
Abstract
Background
Large population-based translational epigenetic studies are emerging due to recent technological advances that have made molecular analyses possible. For example, the Infinium HumanMethylation450 Beadchip (HM450K) has enabled studies of genome-wide methylation on a scale not previously possible. However, application of the HM450K to DNA extracted from formalin-fixed paraffin-embedded (FFPE) tumour material has been more challenging than application to high quality DNA extracted from blood. To facilitate the application of this assay consistently across a large number of FFPE tumour-enriched DNA samples we have devised a modification to the HM450K protocol for FFPE that includes an additional quality control (QC) checkpoint.
Results
QC checkpoint 3 was designed to assess the presence of DNA after bisulfite conversion and restoration, just prior to application of the HM450K assay. DNA was extracted from 474 archival FFPE breast tumour material. Five samples did not have a detectable amount of DNA with an additional 42 failing to progress past QC checkpoint 3. Genome-wide methylation was measured for the remaining 428 tumour-enriched DNA. Of these, only 4 samples failed our stringent HM450K data criteria thus representing a 99 % success rate. Using prior knowledge about methylation marks associated with breast cancer we further explored the quality of the data. Twenty probes in the BRCA1 promoter region showed increased methylation in triple-negative breast cancers compared to Luminal A, Luminal B and HER2-positive breast cancer subtypes. Validation of this observation in published data from The Cancer Genome Atlas (TCGA) Network (obtained from DNA extracted from fresh frozen tumour samples) confirms the quality of the data obtained from the improved protocol.
Conclusions
The modified protocol is suitable for the analysis of FFPE tumour-enriched DNA and can be systematically applied to hundreds of samples. This protocol will have utility in population-based translational epigenetic studies and is applicable to a wide variety of translated studies interested in analysis of methylation and its role in the predisposition to disease and disease progression.
Similar content being viewed by others


Background
Recent technological advances have made genome-wide studies of epigenetics possible on a scale suitable for large population-based studies (akin to what SNP chips enabled for genome-wide association studies). For example, the Infinium HumanMethylation450 Beadchip (HM450K) has enabled studies of genome-wide methylation on a scale not previously possible. However, application of the HM450K to DNA extracted from formalin-fixed paraffin-embedded (FFPE) tumour material has been more challenging than application to high quality DNA extracted from blood.
Archival FFPE tumour material represents a precious resource for many large epidemiological studies. However, the use of this material is often challenging as DNA extracted from FFPE tumour material is frequently of low quantity and highly degraded [1], thus making it difficult to obtain consistent molecular analysis across a large number of samples.
Genome-wide detection methodologies based on next-generation sequencing and microarrays applied to FFPE tumour-enriched DNA have the potential to address many of the current significant research questions being pursued in translational epigenetic studies. However, most of these platforms require large amounts of high quality DNA. Alternate protocols are therefore needed to address the issues of DNA quantity and quality associated with DNA extracted from FFPE tumour material so that the potential of combining these platforms and resources can be realised for translational studies.
We have previously shown that DNA extracted from dried blood spots can be successfully applied to the HM450K platform. High quality and reproducible results were obtained from DNA extracted from matched archival dried blood spot and frozen buffy coat (correlation coefficient r > 0.99) [2]. Although more recent studies have demonstrated the reliable application of the HM450K platform to FFPE tumour-enriched DNA compared with DNA extracted from fresh frozen material [3, 4], they do not address the issues faced by researchers wanting to apply this assay to large population studies. Moran and colleagues measured methylation on the HM450K platform in DNA extracted from matched fresh frozen tumour and newly fixed FFPE tumour material. This work addressed the effect of formalin but it did not consider the possible effect that long term storage of FFPE tumour material might have on DNA quality and assay output [3]. This issue was considered by Dumenil and colleagues who compared DNA extracted from fresh frozen material with FFPE tumour-enriched DNA that was stored between 4 and 19 years (storage conditions unspecified). Based on the signal of the 3000 most differential probes between the two DNA types, they found that the difference in methylation signals between FFPE tumour-enriched DNA and DNA extracted from fresh frozen material correlated with the length of storage time [4].
In both studies, FFPE tumour-enriched DNA was restored with the recommended Infinium HD FFPE Restore protocol (Illumina, San Diego, CA, USA) that has been shown to improve the performance of FFPE tumour-enriched DNA on the HM450K [5, 6]. Highly correlated methylation values were observed between DNA extracted from matched fresh frozen and restored FFPE tumour-enriched DNA compared with unrestored FFPE tumour-enriched DNA (r > 0.91 vs r > 0.81). Additionally, highly reproducible data were obtained from restored FFPE tumour-enriched DNA (r = 0.99) compared with their unrestored counterparts (r = 0.90) [5]. The same observations were reported when Siegel and colleagues compared the restoration protocol to an alternate ligation method (using the REPLI-g ligase) in DNA extracted from FFPE tumour material and matched fresh frozen material [6].
All four studies described above used relatively moderate quantities of DNA (100–500 ng) as starting material for bisulfite conversion, followed by the Infinium restoration and HM450K protocols [3–6]. However, obtaining this amount of DNA is challenging to achieve consistently across a large number of archival FFPE tumour samples. Further, these reports did not measure the presence of FFPE tumour-enriched DNA after bisulfite conversion and restoration, making the subsequent performance of these samples on the HM450K platform unpredictable.
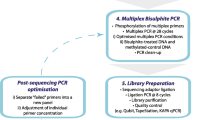
To address these issues, we adopted a modified protocol (Fig. 1) based on the recommended Illumina Infinium workflow for FFPE tumour-enriched DNA which incorporated a third quality control (QC) checkpoint in the form of an in-house designed bisulfite-specific qPCR assay to assess the capacity of the protocol to support large population-based translation epigenetics studies.

Workflow diagram for assessing the suitability of FFPE tumour-enriched DNA on the HM450K platform. A novel checkpoint (QC checkpoint 3) has been incorporated in addition to the standard protocol to assess DNA availability after sodium bisulfite modification and restoration. The number of samples entering and exiting each QC checkpoint is as indicated
Methods
Melbourne Collaborative Cohort Study
The study was performed on material from 474 breast cancer affected female participants of the Melbourne Collaborative Cohort Study (MCCS). The MCCS is a prospective study of more than 40,000 men and women aged 40–69 years at baseline who were recruited between the years 1990–1994 [7]. Pathology material related to each breast cancer case had previously been retrieved from the diagnostic service laboratory and reviewed by qualified pathologists. Representative haematoxylin and eosin (H&E) stained sections and unstained sections (3 μm) had been prepared and stored desiccated at 4 °C for up to 15 years. Immunohistochemical staining and breast cancer subtyping were conducted as described by Blows et al. [8]. Written informed consent was obtained from each participant and the study was approved by the Human Research Ethics Committee of Cancer Council Victoria [7].
DNA extraction from FFPE breast tumour material
The tumour area most suitable for macrodissection was identified by a qualified pathologist (CAM) and recorded by directly marking up the representative H&E stained section. Macrodissection of FFPE breast tumour material was performed on an average of two 3 μm unstained sections (corresponding to the marked H&E stained section). DNA was extracted using the QIAamp DNA FFPE protocol (Qiagen, Hilden, Germany) as per the manufacturer’s instructions except that the tumour material was incubated in Buffer ATL at 56 °C for 48 h, with 20 ul of Proteinase K (20 mg/ml) replenished at 0 and 24 h to increase the digestion of proteins. FFPE tumour-enriched DNA was eluted twice in Buffer ATE to achieve a final elution volume of 20 μl.
QC checkpoint 1: Qubit® dsDNA BR assay
The presence of FFPE tumour-enriched DNA was measured using the Qubit® dsDNA BR Assay kit on the Qubit® Fluorometer (Life Technologies, Carlsbad, CA, USA) as per manufacturer’s instructions. Samples with undetectable DNA amounts were not further progressed through the workflow.
QC checkpoint 2: Infinium HD FFPE QC assay
The quality of FFPE tumour-enriched DNA was assessed using the Infinium HD QC assay (Illumina, San Diego, CA, USA) on the LightCycler® 480 System (Roche, Basel, Switzerland) with subsequent data analysis performed as per the manufacturers’ instructions and as described previously [3, 4]. Taking into consideration the poor quality of our samples, the recommended ∆Cq threshold was relaxed so that samples that had ∆Cq values of ≤6 (instead of the recommended 5) were further processed [3, 4].
Sodium bisulfite conversion
FFPE tumour-enriched DNA samples (16–750 ng) were bisulfite converted using the EZ DNA Methylation-Gold kit (Zymo, Irvine, CA, USA), and eluted in a final volume of 8 μl elution buffer.
Restoration of bisulfite converted FFPE tumour-enriched DNA
Restoration of bisulfite converted FFPE tumour-enriched DNA was performed using the Infinium HD FFPE Restore kit (Illumina, San Diego, CA, USA) as per the manufacturer’s instructions and as previously described [3, 4] except that restored bisulfite converted samples were eluted in a final volume of 10 ul nuclease free water.
QC checkpoint 3: bisulfite-specific qPCR assay
The presence of FFPE tumour-enriched DNA after bisulfite conversion and restoration was determined using an in-house qPCR assay. Primers specific for bisulfite converted DNA were designed using EpiDesigner (http://www.epidesigner.com) (forward sequence: 5′ tAA GGT AtA ATt AGA GGA TGG GAG GGA t; reverse sequence: 5′ aaC AAA CTC Aaa TAa AAT TCT TCC TC) to amplify a 134 bp region (hg19:chr17:41277493-41277626) within the promoter of the breast cancer 1, early onset (BRCA1) gene (GenBank: L78833.1) [9]. Lower-case letters correspond to converted cytosines. The amplicon sequence is as follows: 5′ tAA GGT AtA ATt AGA GGA TGG GAG GGA tAG AAA GAG CCA AGC GTC TCT CGG GGC TCT GGA TTG GCC ACC CAG TCT GCC CCC GGA TGA CGT AAA AGG AAA GAG ACG GAA GAG GAA GAA TTt TAt tTG AGT TTG tt. CpG dinucleotides are in bold. The qPCR primers and the PCR conditions were developed to ensure that products be unaffected by the methylation status of the template (data not shown) [10].
Each reaction consisted of 1X SYBR Green I Master (Roche, Basel, Switzerland), 300 pM each of forward and reverse primers (Integrated DNA technologies, Coralville, IA), and 3 ul diluted restored bisulfite converted FFPE tumour-enriched DNA (diluted 1:3 in nuclease free water). The reaction was equilibrated to 10 μl with nuclease free water (Life Technologies, Carlsbad, CA, USA).
QPCR cycling conditions were as follows: initial polymerase activation for 5 min at 95 °C followed by 40 cycles of DNA denaturation for 10 s at 95 °C, primer annealing for 30 s at 60 °C and extension for 90 s at 72 °C. Subsequent melting of the amplified product was performed from 97 °C to 65 °C for 60 s. Fluorescent data was acquired on the green channel.
All samples were assayed in duplicate and non–bisulfite converted, unrestored U266 multiple myeloma cell line DNA was used as a negative control. Subsequent data analysis was performed similar to QC checkpoint 2 except that the difference in Cq value (ΔCq) was determined by subtracting the average Cq value of each FFPE tumour-enriched DNA sample from the average Cq value of the negative control (ΔCq= Average CqNegative control − Average CqTumour-enriched DNA). Using the Infinium-recommended ΔCq threshold for QC checkpoint 2 as a reference (Cq difference of 5) and taking into account the poor quality and limited quantity of FFPE tumour-enriched DNA, a more relaxed ΔCq threshold for this QC checkpoint was adopted. Only restored bisulfite converted FFPE tumour-enriched DNA with a ΔCq value of ≥4 was assayed on the HM450K platform.
Infinium HumanMethylation450 Beadchip assay
The Infinium HumanMethylation450 Beadchip assay (Illumina, San Diego, CA, USA) was performed as per manufacturer’s instructions specific for formalin-fixed embedded-material. The following replicates were included for every 48 samples to ensure the reproducibility of data: (1) one technical replicate from good quality bisulfite converted cell line DNA (U266 multiple myeloma cell line) to test for possible batch effects between different reagents and beadchips used, and different processing times and (2) one technical replicate from restored bisulfite converted FFPE tumour-enriched DNA, where each replicate was placed on a different beadchip to test for possible chip effects between beadchips.
Data analysis
Data from samples assayed on the HM450K platform were imported into the R environment (R Programming Software version 2.15.1) as previously described [2] and processed using the Minfi package version 1.10.1 [11]. The following criteria were applied to evaluate the overall data quality and performance of individual samples across all probes and individual probes within each sample, respectively: (1) average detection p-value across all probes of p ≤ 0.01; (2) individual probe detection p-value of p ≤ 0.05. Individual FFPE tumour-enriched DNA or probes that failed either criterion were removed from further analysis. Any sample with more than 1 % failed probes from a total of 485,512 probes were also classed as “failed” and excluded for the purposes of this analysis.
Analysis of TCGA HM450K methylation data
Subtype information and associated level 3 methylation signals (calculated beta values mapped to the genome) from 156 breast tumours were obtained from the TCGA Download Portal (https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp) [12]. The data consisted of the following subtypes based on the expression of 50 genes and classified using the prediction analysis of microarray (PAM): 86 Luminal A, 39 Luminal B, 22 basal-like and 9 HER2-enriched [13]. The mean beta value, standard deviation and 95 % confidence interval (CI) were calculated for each of the 48 methylation probes specific to the BRCA1 gene (5 additional probes were classified as ‘NA’ due to having detection p-values >0.05) across all tumours of the same subtype, resulting in 3 values per methylation probe for each subtype.
Results
DNA was macrodissected from marked up areas of 474 FFPE breast tumours. Five samples did not have a detectable amount of DNA at QC checkpoint 1. Of the remaining 469 FFPE tumour-enriched DNA samples that were progressed past QC checkpoint 2, 42 (8.93 %) samples failed to progress past QC checkpoint 3 with ∆Cq values ranging from 0 (no amplification) to 3.77. The methylome of 427 FFPE tumour-enriched DNA were subsequently evaluated on the HM450K platform (Fig. 1). Of these, 4 (<1 %) had an average detection p-value across all probes of ≥0.01 and were removed from further analysis. Based on our criteria, 5 % (24,816) of probes were removed, with 460,696 common probes remaining across 423 FFPE tumour-enriched DNA for downstream data analysis. The average detection p-value across these probes for all remaining samples (n = 423) was 4.54 × 10−4 (Fig. 2a) with high correlations observed between sample replicates (correlation coefficients r > 0.98) (Fig. 2b) thereby confirming the capacity of our protocol to generate reproducible, high quality data.

Post HM450K assay data quality checks of FFPE tumour-enriched DNA. a Average probe detection p-values across all probes for each sample. Each black dot represents a single sample. The average detection p–value across all probes for all samples (n = 423) was 4.54 × 10−4. b Scatterplots of replicates assayed on different beadchips (n = 11). The average correlation coefficient (rave) across all replicates was 0.993
We further evaluated the 4 failed samples on the HM450K platform and 16 randomly selected ‘successful’ samples at the QC checkpoints (Table 1). We did not observe a correlation between age of tumour material (8–20 years), performance at any of the QC checkpoints and subsequent performance on the HM450K platform.
Performance of internal control probes
We assessed the performance of sample-specific and sample-independent internal control probes present on the HM450K beadchip (http://www.illumina.com) between ‘failed’ and ‘successful’ tumour-enriched DNA samples and two unrestored U266 cell line genomic DNA (Additional file 1: Figure S1, Additional file 2: Figure S2). The performance of the controls probes was variable between tumour-enriched samples. The majority of the control probes for samples 2657 and 3282 which had the largest number of failed HM450K probes performed poorly and had the highest level of background signal amongst the 22 samples evaluated.
Methylation at the BRCA1 gene
The HM450K assay measures methylation at 53 CpG sites (53 methylation probes) across the BRCA1 gene. Three probes (cg19088651, cg11126247 and cg16919093) did not meet the QC criteria and were removed from the analysis. The 423 remaining FFPE tumour-enriched breast DNA in our study consisted of the following subtypes: 238 Luminal A, 87 Luminal B, 63 triple-negative and 30 HER2-positive. The mean beta value, standard deviation and 95 % CI for each BRCA1-specific probe across each subtype were calculated and plotted as a line graph. Approximately 20 probes along the BRCA1 promoter region displayed higher mean beta levels in the triple-negative breast tumours compared to other subtypes (student’s t-test, p < 10−5) (Fig. 3a). This probe set overlapped BRCA1 exon 1, exon 1 of its’ neighbouring gene, NBR2 [14] and their shared bi-directional promoter [15].

Probe mean beta values at the BRCA1 gene. a Mean beta values measured from 419 tumour-enriched DNA in our dataset. Twenty probes overlapping exon 1 of BRCA1, exon 1 of NBR2 and their shared bi-directional promoter showed increased methylation in triple-negative breast cancers (TNBC) compared with Luminal A (Lum A), Luminal B (Lum B) and HER2-positive (HER2-pos) subtypes (* denotes probes not present in the TCGA dataset). b Mean beta values measured from 156 tumour-enriched DNA in TCGA. Twenty-two probes overlapping exon 1 of BRCA1, exon 1 of NBR2 and their shared bi-directional promoter showed increased methylation in basal-like breast cancers (TNBC) compared with Luminal A (Lum A), Luminal B (Lum B) and HER2-enriched (HER2-pos) subtypes (+ denotes probes not present in our dataset)
We sought to compare our results against publicly available data from The Cancer Genome Atlas where genome-wide methylation was measured using the same platform and on DNA extracted from fresh frozen material thereby representing good quality DNA [12]. We compared methylation signals at BRCA1 between the basal-like, Luminal A, Luminal B and HER2-enriched breast tumour subtypes and although not at a significant level, we observed a similar trend along the same 20 probes where increased methylation was observed specifically in basal-like breast tumours compared with other subtypes (Fig. 3b). An additional two probes within the same region of BRCA1 which failed in our dataset also displayed the same trend.
Discussion
This protocol facilitated the successful application of the HM450K platform to DNA extracted from FFPE tumour material and is relevant for any large population-based translational epigenetic studies of human disease. This assertion is based on assessment of the quality metrics and consistency of the data with prior research data examining methylation at the BRCA1 promoter.
Methylation of the BRCA1 promoter along with the corresponding loss of BRCA1 expression in mutation-negative breast cancer is well described for both familial and sporadic breast cancer and has been demonstrated using low-throughput loci-specific methods [16–18]. Promoter methylation of this gene has also been associated with the triple-negative breast cancer subtype and/or histological features commonly associated with triple-negative breast cancers [19–22].
In our data 20 probes along the BRCA1 promoter region displayed higher mean beta levels in the triple-negative breast tumours compared to other subtypes. This region encompassed a region of the BRCA1 promoter (CpG sites −44 to +55 relative to the transcription start site and corresponding to the region between probes cg19531713 and cg17301289) that has been specifically measured by MethyLight Real-time PCR in previous studies [20].
The modified protocol described here incorporates an important third QC checkpoint. Assuming all samples that failed the QC would have also failed the HM450K assay, incorporation of this checkpoint increased the success rate of our samples on the HM450K platform from 90 % to the reported 99 %. Using this modified protocol, we were able to identify and remove samples that were likely to fail prior to application on the HM450K platform.
However, there are several aspects to this data that are important to consider further. Firstly, as we excluded samples that fell below the described threshold (ΔCq value ≥ 4) we do not have data that confirms that these samples would not have generated data, on the HM450K platform, that met our criterion for success (average detection p-value across all probes of p ≤ 0.01 and individual probe detection p-value of p ≤ 0.05). The costs associated with running this platform makes this challenging to test, but data from 2 archival FFPE tumour samples which failed QC checkpoint 3 (ΔCq 2.52 and 0) in a subsequent study in our laboratory produced very poor quality data with 61,803 and 200,552 failed probes, respectively [data not shown]. This finding supports our protocol to exclude samples with ΔCq value ≤4 at QC checkpoint 3.
Secondly, the BRCA1 promoter amplicon utilised in QC checkpoint 3 includes CpGs sites. Although our methodology was developed to balance amplification of both the methylated and non-methylated template [10], this is difficult to achieve due to potential PCR bias favouring the amplification of the unmethylated template [23]. Our assay was not designed to differentiate between methylated and unmethylated template but, if this is important to other related applications, it would be more appropriate to select an alternate amplicon (without CpG sites) that would offer a superior assay.
We are also aware that we cannot exclude the possibility of Loss of Heterozygosity or homozygous deletion at the BRCA1 locus being responsible for a proportion of the QC checkpoint failures. Further investigation using other molecular techniques such as Multiplex Ligation-dependent Probe Analysis (MLPA) and Sanger sequencing of heterozygous SNPs flanking this region is certainly warranted but not within the scope of this technical report.
The performance of the HM450K internal control probes did not always correlate with the detection p-values. For instance, the internal control probes for sample 27042 performed satisfactorily although having a high number of probes with detection p-values >0.05. In contrast, sample 35995 passed all our data QC hurdles although some of the bisulfite conversion II control probes showed higher background signal intensities.
This protocol is suitable for the analysis of macrodissected FFPE tumour-enriched DNA samples of variable quantities and quality and can be reliably and systematically applied to hundreds of samples in a standard and controlled manner. Moreover, it has the potential to be translated into molecular pathology diagnostic services as the assessment of methylation becomes more clinically applied in the diagnosis and treatment of disease. This methodology will have utility in population-based translational epigenetic studies and is applicable to a wide variety of translated studies interested in analysis of methylation and its role in the predisposition to disease and disease progression.
Conclusions
This protocol facilitated the successful application of the HM450K platform to DNA extracted from archival FFPE tumour material and is relevant any large population-based translational epigenetic studies of human disease.
Abbreviations
- bp:
-
base pair
- BRCA1 :
-
breast cancer 1, early onset
- CI:
-
confidence interval
- Cq :
-
quantitation cycle
- DNA:
-
deoxyribonucleic acid
- FFPE:
-
formalin-fixed paraffin-embedded
- HM450K:
-
Infinium HumanMethylation450 Beadchip
- H&E:
-
haematoxylin and eosin
- HER2:
-
human epidermal growth factor receptor
- MCCS:
-
Melbourne Collaborative Cohort Study
- NBR2:
-
neighbour of BRCA1 gene 2
- qPCR:
-
quantitative polymerase chain reaction
- PAM:
-
prediction analysis of microarray
- QC:
-
quality control
- SNP:
-
single nucleotide polymorphism
- TCGA:
-
The Cancer Genome Atlas Network
References
Ferrer I, Armstrong J, Capellari S, Parchi P, Arzberger T, Bell J, et al. Effects of formalin fixation, paraffin embedding, and time of storage on DNA preservation in brain tissue: a BrainNet Europe study. Brain Pathol. 2007;17(3):297–303.
Joo JE, Wong EM, Baglietto L, Jung CH, Tsimiklis H, Park DJ, et al. The use of DNA from archival dried blood spots with the Infinium HumanMethylation450 array. BMC Biotechnol. 2013;13:23.
Moran S, Vizoso M, Martinez-Cardus A, Gomez A, Matias-Guiu X, Chiavenna SM, et al. Validation of DNA methylation profiling in formalin-fixed paraffin-embedded samples using the Infinium HumanMethylation450 Microarray. Epigenetics. 2014;9(6):829–33.
Dumenil TD, Wockner LF, Bettington M, McKeone DM, Klein K, Bowdler LM, et al. Genome-wide DNA methylation analysis of formalin-fixed paraffin embedded colorectal cancer tissue. Genes Chromosomes Cancer. 2014;53(7):537–48.
de Ruijter TC, de Hoon JP, Slaats J, de Vries B, Janssen MJ, van Wezel T, et al. Formalin-fixed, paraffin-embedded (FFPE) tissue epigenomics using Infinium HumanMethylation450 BeadChip assays. Lab Invest. 2015;95(7):833–42.
Siegel EM, Berglund AE, Riggs BM, Eschrich SA, Putney RM, Ajidahun AO, et al. Expanding epigenomics to archived FFPE tissues: an evaluation of DNA repair methodologies. Cancer Epidemiol Biomark Prev. 2014;23(12):2622–31.
Giles GG, English DR. The Melbourne Collaborative Cohort Study. IARC Sci Publ. 2002;156:69–70.
Blows FM, Driver KE, Schmidt MK, Broeks A, van Leeuwen FE, Wesseling J, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010;7(5):e1000279.
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71.
Shen L, Guo Y, Chen X, Ahmed S, Issa JP. Optimizing annealing temperature overcomes bias in bisulfite PCR methylation analysis. Biotechniques. 2007;42(1):48–50 (2 passim).
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9.
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70.
Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7.
Xu CF, Brown MA, Nicolai H, Chambers JA, Griffiths BL, Solomon E. Isolation and characterisation of the NBR2 gene which lies head to head with the human BRCA1 gene. Hum Mol Genet. 1997;6(7):1057–62.
Xu CF, Chambers JA, Solomon E. Complex regulation of the BRCA1 gene. J Biol Chem. 1997;272(34):20994–7.
Rice JC, Futscher BW. Transcriptional repression of BRCA1 by aberrant cytosine methylation, histone hypoacetylation and chromatin condensation of the BRCA1 promoter. Nucleic Acids Res. 2000;28(17):3233–9.
Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006;8(4):R38.
Iwamoto T, Yamamoto N, Taguchi T, Tamaki Y, Noguchi S. BRCA1 promoter methylation in peripheral blood cells is associated with increased risk of breast cancer with BRCA1 promoter methylation. Breast Cancer Res Treat. 2011;129(1):69–77.
Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344(8):539–48.
Wong EM, Southey MC, Fox SB, Brown MA, Dowty JG, Jenkins MA, et al. Constitutional methylation of the BRCA1 promoter is specifically associated with BRCA1 mutation-associated pathology in early-onset breast cancer. Cancer Prev Res (Phila). 2011;4(1):23–33.
Hsu NC, Huang YF, Yokoyama KK, Chu PY, Chen FM, Hou MF. Methylation of BRCA1 promoter region is associated with unfavorable prognosis in women with early-stage breast cancer. PLoS One. 2013;8(2):e56256.
Tapia T, Smalley SV, Kohen P, Munoz A, Solis LM, Corvalan A, et al. Promoter hypermethylation of BRCA1 correlates with absence of expression in hereditary breast cancer tumors. Epigenetics. 2008;3(3):157–63.
Wojdacz TK, Hansen LL, Dobrovic A. A new approach to primer design for the control of PCR bias in methylation studies. BMC Res Notes. 2008;1:54.
Authors’ contributions
EMW and JEJ performed laboratory based work to establish the experimental protocol and performed all experiments described in this manuscript. CAM provided histopathological expertise. LB and DRE, GS and JLH made prior contribution to the MCCS resource utilised in this report. LB, MCS and DRE provided grant support for this study. RM and LMF contributed to the MCCS resource and logistical aspects of the project. GGG is the principal investigator of the Melbourne Collaborative Cohort Study. MCS provided additional grant support, the initial idea of the research described and overall supervision of the laboratory based work. EMW, JEJ and MCS wrote the first draft of the manuscript to which all authors contributed. All authors read and approved the final manuscript.
Acknowledgements
This study was made possible by the contribution of many people including those taking part in the Melbourne Collaborative Cohort Study, the original investigators and the diligent team who manage the study and continue working on follow up. Cohort recruitment was funded by VicHealth and Cancer Council Victoria. This work was further supported by the National Health and Medical Research Council (NHMRC; APP1011618 and APP1026892) and The Victorian Breast Cancer Research Consortium. MCS is a NHMRC SRF and VBCRC Group Leader. JLH is a NHMRC SPRF. LMF is the Cancer Council Victoria David Hill Fellow and is funded by a Movember and Cure Cancer Australia Foundation Young Investigator Grant awarded through Prostate Cancer Foundation of Australia’s Research Program. We thank Nicole Wong Doo and Andy Hsu from the Peter MacCallum Cancer Centre for providing the U266 multiple myeloma cell line and Carolyn Staines of Cancer Council Victoria for editing the manuscript.
Previous presentation of work Part of the work described in this manuscript had previously been presented as a poster at the 2014 American Society of Human Genetics annual meeting.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ee Ming Wong and JiHoon E. Joo contributed equally to this work
Additional files
13104_2015_1487_MOESM1_ESM.pdf
Additional file 1: Figure S1. Performance of HM450K sample-independent control probes for failed and a subset of successful samples. Sample-independent control probes measured the staining (A), extension (B), target removal (C) and hybridisation (D) steps in the HM450K assay on the red and green channels. Failed samples are boxed.
13104_2015_1487_MOESM2_ESM.pdf
Additional file 2: Figure S2. Performance of HM450K sample-dependent control probes for failed and a subset of successful samples. Sample-dependent control probes measured background signal levels (negative; A), efficiency of bisulfite conversion using Infinium type I (B) and type II (C) probe designs, allele-specific extension of Infinium I and II probes (specificity; D) and restoration (E) and overall assay performance (non-polymorphic; F) on the red and green channels. Failed samples are boxed.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wong, E.M., Joo, J.E., McLean, C.A. et al. Tools for translational epigenetic studies involving formalin-fixed paraffin-embedded human tissue: applying the Infinium HumanMethyation450 Beadchip assay to large population-based studies. BMC Res Notes 8, 543 (2015). https://doi.org/10.1186/s13104-015-1487-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-015-1487-z