through sequence analysis of the mtDNA cytochrome b gene [13]. Biological samples were also collected from national parks and captive populations of sambar after obtaining permission from the Principal Chief Conservation of Forests (Wildlife) and Chief Wildlife Warden of the States of Uttarakhand, Madhya Pradesh, Andhra Pradesh, Kerala, Tamil Nadu, Karnataka and Goa. International regulations such as the Convention on the Trade in Endangered Species of Wild Fauna and Flora (CITES, http://www.cites.org) and Convention on Biological Diversity (CBD, http://www.cbd.int/convention) were also complied with during the study. High standards were maintained during sample collection, and no animals were harmed during this study. Six tissue samples and one shed antler sample were collected from predator kills from Panna Tiger Reserve, Madhya Pradesh. Seven and four individual’s hair samples were collected from confined populations in deer parks in Andhra Pradesh and Goa, respectively. These hair samples were collected from the metal grills and concrete wall of the captive area. Ten faecal samples were collected from forests in Kerala and Tamil Nadu. Since no animal handling was involved, approval of animal ethical committees was not required. Whole genomic DNA was extracted using phenol–chloroform [14]. A standardized protocol developed for extraction of DNA from non-invasive sampling was followed [15].

Locations of sampling site are shown with square from south India, which has 40 bp insertion, and triangle for populations from central and north India those do not have this insertion. Gray and yellow sheds represent tentative sites for insertion positive and negative populations, respectively. Source of the base map of India was Survey of India, Government of India and it was procured by Wildlife Institute of India for academic research and illustration purpose
PCR amplification
The DNA extracted from above samples was used for PCR amplification of an approximately 600 bp long sequence of the mtDNA control region. A primer pair specific to the family Cervidae (deer) was used [10]. The sequences of the primer are CervtPro “CCACYATCAACACCCAAAGC” and CervCRH “GCCCTGAARAAAGAACCAGATG”. PCR reactions were carried out in 20 µl reaction volumes using 1 × PCR buffer (10 mM Tris–HCl, pH 8.3, and 50 mM KCl), 1.5 mM MgCl2, 1 × BSA, 100 µM dNTPs, 4 pmol of each primer, 0.5 U AmpliTaq Gold DNA polymerase enzyme (Invitrogen Inc.) and 1 µl (~30 ng) of template DNA. The PCR conditions were initial denaturation at 95 °C for 10 min, followed by 35 cycles of denaturation at 95 °C for 45 sec, annealing at 54 °C for 40 sec and extension at 72 °C for 75 sec. The final extension was at 72 °C for 10 min. The efficiency and reliability of the PCR reactions were monitored using control reactions. PCR amplification was confirmed by electrophoresis on 2.2 % agarose gel stained with ethidium bromide (0.5 mg/ml) and visualised under a UV transilluminator.
DNA sequencing and analysis
The PCR products were treated with exonuclease-I and shrimp alkaline phosphatase for 15 min each at 37 and 80 °C. A BigDye terminator kit (version3.1) and an ABI 3130 Genetic Analyzer (Applied Biosystems) were used to generate DNA sequences from both the directions. The generated sequences were aligned by eye using Clustal W [16], available in the BioEdit package [17]. Since the primer pair used in this study amplifies approximately 40–50 bases of Proline tRNA at the beginning of the PCR reaction, the initial sequence of Proline tRNA was deleted from the aligned sequences. Hence, the start sequence examined in this study was the first nucleotide of the control region. The aligned sequences of the control region were subjected to haplotype analysis using DnaSP [18] (Table 1). A percentage similarity matrix and pairwise distance matrix were generated using Clustal W [16] and MEGA 5 [19] (Table 2). Network 4.613 software (http://www.fluxus-engineering.com) was used to generate median-joining network tree (Fig. 3).
Abbreviations
- CBD:
-
Convention on Biological Diversity
- CITES:
-
Convention on the Trade in Endangered Species of Wild Fauna and Flora
- HVR-I :
-
hypervariable region I
- INDEL:
-
insertion–deletion
- KMTR:
-
Kalakkad-Mundunthurai Tiger Reserve
- MJ:
-
median-joining
- mtDNA:
-
mitochondrial DNA
- nt:
-
nucleotide
References
Leslie DM. Rusa unicolor (Artiodactyla: Cervidae). Mamm Species. 2011;43:1–30.
Luo SJ, Kim JH, Johnson WE, van der Walt J, Martenson J, Yuhki N, Miquelle DG, Uphyrkina O, Goodrich JM, Quigley HB, Tilson R, Brady G, Martelli P, Subramaniam V, McDougal C, Hean S, Huang S-Q, Pan W, Karanth UK, Sunquist M, Smith JLD, O’Brien SJ. Phylogeography and genetic ancestry of tigers (Panthera tigris). PLoS Biol. 2004;2:2275–93.
Larson G, Dobney K, Albarella U, Matisoo-Smith E, Robins J, Lowden S, Rowley-Conwy P, Andersson L, Cooper A. Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Sci. 2005;307:1618–21.
Douzery E, Randi E. The mitochondrial control region of Cervidae: evolutionary patterns and phylogenetic content. Mol Bio Evol. 1997;14:1154–66.
Cook CE, Wang Y, Sensabaugh G. A Mitochondrial control region and cytochromeb phylogeny of sika deer (Cervus nippon) and report of tandem repeats in the control region. Mol Phylo Evol. 1999;12:47–56.
Vernesi C, Pecchioli E, Caramelli D, Tiedemann R, Randi E, Bertorelle G. The genetic structure of natural and reintroduced roe deer (Capreolus capreolus) populations in the Alps and central Italy, with reference to the mitochondrial DNA phylogeography of Europe. Mol Ecol. 2002;11:1285–97.
Moscarella RA, Aguilera M, Escalante AA. Phylogeography, population structure, and implications for conservation of white-tailed deer (Odocoileus virginianus) in Venezuela. J Mammal. 2003;84:1300–15.
Hu J, Fang SG, Wan QH. Genetic diversity of Chinese water deer (Hydropotes inermis inermis): implications for conservation. Biochem Genet. 2006;44:161–72.
Wu HL, Fang SG. Mitochondrial DNA genetic diversity of black muntjac (Muntiacus crinifrons), an endangered species endemic to China. Biocheml Genet. 2005;43:407–16.
Balakrishnan CN, Monfort SL, Gaur A, Singh L, Sorenson MD. Phylogeography and conservation genetics of Eld’s deer (Cervus eldi). Mol Ecol. 2003;12:1–10.
Thangaraj K, Sridhar V, Kivisild T, Reddy AG, Chaubey G, Singh VK, Kaur S, Agarawal P, Rai A, Gupta J, Mallick CB, Kumar N, Velavan TP, Suganthan R, Udaykumar D, Kumar R, Mishra R, Khan A, Annapurna C, Singh L. Different population histories of the Mundariand Mon-Khmer-speaking Austro-Asiatic tribes inferred from the mtDNA 9-bp deletion/insertion polymorphism in Indian populations. Hum Genet. 2005;116:507–17.
da Costa Francez PA, Rodrigues EM, de Velasco AM, dos Santos SE. Insertion–deletion polymorphisms—utilization on forensic analysis. Int J Leg Med. 2012;126:491–6.
Gupta SK, Kumar A, Hussain SA. Novel primers for sequencing of the complete mitochondrial cytochrome b gene of ungulates using non-invasive and degraded biological samples. Conser Genet Res. 2014;6:499–501.
Sambrook J, Fritsch EF, Maniatis TL. Molecular cloning: a laboratory manual, vol. 2nd edn. New York: Cold Spring Harbor Press; 1989. p. 40–1.
Gupta SK, Kumar A, Hussain SA. Extraction of PCR-amplifiable DNA from a variety of biological samples with uniform success rate. Conser Genet Res. 2013;5:215–7.
Thompson JD, Higgins DG, Gibson TJ. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–8.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Bio Evol. 2011;28:2731–9.
Authors’ contributions
SKG and SAH conceived and designed this study. SKG, AK and AG carried out the sample collection. SKG and AK performed sequencing. SKG executed statistical analysis. All authors read and approved the final manuscript.
Acknowledgements
This work was funded by the WII and Scientific & Engineering Research Board, Department of Science and Technology, Govt. of India. We thank the Director and Dean, WII for their support. We also thank Dr. S. Shivaji, Former Outstanding Scientist, CCMB; Dr. Y.V. Jhala, Scientist G and Prof. Qamar Qureshi, Scientist G of WII for their support. The IT and RS & GIS Cell at WII is acknowledged for their support in the preparation of map. We sincerely acknowledge the help of Forest Departments of Kerala, Madhya Pradesh, Uttarakhand, Goa, Karnataka, Andhra Pradesh, Rajasthan, Assam and Tamil Nadu States for facilitating the sample collection.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gupta, S.K., Kumar, A., Gaur, A. et al. Detection of 40 bp insertion-deletion (INDEL) in mitochondrial control region among sambar (Rusa unicolor) populations in India. BMC Res Notes 8, 581 (2015). https://doi.org/10.1186/s13104-015-1573-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-015-1573-2