
Abstract
Background
Human urogenital schistosomiasis caused by Schistosoma haematobium is widely distributed across Africa and is increasingly targeted for control and regional elimination. The development of new high-throughput, cost-effective molecular tools and approaches are needed to monitor and evaluate the impact of control programs on the parasite populations. Microsatellite loci are genetic markers that can be used to investigate how parasite populations change over time and in relation to external influences such as control interventions.
Findings
Here, 18 existing S. haematobium microsatellite loci were optimised to enable simultaneous amplification across two novel multiplex microsatellite PCR’s, each containing nine loci. Methods were developed for the cost effective and rapid processing and microsatellite analysis of S. haematobium larval stages stored on Whatman-FTA cards and proved robust on miracidia and cercariae collected from Zanzibar and Niger.
Conclusion
The development of these novel and robust multiplex microsatellite assays, in combination with an improved protocol to elute gDNA from Whatman-FTA fixed schistosome larval stages, enables the high-throughput population genetic analysis of S. haematobium. The molecular resources and protocols described here advance the way researchers can perform multi locus-based population genetic analyses of S. haematobium as part of the evaluation and monitoring of schistosomiasis control programmes.
Similar content being viewed by others


Findings
Introduction
Infection with the blood fluke Schistosoma haematobium causes human urogenital schistosomiasis throughout Africa, parts of the Middle East, Madagascar and the Indian Ocean Islands, with an estimated 110 million people infected [1]. Several efforts are underway to control morbidity and ultimately to eliminate S. haematobium infection predominantly through the large-scale administration of the drug praziquantel (PZQ) [1]. The development of new high-throughput, low cost, molecular tools and approaches are now imperative, not only to elucidate the epidemiology and evolution of schistosomiasis but also to monitor and evaluate the impact of progressing control programs [2]. Here we present an enhanced method enabling the high-throughput and cost effective preparation of gDNA from individual schistosome larval stages facilitating multi-loci genetic analysis together with two novel S. haematobium multiplex microsatellite PCRs. Microsatellite loci are highly variable DNA markers in widespread use within the schistosomiasis research community as they enable population-level analysis [3]. The principal drawback of microsatellite markers has been the cost and labour associated with the need to genotype multiple loci. Significant cost and timesaving can be achieved by developing multiplex PCR systems that amplify multiple microsatellite loci in single reactions. The methods outlined here facilitate the high-throughput microsatellite-based population genetic analyses of S. haematobium.
Microsatellite multiplex design and optimisation
S. haematobium microsatellite loci were available from [4] and [3]. Loci that were di, tri or tetra-mer repeats, non-compound, robust and had multiplexing potential were selected for further optimisation. Eighteen loci were chosen in total (15 from [3] and three from [4], Table 1). Initially the functionality and specificity of all the primer pairs were confirmed by amplifying all the loci in singleplex 12.5 μl reactions using 10 ng of S. haematobium reference gDNA obtained from the Schistosomiasis Collection at the Natural History Museum (SCAN [5]) and the Type-it Microsatellite PCR Kit (Qiagen) according to the manufacturer’s protocol.
The loci were successfully divided into two multiplex panels each incorporating nine loci that gave the maximum size difference between each locus and a maximum of four overlapping loci at any size range, together with minimal variance of the annealing temperature of all the primers (T m) (Table 1). Within each panel the forward primer for each locus was 5' labelled with a fluorescent reporter dye according to the 5-dye detection system. Overlapping fragments were assigned a different dye and the maximum distance was maintained between fragments labelled with the same dye to enable accurate identification. The multiplex microsatellite PCRs for each panel were carried out in 12.5 μl reactions using 10 ng of S. haematobium reference gDNA and the Type-it Microsatellite PCR Kit (Qiagen) according to the manufacturer’s protocol. Different T m values were tested with the optimal T m that gave uniform and specific amplification for all loci in each panel determined at 54 °C. Singleplex and multiplex amplicons were visualised on 3 % gel red agarose gels before 2 μl of 1: 50 dilutions were mixed with 0.35 μl of GS500Liz size standard (Applied Biosystems) before being denatured for 5 mins at 95 °C and injected at a 10 s injection speed into an Applied Biosystems 3130xl DNA Analyser. Allele peaks were visualised in Geneious version 6.1.4 (www.geneious.com [6]) using the microsatellite plugin. The multiplex PCRs proved robust giving identical peak scores in repeated reactions, in singleplex versus multiplex reactions, and significant stutter peaks, n-1 products and allelic drop-out were not observed.
Multiplex PCR optimisation and application on field-collected S. haematobium miracidia and cercaria
A novel, high-throughput and cost effective non-wash Whatman-FTA alkaline DNA elution protocol has been developed which provides ~38 μl of eluted DNA from a single schistosome larval stage which has been fixed on a classic indicating Whatman-FTA card. This three-step protocol is very simple, quick and is suitable for multi-well processing. Individual larval DNA is alkaline eluted from a single 2.0 mm Whatman-FTA punch and subsequently neutralised, providing usable DNA for many downstream applications including microsatellite and fragment analysis, mitochondrial and nuclear DNA/gene amplification (http://www.gelifesciences.com). The solutions (1 and 2) needed for the DNA elution steps can be easily made with standard laboratory chemicals at an insignificant cost, especially compared to alternative DNA preparation methods.
Individual S. haematobium miracidia were collected directly from individual urine samples of infected children in Niger and Pemba Island (Zanzibar, United Republic of Tanzania [7]). S. haematobium cercariae were also obtained from naturally infected Bulinus globosus snails from Niger. All samples were collected and individually preserved on Whatman-FTA cards [8, 9].
DNA elutions were carried out in low profile 1.2 ml 96 square well storage microplates with 96 square well sealing cap mats which facilitates DNA elution. The 2.0 mm Whatman-FTA punch containing the DNA from a single larval stage was incubated at room temperature in 14 μl of Solution 1 (0.1 M NaOH, 0.3 mM EDTA, pH13.0) for 5 mins. Subsequently, 26 μl of Solution 2 (0.1 M Tris–HCl, pH7.0) was added, the mixture was pulse vortexed three times, incubated for a further ten minutes at room temperature and then pulse vortexed ten times. The eluted DNA was then transferred to a 96 well storage plate and either used immediately or stored at -20 °C for future use.
The two multiplex microsatellite PCRs were performed on each available sample in 12.5 μl reactions using 2 μl of the eluted DNA and the Type-it Microsatellite PCR Kit (Qiagen) according to the manufacturer’s protocol with the addition of 1.25 μl of the Type-it Microsatellite PCR Kit Q-Solution. Optimal cycling parameters were, an initial denaturing step of 95 °C for 5 mins followed by 32 cycles of 95 °C for 30 s, 54 °C for 90 s, 72 °C for 3 mins and followed by a final elongation step of 60 °C for 30 mins. Reactions were checked by 3 % agarose gel electrophoresis and then diluted 1 in 10 before being denatured and injected at an optimal speed of 12 s into the Applied Biosystems 3130xl DNA analyser for analysis.
Allele peaks were checked and edited using Geneious 6.1.4 (www.geneious.com [6]) before being placed into amplicon size “bins” and exported for analysis. Panel 1 and 2 allele data were compiled for each sample for analysis (Additional file 1: Table S1). Data were analysed from ten miracidia, from five children from Koutoukale Zeno (Lat. 13.680, Long. 1.738) in Niger, five children from Chambani school (Lat. 5.33457 Long. 39.77256) on Pemba Island, Zanzibar, United Republic of Tanzania and also from 16 cercariae from two infected Bulinus snails from Niger.
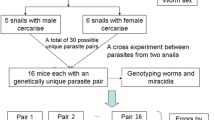
All loci amplified successfully with no significant stutter peaks or n-1 products. Whilst low peak height was often observed in the loci Sh7 (Panel 1) compared to the other loci and was lower in samples from Niger compared to Pemba, the data were still scorable. Genetic diversity indices were calculated using the program GenAlEx 6.5 [10] and the presence of null alleles and allele dropout was evaluated using Micro-Checker [11]. The numbers of alleles observed across the loci ranged from 2 to 33 with loci C131 being the least diverse. Higher genetic diversity was observed in the Pembamiracidial population compared to that from Niger (Table 1). Cercariae obtained from each individual snail had identical genotypes, showing they were clonal, derived from a single miracidium.
Inter-species specificity
The cross-reactivity of the multiplex microsatellite PCRs was also assessed on S. mansoni, which causes intestinal schistosomiasis and is very common throughout Africa and can sometimes be found ectopically excreted in urine samples in endemic co-infection foci [12]. Singleplex and multiplex reactions were performed on 10 ng of reference gDNA from individual S. mansoni male worms obtained from the Schistosomiasis Collection at the Natural History Museum (SCAN [5]). Cross-reactivity was found to be low: seven loci failed to amplify, six gave low and/or non-specific amplification, two exhibited a size shift and only three among the total of 18 loci amplified well and were within the size range expected (Table 2).
In conclusion, this study describes two novel robust and informative multiplex microsatellite assays enabling the simultaneous amplification of 18 individual loci; facilitating population genetic analysis of all S. haematobium life-cycle stages. Protocols are presented that facilitate high-throughput, and cost effective processing and robust genetic analysis of S. haematobium larval stages. Such tools can greatly assist large-scale population genetic analysis of human schistosome populations such as that now underway within the SCORE programme (http://score.uga.edu). The alkaline elution of larval schistosome DNA from Whatman-FTA stored samples is simple, quick, high-throughput and low cost, providing adequate amounts of gDNA preparations for multiple molecular analyses and repeats, significantly overcoming the limitations encountered from the standard Whatman-FTA preparations [2]. Additionally, the multiplexing of the microsatellite loci significantly reduces the resources associated with genotyping multiple microsatellite loci for analysis.
Ethics statement
For the Niger sample collection, ethical approval was obtained from the St Mary’s Hospital Local Ethics Research Committee (part of the Imperial College London Research Ethics Committee (ICREC; (EC NO: 03.36. R&D No: 03/SB/033E)) in London, United Kingdom. For the Zanzibar sample collection, ethical approval was obtained from the Zanzibar Medical Research and Ethics Committee (ZAMREC, reference no. ZAMREC 0003/Sept/011) in Zanzibar, United Republic of Tanzania, the “Ethikkomission beider Basel” (EKBB, reference no. 236/11) in Basel, Switzerland, and the Institutional Review Board of the University of Georgia (project no. 2012-10138-0). Within both Niger and Zanzibar, all aspects of sample collections were carried out in the framework of the disease control activities implemented and approved by the local Ministry of Health (MoH) and adopted by regional and local administrative and health authorities. The study participants were informed about the study objectives and procedures. Written consent was obtained from parents prior to sample collection from children. Participation was voluntary and children could withdraw or be withdrawn from the study at any time without obligation. All children were offered PZQ (40 mg/kg single oral dose) treatment in the frame of the following school-based or community-wide treatment carried out by the MoH.
Abbreviations
- SCORE:
-
Schistosomiasis Consortium for Operational Research and Evaluation
- PCR:
-
Polymerase chain reaction
- gDNA:
-
Genomic deoxyribonucleic acid
- PZQ:
-
Praziquantel
References
World Health Organization. Accelerating work to overcome the global impact of neglected tropical diseases: a roadmap for implementation. 2012. Available: http://www.who.int/neglected_diseases/NTD_RoadMap_2012_Fullversion.pdf
Webster JP, Gower CM, Norton AJ. Evolutionary concepts in predicting and evaluating the impact of mass chemotherapy schistosomiasis control programmes on parasites and their hosts. Evol Appl. 2008;1:66–83.
Glenn TC, Lance SL, McKee AM, McKee AM, Webster BL, Emery AM, et al. Significant variance in genetic diversity among populations of Schistosoma haematobium detected using microsatellite DNA loci from a genome-wide database. Parasites & Vectors. 2013;6:300.
Gower CM, Gabrielli AF, Sacko M, Dembelé R, Golan R, Emery AM, et al. Population genetics of Schistosoma haematobium: development of novel microsatellite markers and their application to schistosomiasis control in Mali. Parasitology. 2011;138:978–94.
Emery AM, Allan FE, Rabone ME, Rollinson D. Schistosomiasis collection at NHM (SCAN). Parasites & Vectors. 2012;5:185.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Knopp S, Mohammed KA, Ali SM, Khamis IS, Ame SM, et al. Study and implementation of urogenital schistosomiasis elimination in Zanzibar (Unguja and Pemba islands) using an integrated multidisciplinary approach. BMC Public Health. 2012;12:930.
Webster BL, Emery A, Webster JP, Gouvras A, Garba A, Diaw O, et al. Genetic diversity within Schistosoma haematobium: DNA barcoding reveals two distinct groups. PLoS Neg Trop Dis. 2012;6:e1882.
Gower CM, Shrivastava J, Lamberton PHL, Rollinson D, Webster BL, Emery A, et al. Development and application of an ethical and epidemiologically appropriate assay for the multi-locus microsatellite analysis of Schistosoma mansoni. Parasitology. 2007;134:523–36.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics. 2012;28:2537–9.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. MICRO-CHECKER: software fro identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004;4:535–8.
Meurs L, Mbow M, Vereecken K, Menten J, Mboup S, Polman K. Epidemiology of mixed Schistosoma mansoni and Schistosoma haematobium in Northern Senegal. Int J for Parasitology. 2012;42:305–11.
Acknowledgments
This study received financial support from the University of Georgia Research Foundation, which is funded by the Bill and Melinda Gates Foundation for the SCORE project (Prime award number 50816, sub-award numbers: RR374-053/4785426 (Niger), RR374-053/4893206 (Zanzibar). Stefanie Knopp is financially supported by sub-award no. RR374-053/4893196. Fiona Allan and Muriel Rabone are financially supported by the Wellcome Trust grant no. 104958/Z/14/Z. The authors would like to thank everyone that was involved in any of the field collections (specifically Mtumweni A. Mubsin and Khamis Rashid from Pembaand Bassirou Mamane Madougou from Niger) and the people that provided samples. Julia Llewellyn-Hughes, Claire Griffiths and Lisa Smith in the Natural History Museum sequencing facility for the fragment analysis. Royal veterinary College manuscript reference number PPB_00991.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BW, FA and AE refined the FTA elution protocol. BW designed the experiments and carried out all the laboratory work. BW, MR and TP carried out the data analysis. BW, JW and DR wrote the manuscript with comments and editing from all of the co-authors. All other authors were involved in the fieldwork and/or sample collection/storage. All authors read and approved the final version of the manuscript.
An erratum to this article is available at http://dx.doi.org/10.1186/s13071-015-1134-5.
Additional file
Additional file 1: Table S1.
Allele sizes for all 18 loci for 50 miracidia from both Niger and Pemba (Zanzibar). (CSV 15 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Webster, B.L., Rabone, M., Pennance, T. et al. Development of novel multiplex microsatellite polymerase chain reactions to enable high-throughput population genetic studies of Schistosoma haematobium . Parasites Vectors 8, 432 (2015). https://doi.org/10.1186/s13071-015-1044-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-015-1044-6