
Abstract
Background
Design and synthesis of new inhibitor agents to deal with pathogenic microorganisms is expanding. In this project, an efficient, environmentally friendly, economical, rapid and mild procedure was developed for the synthesis of novel functionalized isoxazole derivatives as antimicrobial potentials.
Methods
Multicomponent reaction between malononitrile (1), hydroxylamine hydrochloride (2) and different aryl or heteroaryl aldehydes 3a–i afforded novel 5-amino-isoxazole-4-carbonitriles 4a–i in good product yields and short reaction times. Deep eutectic solvent K2CO3/glycerol was used as catalytic reaction media. Structure of all molecules were characterized by different analytical tools. In vitro inhibitory activity of all derivatives was evaluated against a variety of pathogenic bacteria including both Gram-negative and Gram-positive strains as well as some fungi. In addition, their free radical scavenging activities were assessed against DPPH.
Results
Broad-spectrum antimicrobial activities were observed with isoxazoles 4a, b, d. In addition, antioxidant activity of isoxazole 4i was proven on DPPH.
Conclusions
In this project, compounds 4a, b, d could efficiently inhibit the growth of various bacterial and fungal pathogens. Antioxidant properties of derivative 4i were also significant. These biologically active compounds are suitable candidates to synthesize new prodrugs and drugs due to the presence of different functional groups on their rings.

Similar content being viewed by others

Background
Isoxazoles are five-membered aromatic heterocycles containing adjacent oxygen and nitrogen atoms. The isoxazole ring system is found in a variety of naturally occurring compounds and biologically active molecules [1]. They are especially useful in medicine, since many antifungal drugs belong to the isoxazole class [2]. Sulfisoxazole and sulfamethoxazole are two bacteriostatic sulfonamide antibiotics that applied alone or combined with others in the treatment of infections caused Gram-positive and Gram-negative bacteria [3, 4]. Acivicin is a γ-glutamyl transferase inhibitor with anticancer, anti-parasitic and antileishmania activities [5]. Isoxazole derivatives possess a broad variety of biological activities viz. antifungal, anti-inflammatory, antiplatelet, anti-HIV, anti-Alzheimer and analgesic [6,7,8,9,10,11].
Cycloisomerization of α,β-acetylenic oximes [12], cycloaddition of aldoxime and alkynes [13], reaction of alkyl nitriles and α-chlorooximes [14], 1,3-dipolar cycloaddition of in situ generated nitrile oxides and terminal acetylenes [15, 16], addition of hydroxylamine to α-cyano ketones [17] and palladium-catalyzed four-component coupling of a terminal alkyne, hydroxylamine and carbon monoxide [18] are some recently developed methods for isoxazole synthesis. Furthermore, multicomponent reaction of active methylene compounds, aldehydes and hydroxylamine derivatives were well studied under different conditions [19,20,21,22,23].
Deep eutectic solvents (DES) play an essential key in green chemistry. They can be used as safe, low-cost, non-toxic, reusable, catalytic and environmentally friendly media in the most reactions [24]. Their applications are expanding in the field of materials, energy and environmental science [25]. Glycerol is a valuable green, nontoxic, low flammable and available solvent that applied as anti-freezer, sweetener, humectant, lubricant and thickener in industry [26]. This natural polyol as hydrogen bond donor is present in DESs with hydrogen bond acceptors such as choline chloride, methyl triphenyl phosphonium bromide, benzyl triphenyl phosphonium chloride, allyl triphenyl phosphonium bromide, N,N-diethylethanolammonium chloride, and tetra-n-butylammonium bromide [27]. Glycerol/potassium carbonate is a low cost and environmentally friendly DES that recently its efficiently was proven in the preparation of pyrazole derivatives [28].
In order to develop applications of Gly/K2CO3 to other heterocycles, it was successfully used as catalytic media in the synthesis of novel 5-amino-isoxazole-4-carbonitrile derivatives via multicomponent reaction of malononitrile, hydroxylamine and various aryl aldehydes (Fig. 1). In vitro inhibitory activity of all derivatives was evaluated against some pathogenic bacteria including both Gram-negative and Gram-positive strains as well as some fungi. In addition, their antioxidant potentials were assessed against DPPH.

Schematic representation of isoxazole skeletons with antimicrobial and antioxidant activity
Results
Characterization of isoxazoles 4a–i
Multicomponent reaction of malononitrile (1), hydroxylamine hydrochloride (2) and aryl or heteroaryl aldehydes 3a–i afforded 5-amino-isoxazole-4-carbonitriles 4a–i in 70–94% yields (Scheme 1). Products were obtained in glycerol/potassium carbonate (4:1) at room temperature for 20–120 min.

Multicomponent synthesis of 5-amino-isoxazole-4-carbonitriles
Evaluation of the bioactivity of isoxazoles 4a–i
All synthesized compounds were assessed for their antimicrobial efficiency as well as antioxidant activity. Inhibitory effects of isoxazoles 4a–i were presented as MIC, MBC and MFC values in Tables 1 and 2.
Discussion
Chemistry
The effects of variations in solvent, temperature and order mixing reactants were studied on product yield and reaction time. Aldoximes were produced as major products in glycerol at different conditions. They were also formed in Gly/K2CO3 deep eutectic solvents under one-pot two-step procedures involving initial mixing hydroxylamine and aldehydes, followed by malononitrile. In addition, oximes were present as by-products in one-pot two-step processes involving initial mixing malononitrile and aldehydes. There are two possible mechanisms to form the products (Scheme 2). A reaction pathway, that does not lead to the target products, includes the reaction of aldoximes produced from aldehydes and hydroxylamine with malononitrile. On another path, the Knoevenagel condensation of aldehydes with malononitrile gives arylidene malononitriles, which react with hydroxylamine to form isoxazoles. The best results were obtained via simultaneous reaction of reagents in Gly/K2CO3 (4:1 molar) as green catalytic media at room temperature, which considered as optimal conditions. Increase in Gly/K2CO3 ratio and temperature led to a decrease in yields.

Proposed mechanisms for the formation of isoxazoles 4a–i
Multicomponent reaction of hydroxylamine derivatives, aldehydes and active methylene compounds is an efficient procedure to synthesize isoxazoles. Some recently proposed protocols were presented in Table 3. According to the data in the Table 3, reaction times decreased in the presence of catalysts at room temperature or under heating or UV radiation. It seems that basic catalysts are more effective than acidic equivalents. Our newly modified process provides an efficient, simple, economical, safe and eco-friendly reaction under mild conditions at acceptable products yields.
The chemical structure of isoxazoles 4a–i was characterized by spectral data. Nitrile groups were detected by FT-IR (~ 2220 cm−1) and 13C NMR (~ 115 ppm). Amino groups were also identified based on their absorption bands in region of ~ 3430–3330 cm−1 and proton chemical shifts appeared approximately 8.50 ppm.
Biological evaluation
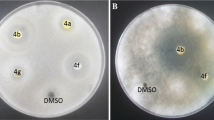
Based on the results obtained, isoxazoles 4a, b, d, e showed broad-spectrum inhibitory activates against both Gram-positive and Gram-negative bacteria. These compounds respectively include p-tolyl, 4-hydroxyphenyl, 2,4-dichlorophenyl and 2,6-dichlorophenyl substituents in 3-position on isoxazole ring. Heterocycle 4b was the only effective antibacterial agent on Shigella flexneri. Similarity, Shigella dysenteriae and Escherichia coli were blocked only with isoxazole 4d. Derivatives 4c, f, g, h, i were effective only against Gram-positive pathogens. All derivative could inhibit the growth of Gram-positive Listeria monocytogenes. No antifungal activity was observed with heterocyclic compounds 4c, e, f, g, h, i. Isoxazoles 4b, d were effective on all tested pathogenic fungi.
Free radical scavenging ability of methanolic solutions of all synthesized compounds against DPPH was determined spectrophotometrically at 517 nm. However, notable in vitro antioxidant activity was only observed in isoxazole 4i, including pyridine-4yl substituent, with an IC50 = 67.51 μg ml−1. These effects are comparable to the effects of isoxazole derivatives with IC50 in the range 62.76–100.73 μg ml−1 [29].
Conclusion
In summary, some novel 5-amino-isoxazole-4-carbonitriles were prepared via a green and efficient multicomponent procedure in acceptable product yields and short reaction times. Antimicrobial activity of isoxazoles was studied against a variety of bacterial and fungal pathogens. Significant inhibitory potentials were observed with compounds 4a, b, d. Isoxazole 4i also showed considerable antioxidant activities. These functionalized biologically active compounds could applied as prodrugs in future researches.
Methods
Materials
All reagents, solvents, antibiotics, DPPH and antifungal agents were purchased from commercial sources (Merck, Sigma and Aldrich), and used without further purification. The bacterial and fungal culture media were obtained from (HiMedia). Melting points were determined with Kruss type KSP1N melting point meter and are uncorrected. Reaction progress was monitored by aluminium TLC plates pre-coated by SiO2 with fluorescent indicator F254 using CHCl3/CH3OH (9:1, v/v) as mobile phase, which were visualized under UV radiation (254 nm). The absorption spectra were determined using a UV-2100 RAY Leigh UV–Vis spectrophotometer. FT-IR spectra of the products were collected using a Bruker Tensor-27 FT-IR spectrometer. 1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively, on a Bruker FT-NMR Ultra Shield-400 spectrometer. Elemental analyses (CHNS/O) were performed on a Thermo Finnigan Flash EA microanalyzer. DESs were prepared in various ratios of glycerol/K2CO3 according to the published procedure [30] (Additional file 1).
General procedure for the synthesis of isoxazoles 4a–i
A mixture of K2CO3 (0.140 g, 0.001 mol) and glycerol (0.360 g, 0.004 mol) was stirred at 80 °C for 2 h to form a homogenous colorless liquid. After cooling DES to room temperature, 0.001 mol each of malononitrile (1) (0.660 g), hydroxylamine hydrochloride (2) (0.070 g) and benzaldehydes 3a–i (3a: 0.120 g, 3b: 0.122 g, 3c: 0.151 g, 3d: 0.175 g, 3e: 0.175 g, 3f: 0.152 g; 3 g: 0.096 g; 3 h: 0.112 g; 3i: 0.107 g) was simultaneously added to it. The reaction mixture was stirred for 20–120 min. 1 ml each of ethanol and water was added to it. The resulting precipitates were collected by filtration, washed respectively with water (5 ml) and ethanol (5 ml), and recrystallized from methanol to give pure isoxazoles 4a–i.
5-Amino-3-(p-tolyl)isoxazole-4-carbonitrile (4a)
Yield: 0.14 g, 70%; mp: 135–137 °C; reaction time: 40 min; IR (KBr) ν: 3408, 3337 (NH2), 2223 (C≡N), 1605 (C=N), 1221 (C–O–N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 2.37 (s, 3H, CH3), 7.39 (d, J = 7.2 Hz, 2H, H-3ʹ,5ʹ), 7.82 (d, J = 7.2 Hz, 2H, H-2ʹ,6ʹ), 8.44 (s, 2H, NH2); 13C NMR (100 MHz, DMSO-d6) δ: 21.90 (CH3), 80.31 (C-4), 113.88 (C-1ʹ), 114.84 (C≡N), 129.17 (C-4ʹ), 130.58 (C-3ʹ,5ʹ), 131.12 (C-2ʹ,6ʹ), 146.12 (C-5), 161.70 (C-3); Anal. Calcd. for C11H9N3O: C 66.32, H 4.55, N 21.09. Found: C 66.28, H 4.52, N 21.15.
5-Amino-3-(4-hydroxyphenyl)isoxazole-4-carbonitrile (4b)
Yield: 0.19 g, 94%; mp: 118–120 °C; reaction time: 30 min; IR (KBr) ν: 3509 (OH), 3426, 3335 (NH2), 2227 (C≡N), 1611 (C=N), 1263 (C–O–N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 6.95 (d, J = 8.3 Hz, 2H, H-3ʹ,5ʹ), 7.85 (d, J = 7.2 Hz, 2H, H-2ʹ,6ʹ), 8.25 (s, 2H, NH2), 11.06 (s, 1H, OH); 13C NMR (100 MHz, DMSO-d6) δ: 75.53 (C-4), 114.60 (C≡N), 115.51 (C-1ʹ), 117.03 (C-3ʹ,5ʹ), 123.21 (C-5), 134.30 (C-2ʹ,6ʹ), 160.90 (C-4ʹ), 164.30 (C-3); Anal. Calcd. for C10H7N3O2: C 59.70, H 3.51, N 20.89. Found: C 59.67, H 3.58, N 20.83.
5-Amino-3-(4-nitrophenyl)isoxazole-4-carbonitrile (4c)
Yield: 0.21 g, 92%; mp: 183–184 °C; reaction time: 35 min; IR (KBr) ν: 3417, 3379 (NH2), 2220 (C≡N), 1603 (C=N), 1541, 1361 (NO2), 1289 (C–O–N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 7.92 (d, J = 9.4 Hz, 2H, H-2ʹ,6ʹ), 8.32 (s, 2H, NH2), 8.41 (m, 4H, H-3ʹ,5ʹ, NH2); 13C NMR (100 MHz, DMSO-d6) δ: 80.63 (C-4), 115.05 (C≡N), 124.23 (C-2ʹ,6ʹ), 130.97 (C-3ʹ,5ʹ), 135.98 (C-1ʹ), 146.36 (C-5), 148.00 (C-4ʹ), 152.36 (C-3); Anal. Calcd. for C10H6N4O3: C 52.18, H 2.63, N 24.34. Found: C 52.24, H 2.59, N 24.37.
5-Amino-3-(2,4-dichlorophenyl)isoxazole-4-carbonitrile (4d)
Yield: 0.23 g, 92%; mp: 119–120 °C; reaction time: 60 min; IR (KBr) ν: 3426, 3347 (NH2), 2228 (C≡N), 1648 (C=N), 1290 (C–O–N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 7.64 (m, 1H, H-5ʹ), 7.86 (s, 1H, H-3ʹ), 8.01 (d, J = 7.9 Hz, 1H, H-6ʹ), 8.58 (s, 2H, NH2); 13C NMR (100 MHz, DMSO-d6) δ: 87.50 (C-4), 113.76 (C≡N), 128.28 (C-5ʹ), 128.75 (C-1ʹ), 129.69 (C-6ʹ), 130.47 (C-3ʹ), 131.38 (C-2ʹ) 139.18 (C-4ʹ), 144.13 (C-5), 157.13 (C-3); Anal. Calcd. for C10H6N4O3: C 52.18, H 2.63, N 24.34. Found: C 52.24, H 2.59, N 24.37.
5-Amino-3-(2,6-dichlorophenyl)isoxazole-4-carbonitrile (4e)
Yield: 0.22 g, 88%; mp: 150–152 °C; reaction time: 50 min; IR (KBr) ν: 3432, 3358 (NH2), 2221 (C≡N), 1647 (C=N), 1299 (C–O–N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 7.38 (d, J = 7.1 Hz, 1H, H-4ʹ), 7.48 (d, J = 7.1 Hz, 2H, H-3ʹ,5ʹ), 8.18 (s, 2H, NH2); 13C NMR (100 MHz, DMSO-d6) δ: 82.57 (C-4), 113.10 (C≡N), 129.31 (C-3ʹ,5ʹ), 129.78 (C-1ʹ), 131.37 (C-2ʹ,6ʹ), 134.32 (C-4ʹ), 144.20 (C-5), 155.25 (C-3); Anal. Calcd. for C10H6N4O3: C 52.18, H 2.63, N 24.34. Found: C 52.20, H 2.66, N 24.29.
5-Amino-3-(2-hydroxy-3-methoxyphenyl)isoxazole-4-carbonitrile (4f)
Yield: 0.17 g, 75%; mp: 220–222 °C; reaction time: 120 min; IR (KBr) ν: 3509 (OH), 3408, 3341 (NH2), 2230 (C≡N), 1606 (C=N), 1287 (C–O–N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.87 (s, 3H, CH3), 7.27–7.39 (m, 3H, H-4ʹ,5ʹ,6ʹ), 8.38 (s, 2H, NH2), 10.31 (s, 1H, OH); 13C NMR (100 MHz, DMSO-d6) δ: 56.67 (CH3), 102.74 (C-4), 114.97 (C≡N), 117.82 (C-4ʹ), 118.37 (C-1ʹ), 121.16 (C-5ʹ), 125.82 (C-6ʹ), 143.68 (C-2ʹ), 146.87 (C-5), 154.08 (C-3ʹ), 157.00 (C-3); Anal. Calcd. for C11H9N3O3: C 57.14, H 3.92, N 18.17. Found: C 57.19, H 3.94, N 18.13.
5-Amino-3-(furan-2-yl)isoxazole-4-carbonitrile (4g)
Yield: 0.13 g, 85%; mp: 270–272 °C (dec.); reaction time: 25 min; IR (KBr) ν: 3425, 3369 (NH2), 2221 (C≡N), 1601 (C=N), 1289 (C–O–N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 6.77 (m, 1H, H-3ʹ), 7.23 (m, 1H, H-2ʹ), 8.02 (m, 1H, H-4ʹ), 8.30 (s, 2H, NH2); 13C NMR (100 MHz, DMSO-d6) δ: 76.90 (C-4), 109.35 (C-2ʹ), 113.05 (C-3ʹ), 115.52 (C≡N), 135.12 (C-4ʹ), 146.31 (C-3), 153.00 (C-1ʹ), 160.29 (C-5); Anal. Calcd. for C8H5N3O2: C 54.86, H 2.88, N 23.99. Found: C 54.81, H 2.90, N 24.03.
5-Amino-3-(thiophen-2-yl)isoxazole-4-carbonitrile (4h)
Yield: 0.15 g, 79%; mp: 249–251 °C (dec.) (Lit. [31]: 225–226 °C); reaction time: 60 min; IR (KBr) ν: 3425, 3363 (NH2), 2204 (C≡N), 1600 (C=N), 1281 (C–O–N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 7.25 (m, 1H, H-3ʹ), 7.45 (m, 1H, H-2ʹ), 7.87 (m, 1H, H-4ʹ), 8.34 (s, 2H, NH2); 13C NMR (100 MHz, DMSO-d6) δ: 80.52 (C-4), 115.26 (C≡N), 128.16 (C-2ʹ), 130.63 (C-3ʹ), 131.21 (C-4ʹ), 141.09 (C-1ʹ), 152.56 (C-3), 161.60 (C-5); Anal. Calcd. for C8H5N3OS: C 50.25, H 2.64, N 21.98, S 16.77. Found: C 50.31, H 2.61, N 22.01, S 16.71.
5-Amino-3-(pyridin-4-yl)isoxazole-4-carbonitrile (4i)
Yield: 0.17 g, 91%; mp: 255–257 °C (dec.); reaction time: 20 min; IR (KBr) ν: 3434, 3356 (NH2), 2216 (C≡N), 1602 (C=N), 1288 (C–O–N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 7.37–7.55 (m, 2H, H-2ʹ,6ʹ), 8.45 (s, 2H, NH2), 8.76 (d, J = 7.5 Hz, 2H, H-3ʹ,5ʹ); 13C NMR (100 MHz, DMSO-d6) δ: 80.03 (C-4), 114.82 (C≡N), 123.80 (C-2ʹ,6ʹ), 142.69 (C-1ʹ), 150.39 (C-3ʹ,5ʹ), 152.43 (C-3), 161.23 (C-5); Anal. Calcd. for C9H6N4O: C 58.06, H 3.25, N 30.09. Found: C 58.01, H 3.27, N 30.15.
Biological assay
Gram-negative bacterial strains including Pseudomonas aeruginosa (PTCC 1310), Shigella flexneri (PTCC 1234), Shigella dysenteriae (PTCC 1188), Klebsiella pneumoniae (PTCC 1290), Acinetobacter baumannii (PTCC 1855), Escherichia coli (PTCC 1399), Gram-positive bacterial strains including Streptococcus pyogenes (PTCC 1447), Streptococcus agalactiae (PTCC 1768), Streptococcus pneumoniae (PTCC 1240), Staphylococcus epidermidis (PTCC 1435), Rhodococcus equi (PTCC 1633), Listeria monocytogenes (PTCC 1297), Streptococcus equinus (PTCC 1445), Bacillus subtilis subsp. spizizenii (PTCC 1023), Bacillus thuringiensis subsp. kurstaki (PTCC 1494), Staphylococcus aureus (PTCC 1189), Bacillus cereus (PTCC 1665) and fungi including Aspergillus fumigatus (PTCC 5009), Candida albicans (PTCC 5027) and Fusarium oxysporum (PTCC 5115) were prepared from the Persian Type Culture Collection (PTCC), Karaj, Iran. All biological tests were repeated at least three times. The results were reported as the mean of three independent experiments.
MIC determination
Broth microdilution methods according to CLSI guidelines M07-A9 and M27-A2 were used for the determination of MIC values [32, 33]. Bacterial and fungal suspensions at 0.5 McFarland standard were prepared in MHB and SDB, respectively. They were diluted to 150 and 250 times with MHB and SDB, respectively. 20 μl of each isoxazoles 4a–i with concentration of 20,480 μg ml−1 in DMSO was added to first and second wells in a row of a 96-well microtiter plate. 20 μl DMSO was added to wells 2–12, and two-fold serial dilutions were carried out in them. 170 μl of MHA or SDB with 10 μl of diluted microbial suspensions were added to all wells. Finally, a concentration range of 2048–1 μg ml−1 of the derivatives was prepared in each row; in addition, the concentration of DMSO did not exceed 10% (v/v). Microtiter plates were incubated with shaking at 100 rpm at 37 °C for 24 h. Fungi must be incubated in the relative humidity (45–55%). The lowest concentration of derivatives that resulted in no visible turbidity was considered as the MIC value.
MBC and MFC determination
Time-kill test according to CLSI guideline M26-A was applied to determine MBC and MFC values [32, 33]. Samples of all wells that showed no growth in the MIC test, were cultured in MHA or SDA media plates. Dishes were incubated at 37 °C for another 24 h under similar conditions. The MBC or MFC was identified as the lowest concentration of derivatives at which no microorganisms survived.
IC50 identification
Free radical scavenging activity of all synthesized heterocycles were evaluated against DPPH [34]. 1 ml of various concentrations of all compounds (25, 50, 75, and 100 µg ml−1) in methanol was added to 4 ml of 0.004% (w/v) methanolic solution of freshly prepared DPPH. Solutions were shaken and left to stand for 30 min at room temperature in darkness. A solution including 1 ml of methanol and 4 ml of 0.004% (w/v) methanolic solution of DPPH was considered as blank sample. The absorbance was read at 517 nm against methanol. It should be noted that the concentration of solute is decreased to one-fifth after a dilution. The inhibition percentage (I%) for scavenging DPPH free radical was calculated according to the following equation:
where “A blank” and “A sample” are the absorbance of control and sample solutions, respectively. A graph of inhibition percentage vs concentration (where X axis is concentration and Y axis is I%). Equation of straight lines was determined. The half maximal inhibitory concentration (IC50) is “x” in equation y = mx + b while y = 50.
Abbreviations
- MHB:
-
Mueller–Hinton broth
- SDB:
-
sabouraud dextrose broth
- MHA:
-
Mueller–Hinton agar
- SDA:
-
sabouraud dextrose agar
- DPPH:
-
1,1-diphenylpicrylhydrazyl
- HIV:
-
the human immunodeficiency virus
- DES:
-
deep eutectic solvent
- MIC:
-
the minimum inhibitory concentration
- MBC:
-
the minimum bactericidal concentration
- MFC:
-
the minimum fungicidal concentration
- FT-IR:
-
Fourier Transform infrared
- 1H NMR:
-
proton nuclear magnetic resonance
- 13C NMR:
-
carbon-13 nuclear magnetic resonance
- UV:
-
ultraviolet
- IC50 :
-
the half maximal inhibitory concentration
- PTCC:
-
Persian Type Culture Collection
- CLSI:
-
Clinical and Laboratory Standards Institute
References
Barmade MA, Murumkar PR, Sharma MK, Yadav MR (2016) Medicinal chemistry perspective of fused isoxazole derivatives. Curr Top Med Chem 16:2863–2883
Bormann AM, Morrison VA (2009) Review of the pharmacology and clinical studies of micafungin. Drug Des Dev Ther 3:295–302
Jorgensen JH, Crawford SA, Fiebelkorn KR (2005) Susceptibility of Neisseria meningitidis to 16 antimicrobial agents and characterization of resistance mechanisms affecting some agents. J Clin Microbiol 43:3162–3171
Fiori ATM, Nakahata DH, Cuin A, Lustri WR, Corbi PP (2017) Synthesis, crystallographic studies, high resolution mass spectrometric analyses and antibacterial assays of silver(I) complexes with sulfisoxazole and sulfadimethoxine. Polyhedron 121:172–179
Kreuzer J, Bach NC, Forler D, Sieber SA (2015) Target discovery of acivicin in cancer cells elucidates its mechanism of growth inhibition. Chem Sci 6:237–245
Jin RY, Sun XH, Liu YF, Long W, Chen B, Shen SQ, Ma HX (2016) Synthesis, crystal structure, biological activity and theoretical calculations of novel isoxazole derivatives. Spectrochim Acta Part A 152:226–232
Panda SS, Chowdary PVR, Jayashree BS (2009) Synthesis, antiinflammatory and antibacterial activity of novel indolyl-isoxazoles. Indian J Pharm Sci 71:684–687
Gutiérrez M, Amigo J, Fuentes E, Palomo I, Astudillo L (2014) Synthetic isoxazole as antiplatelet agent. Platelets 25:234–238
Ali MA, Ismail R, Choon TS, Yoon YK, Wei AC, Pandian S, Samy JG, De Clercq E, Pannecouque C (2011) Synthesis and anti-HIV activity of novel 3-substituted phenyl-6,7-dimethoxy-3a,4-dihydro-3H-indeno[1,2-c]isoxazole analogues. Acta Pol Pharm 68:343–348
Filali I, Romdhane A, Znati M, Jannet HB, Bouajila J (2016) Synthesis of new harmine isoxazoles and evaluation of their potential anti-Alzheimer, anti-inflammatory, and anticancer activities. Med Chem 12:184–190
Abu-Hashem AA, El-Shazly M (2018) Synthesis of new isoxazole-, pyridazine-, pyrimidopyrazines and their anti-inflammatory and analgesic activity. Med Chem. https://doi.org/10.2174/1573406414666180112110947
Tu KN, Hirner JJ, Blum SA (2016) Oxyboration with and without a catalyst: borylated isoxazoles via B-O σ-bond addition. Org Lett 18:480–483
Kadam KS, Gandhi T, Gupte A, Gangopadhyay AK, Sharma R (2016) Alkyl nitrites: novel reagents for one-pot synthesis of 3,5-disubstituted isoxazoles from aldoximes and alkynes. Synthesis 48:3996–4008
Bourbeau MP, Rider JT (2006) A convenient synthesis of 4-alkyl-5-aminoisoxazoles. Org Lett 8:3679–3680
Hansen TV, Wu P, Fokin VV (2005) One-pot copper (I)-catalyzed synthesis of 3,5-disubstituted isoxazoles. J Org Chem 70:7761–7764
Cecchi L, De Sarlo F, Machetti F (2006) 1,4-Diazabicyclo[2.2.2]octane (DABCO) as an efficient reagent for the synthesis of isoxazole derivatives from primary nitro compounds and dipolarophiles: the role of the base. Eur J Org Chem 2006:4852–4860
Johnson L, Powers J, Ma F, Jendza K, Wang B, Meredith E, Mainolfi N (2013) A reliable synthesis of 3-amino-5-alkyl and 5-amino-3-alkyl isoxazoles. Synthesis 45:171–173
Ahmed MSM, Kobayashi K, Mori A (2005) One-pot construction of pyrazoles and isoxazoles with palladium-catalyzed four-component coupling. Org Lett 7:4487–4489
Mirzadeh M, Mahdavinia GH (2012) Fast and efficient synthesis of 4-arylidene-3-phenylisoxazol-5-ones. J Chem. 9:425–429
Madabhushi S, Godala KR, Jillella R, Mallu KKR, Chinthala N (2014) A new multicomponent approach to highly functionalized 2,3-dihydroisoxazoles. Tetrahedron Lett 55:514–516
Saikh F, Das J, Ghosh S (2013) Synthesis of 3-methyl-4-arylmethylene isoxazole-5(4H)-ones by visible light in aqueous ethanol. Tetrahedron Lett 54:4679–4682
Kiyani H, Ghorbani F (2017) Potassium phthalimide as efficient basic organocatalyst for the synthesis of 3,4-disubstituted isoxazol-5(4H)-ones in aqueous medium. J Saudi Chem Soc 21:S112–S119
Kiyani H, Ghorbani F (2015) Boric acid-catalyzed multi-component reaction for efficient synthesis of 4H-isoxazol-5-ones in aqueous medium. Res Chem Intermed 41:2653–2664
Xu P, Zheng G-W, Zong M-H, Li N, Lou W-Y (2017) Recent progress on deep eutectic solvents in biocatalysis. Bioresour Bioprocess 4:34
Ge X, Gu C, Wang X, Tu J (2017) Deep eutectic solvents (DESs)-derived advanced functional materials for energy and environmental applications: challenges, opportunities, and future vision. J Mater Chem A 5:8209–8229
García JI, García-Marína H, Pires E (2010) Glycerol based solvents: synthesis, properties and applications. Green Chem 12:426–434
AlOmar MK, Hayyan M, Alsaadi MA, Akib S, Hayyan A, Hashim MA (2016) Glycerol-based deep eutectic solvents: physical properties. J Mol Liq 215:98–103
Beyzaei H, Motraghi Z, Aryan R, Zahedi MM, Samzadeh-Kermani A (2017) Green one-pot synthesis of novel polysubstituted pyrazole derivatives as potential antimicrobial agents. Acta Chim Slov 64:911–918
Padmaja A, Rajasekhar C, Muralikrishna A, Padmavathi V (2011) Synthesis and antioxidant activity of oxazolyl/thiazolylsulfonylmethyl pyrazoles and isoxazoles. Eur J Med Chem 46:5034–5038
Naser J, Mjalli F, Jibril B, Al-Hatmi S, Gano Z (2013) Potassium carbonate as a salt for deep eutectic solvents. Int J Chem Eng Appl 4:114–118
El-Shafei AK, Sultan AA, Soliman AM, Meetzger J (1992) Synthesis and reactions of some 1,3-(2-thienyl)-1,3 oxopropane nitrile derivatives. Phosphorus Sulfur Silicon Relat Elem 72:121–126
Beyzaei H, Moghaddam-Manesh M, Aryan R, Ghasemi B, Samzadeh-Kermani A (2017) Synthesis and in vitro antibacterial evaluation of 6-substituted 4-aminopyrazolo[3,4-d]pyrimidines. Chem Pap 71:1685–1691
Arikan S (2007) Current status of antifungal susceptibility testing methods. Med Mycol 45:569–587
Bhaskara Reddy MV, Srinivasulu D, Peddanna K, Apparao Ch, Ramesh P (2015) Synthesis and antioxidant activity of new thiazole analogues possessing urea, thiourea, and selenourea functionality. Synth Commun 45:2592–2600
Authors’ contributions
HB: design of target compounds and supervision of synthetic part. MKD: synthesis of title compounds and collaboration in the antimicrobial and antioxidant tests. RA: design of target compounds and supervision of synthetic part. BG: supervision of pharmacological part. MMZ: collaboration in the synthetic part. MMM: collaboration in the synthetic part. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank the members of the University of Zabol for their support and assistance at the various stages this project.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All main data were presented in the form of tables and figures. Meanwhile, copies of 1H NMR and 13C NMR spectra for the title compounds were presented in the Additional file 1.
Funding
This work was supported by the [University of Zabol] under Grant [Number UOZ-GR-9517-15].
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1.
The copies of 1H NMR and 13C NMR spectra for isoxazoles 4a–i.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Beyzaei, H., Kamali Deljoo, M., Aryan, R. et al. Green multicomponent synthesis, antimicrobial and antioxidant evaluation of novel 5-amino-isoxazole-4-carbonitriles. Chemistry Central Journal 12, 114 (2018). https://doi.org/10.1186/s13065-018-0488-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0488-0