
Abstract
Background
Usher syndrome (USH) is the most common disease causing combined deafness and blindness. It is predominantly an autosomal recessive genetic disorder with occasionally digenic cases. Molecular diagnosis of USH patients is important for disease management. Few studies have tried to find the genetic cause of USH in Chinese patients. This study was designed to determine the mutation spectrum of Chinese USH patients.
Methods
We applied next generation sequencing to characterize the mutation spectrum in 67 independent Chinese families with at least one member diagnosed with USH. Blood was collected at Peking Union Medical College Hospital. This cohort is one of the largest USH cohorts reported. We utilized customized panel and whole exome sequencing, variant analysis, Sanger validation and segregation tests to find disease causing mutations in these families.
Results
We identified biallelic disease causing mutations in known USH genes in 70 % (49) of our patients. As has been previously reported, MYO7A is the most frequently mutated gene in our USH type I patients while USH2A is the most mutated gene in our USH type II patients. In addition, we identify mutations in CLRN1, DFNB31, GPR98 and PCDH15 for the first time in Chinese USH patients. Together, mutations in CLRN1, DNFB31, GPR98 and PCDH15 account for 11.4 % of disease in our cohort. Interestingly, although the spectrum of disease genes is quite similar between our Chinese patient cohort and other patient cohorts from different (and primarily Caucasian) ethnic backgrounds, the mutations themselves are dramatically different. In particular, 76 % (52/68) of alleles found in this study have never been previously reported. Interestingly, we observed a strong enrichment for severe protein truncating mutations expected to have severe functional consequence on the protein in USH II patients compared to the reported mutation spectrum in RP patients, who often carry partial protein truncating mutations.
Conclusions
Our study provides the first comprehensive genetic characterization of a large collection of Chinese USH patients. Up to 90 % of USH patients have disease caused by mutations in known USH disease genes. By combining NGS-based molecular diagnosis and patient clinical information, a more accurate diagnosis, prognosis and personalized treatment of USH patients can be achieved.
Similar content being viewed by others
Background
Usher Syndrome (USH) is the most common disease of combined deafness and blindness. It is characterized by sensorineural hearing loss (SNHL), retinitis pigmentosa (RP), and manifests with or without vestibular dysfunction. Depending on the age of onset, the severity of the retinal and hearing phenotypes and the presence or absence of vestibular dysfunction, USH is classified into three major categories. USH I is characterized by congenital deafness with severe balance problems. USH I patients develop vision problems in early childhood. USH II patients are born with moderate to severe hearing loss, normal balance, and usually develop RP during adolescence. USH III patients have normal or near normal balance, progressive hearing loss, and vision problems varying in severity which usually develop during adolescence [1]. Genetically, USH is genetically heterogeneous, with 12 known disease genes and 3 additional loci having been identified so far [1–4]. USH is predominantly a recessive disease, though other inheritance patterns occur at lower frequency. For instance, a digenic USH can be caused by simultaneous mutations in both PCDH15 and CDH23 [1]. In addition, truncation of PDZD7 has been reported as a modifier of GPR98 and USH2A mutations [5]. It is also worth noting that mutations in many of the USH genes can also lead to deafness without a retinal phenotype [6–9].
Given the high clinical and genetic complexity underlying USH, molecular screening for mutations in USH genes significantly improves diagnosis. Next generation sequencing (NGS) is emerging as a cost-efficient technology for sequencing a large number of genes [10, 11]. NGS is ideally suited for molecular diagnosis of USH for two reasons. First, many USH disease genes have many isoforms with a large number of exons. In total, more than 400 coding exons have been annotated in known USH genes. As a result, mutation screening for all coding exons by Sanger sequencing is cost prohibitive while NGS is feasible. Second, a large diversity of pathogenic alleles of various types has been reported and novel mutations are frequent, making array based diagnosis inaccurate. Indeed, in a recent report a European USH patient cohort was screened for mutations by Sanger sequencing. Interestingly, 48 % of the alleles identified were novel [12]. The rate of novel mutations is expected to be even higher in poorly studied populations like our Chinese cohort. Therefore, a sequence-based approach is necessary to achieve high diagnosis rate.
Although USH patients of European descent have been under intense investigation [1], only a small number of studies have been published on Chinese USH cohorts, each with a no more than 10 cases [13–17]. Mutations in Chinese USH patients from these studies occurred only in two genes, MYO7A and USH2A. Furthermore, founder mutations specific for many ethnic groups have been identified. For example, the founder mutation c.8559-2A > G inUSH2A accounts for 26 % of all Western Japanese USH patients but was never observed in Europeans. Similarly, the most prevalent mutation in European populations, c.2299delG in USH2A, has never been observed in Asian patients [18, 19]. To gain insight into the molecular basis of Chinese USH patients we performed comprehensive NGS of all known USH genes in a cohort of 67 probands and their families. Indeed, our cohort has a different mutation spectrum than that of patients of European descent.
Methods
Clinical diagnosis of USH and sample collection
All subjects were initially enrolled at Peking Union Medical College Hospital (PUMCH). During their initial visit, a full medical and family history was recorded, pedigrees were drawn, and ophthalmological examinations were performed. Each patient underwent a standard ophthalmic examination including best correct visual acuity (BCVA) according to projected Snellen charts, slit-lamp biomicroscopy, dilated indirect ophthalmoscopy, fundus photography if possible, and visual field tests (Octopus,Interzeag, Schlieren, Switzerland). Retinal structure was examined by optical coherence tomography (OCT) (Topcon, Tokyo, Japan). Electroretinograms (ERGs) were performed (RetiPort ERG system, Roland Consult, Wiesbaden, Germany) using corneal “ERGjet” contact lens electrodes. The ERG protocol complied with the standards published by the International Society for Clinical Electrophysiology of Vision. Auditory examinations including otoscopic exploration, pure-tone and speech audiometry were conducted by Otolaryngologists.
The diagnosis of USH was based on previously reported criteria [20]. Written informed consent was obtained from all participating individuals or their guardians. Genomic DNA was isolated from peripheral leukocytes using QIA amp DNA Blood Midi Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol. This study was approved by the Institutional Review Board of PUMCH and adhered to the tenets of the Declaration of Helsinki and the Guidance on Sample Collection of Human Genetic Diseases by the Ministry of Public Health of China.
Design of retinal disease capture panel
A capture panel of retinal disease genes was developed and assessed by our group [10, 11]. All annotated coding exons and flanking splicing sites for 9 USH genes (MYO7A, PCDH15, CDH23, USH1C, USH1G, USH2A, GPR98, DNFB31, and CLRN1) and one USH modifier gene PDZD7 were included in the capture design. In total, the panel included 196 known retinal disease genes (Additional file 1: Table S1). For all patients without a positive molecular diagnosis, mutations in other recently reported USH disease genes, CIB2, HARS and ABHD12, were screened by whole exome sequencing. Whole exome sequencing was performed as described previously [21].
Panel capture sequencing
About 50 pre-capture libraries were pooled together for one panel capture reaction. Agilent Hybridization and Wash Kits were used for panel capture, following the standard manufacturer’s protocol. Captured libraries were sequenced on the Illumina HiSeq 2000 as 100-bp paired-end reads, following the manufacturer's protocols. Whole exome sequencing library construction, capture and sequencing was performed as previously described [21].
Bioinformatics analysis of sequencing results and pathogenic mutation identification
Sequence data was processed through an automated pipeline developed in house as previously described [10, 11]. Briefly, raw reads were mapped to the hg19 reference genome followed by variant calling including SNPs and indels. Variants were then filtered against both publicly available databases and internal databases with a cut off frequency of less than 0.5 % in the general population. The HGMD professional database (http://www.biobase-international.com/product/hgmd) and USH bases (https://grenada.lumc.nl/LOVD2/Usher_montpellier/USHbases.html) [22] were used to search for known pathogenic mutations. We utilized a previously described stepwise strategy to systematically identify the putative pathogenic mutations for each USH family. Mutations in 9 known USH genes were checked for, in order, known pathogenic mutations, novel loss-of-function mutations, and novel missense mutations. In cases where missense mutations segregated with disease, they were considered as pathogenic even if their functional predictions were neutral. We also considered reported digenic inheritance of PCDH15/CDH23, PDZD7/GPR98 and PDZD7/USH2A. We only considered monoallelic mutations if they were reported pathogenic missense mutations, nonsense mutations, frameshift mutation and splice site mutations in known USH genes. The same prioritizing strategy was applied to other retinal disease genes and we only picked mutations that fit the disease model of a gene. Sanger validation was performed for all putative causal pathogenic mutations. Segregation tests were performed when additional family members were available.
Results
Recruitment of 67 USH families and clinical diagnosis
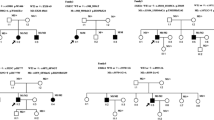
In this study, we recruited a total of 70 patients from 67 unrelated USH families from different regions of China. This group contained 14 patients diagnosed with USH type I, 54 patients as USH type II or USH type II-like, 1 patient as USH type III, and 1 patient with an undetermined subtype. In most families, the proband was the only affected member in the family, including three patients from consanguineous marriages (USHsrf2, USHsrf38, and USHsrf56) (Fig. 1). Two families, USHsrf24 and USHsrf66, have multiple affected members. In family USbHsrf24, both the father and the daughter were diagnosed with USH II. As shown in Fig. 1, the USHsrf66 family is a large family with five affected members, including USHsrf66, USHsrf68, and USHsrf59 who were recruited for this study. Detailed clinical information pertaining to these families is included in Additional file 1: Table S3. All our patients exhibited phenotypes consistent with USH syndrome [20]. All patients’ clinical phenotypes are listed in Additional file 1: Table S3, while representative fundus images and hearing test results are shown in Fig. 2.

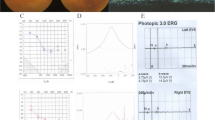
Pedigrees of non-simplex and consanguineous families and sample example figures of clinical data. a Pedigrees of non-simplex and consanguineous families. USH patients are illustrated by squares or circles in black while the unaffected family members are in white. Patients with DNA sequenced by panel or whole exome sequencing in our project are indicated by an arrow. b Fundus of left eye of USHsrf59 at age 31. The fundus showed salt and pepper pigmentation variation in the periphery retina and attenuation of the retinal vessels. c OCT of left eye of USHsrf59 at age 31. OCT showed lack of IS/OS except macula fovea in photoreceptor layer. Her visual acuity is 0.8/0.5 at age 31. This patient was diagnosed with USH II. Her hearing loss began at age 5 and vision loss began at age 12. d Fundus of right eye of USHsrf66 at age 57. The fundus showed bone spicule pigmentation variation and attenuation of the retinal vessels. e OCT of left eye of USHsrf66 at age 57. Her visual acuity is 0.06/0.06 at age 57. OCT showed a thinned retinal pigment epithelium and a photoreceptor layer (lack of IS/OS). This patient was diagnosed with USH II. Her hearing loss began at age 8 and vision loss began at age 30 with night blindness starting from school age. f Hearing test on left ear of USHsrf66

Another sample figure title Summary of mutations identified in USH genes. a Genes mutated in USH I patients. b Genes mutated in USH II patients
Mutation screening for known USH and other eye disease genes
A gene capture panel containing196 known retinal disease genes was developed in our lab and has effectively identified mutations in known retinal disease genes [10, 11]. We applied this retinal disease gene panel to our USH patients and excellent coverage was achieved with an average coverage of 109X for target genes (Additional file 1: Table S2). On average, 96.8 % of the target region was sequenced with more than 10X coverage which is sufficient to call heterozygous mutations.
An in-house automatic variant calling, filtering, and annotation pipeline was used to analyse the sequencing data. Filtering against multiple public and internal databases, only rare SNPS and indels (defined as a frequency < 0.5 % in 20,000 controls) were retained for each patient. Each variant was further annotated and the ones that do not affect protein coding were further removed. As a result, on average 15 rare variants in all known retinal disease genes, including 3 in the USH disease genes, were identified per patient. The pathogenicity of these variants was further evaluated as described in the materials and methods section.
Mutations were identified in 10 out of 14 USH I patients
Biallelic mutations in known USH genes were detected in 10 USH I patients (Fig. 2a). Consistent with previous reports, MYO7A was the most frequently mutated gene with 13 different pathogenic mutations found in 8 patients (Table 1). Among them, 3 have been previously reported as pathogenic alleles while the other 10 alleles are novel, including 3 frameshift mutations, 2 splicing site mutations, 3 nonsense mutations, and 2 missense mutations (Table 1). Both novel missense alleles, MYO7A: c. 2837 T > G: M946R and MYO7A: c. 5396 T > C: P.L1799P, are likely to be pathogenic based on the following evidence. First, both are extremely rare in the control population and have not been observed in any of the public or our internal variant databases that together contain around 20,000 individuals. Second, the amino acids M946 and L1799 are highly conserved across vertebrates and all the way to invertebrates (Additional file 2: Figure S1). Third, in silico prediction of M946R and L1799P variants suggests that they are likely to be detrimental (Additional file 1: Table S6). Finally, both variants segregated with disease within the families in which they occurred.
Additional putatively pathogenic mutations were identified in PCHD15 and CLRN1 in this group of USH I patients. Patient USHsrf8 was found to carry compound heterozygous mutations in PCDH15, consisting of the novel frameshift mutation PCDH15:c.1799_1800insTA:p.S600fs and the novel nonsense mutation PCDH15:c. 2893A > T: p.R965X (Table 1). Interestingly, patient USHsrf14 had an unexpected molecular diagnosis because patient USHsrf14 was diagnosed with USH I but has mutations in CLRN1 which have previously been reported to cause mostly USH III [23]. In one previous case, a patient with USH I was reported to have a CLRN1 frameshift mutation [24]. Together with our study, this may indicate certain CLRN1 mutations can cause USH I. This patient had severely impaired hearing at a very young age and got cochlear implants around age of 6. She experienced poor night vision and wore glasses before she turned 10. This patient was found to carry novel nonsense mutation CLRN1:c. 658C > T:p. R220X and novel missense mutation CLRN1:c. 190G > A:pG64R, which is predicted to be pathogenic (Additional file 1: Table S6). The nonsense mutation is from the patient’s father while the missense mutation is from the patient’s mother, and the mutation thus segregated with disease in the small pedigree.
Mutations were identified in 39 of 54 USH II and atypical patients
Biallelic mutations were detected in 39 USH II or USH II-like patients (Table 2), with USH2A mutated in 32 patients, GRP98 mutated in 3 patients, CLRN1 mutated in 2 patients, MYO7A mutated in 1 patient, and DFNB31 mutated in 1 patient (Fig. 2b).
Consistent with previous reports, we found that USH2A was the most frequently mutated gene in USH II patients, accounting for about 60 % (32 out of 54) of patients in this cohort. A total of 40 different mutations were identified in USH2A, including 27 novel alleles. The vast majority of the novel alleles (21/27) are clearly null mutations, including frameshift, splice site, and nonsense mutations (Table 2). In addition, we identified 6 novel missense mutations predicted to be pathogenic (Table 2). It is worth noting that these novel mutations are mostly private and only two alleles, p.S2251X and p.1912_1912delfs, were observed in two probands. GPR98 is the second most frequently mutated gene in our USH II patients, with pathogenic mutations occurring in 3 patients. Two homozygous mutations in USH type III gene CLRN1 were found in 2 USH II patients. Compound heterozygous missense variants in USH type I gene MYO7A was identified in USH II patient USHsrf40, who carries two missense variants c.4951G > A: p.D1651N and c. 4360G > A: p.V1454I. Both of these variants are absent in the control database and segregate with disease in the family (Fig. 3). Novel homozygous splicing site mutation c.963 + 1G > A in DFNB31 was found in a USH II patient from a consanguineous family, which was confirmed by segregation tests.

Double compound heterozygous mutations in patient USHsrf40. Patient USHsrf40 carries compound heterozygous mutations in two genes MYO7A and CGNA1: two missense mutation in MYO7A and frameshift and missense mutations in CNGA1. Mutations segregate in this family
Patient USHsrf26 was the only patient in our cohort with USH type III, while patient USHsrf3 that does not fit well in any Usher subtype. Patient USHsrf3 was a student in a boarding school for disabled kids. There is no detailed medical record of him and his guardian, a teacher, didn’t know his past medical history. The primary diagnosis of Usher syndrome was reached because he had hearing problems and a retinal phenotype.
No putative mutations in known USH disease genes have been found for either of these two patients.
Biallelic mutations in 3 patients were found in retinal disease genes previously un-associated with USH
We reasoned that mutations in other known retinal disease genes might account for the clinical phenotype observed in some of the patients in our cohort for several reasons. First, some of our patients may have a different syndromic disease whose phenotype is similar to USH. Second, it other retinal disease genes may cause USH syndrome. Third, the hearing loss and visual defects could co-occur in one individual as a result of mutations in multiple genes, one causing eye disease and a second gene causing hearing defects. Finally, it is possible that only part of the clinical phenotype of a patient has genetic cause.
To test these hypotheses, we checked whether our patients bear mutations in other known retinal disease genes. Interestingly, two patients, USHsrf62 and USHsrf5, were found to carry mutations in EYS that have been associated with RP [25]. Patient USHsrf62 is homozygous for a novel frameshift mutation c.910delT:p.W304fs in EYS (Additional file 1: Table S5). Patient USHsrf5 carries a novel frameshift mutation, c.8392delG:p.D2798fs and a known missense mutation in EYS. Therefore, it is likely that the RP phenotype in these two patients is due to their mutations in EYS. As many patients with mutations in EYS have been reported and none of them showed hearing loss, it is likely that these patients’ hearing problems are independent of the retinal phenotype. We did observe a splice site mutation and a missense mutation in LOXHD1 in USHsrf62 in our whole exome sequencing data. Mutations in this gene are associated with non-syndromic hearing loss, making it likely that the patient’s visual and auditory problems have independent genetic origins.
Our molecular diagnosis also suggests that patient USHsrf40’s hearing loss and RP might be explained by mutations in multiple genes. This patient was diagnosed with USH II based on his clinical phenotype. Interestingly, compound heterozygous mutations were identified in both MYO7A and CNGA1 (Fig. 3). Patients with mutations in MYO7A exhibited a spectrum of phenotypes ranging from USH I to USH III to atypical USH consisting of non-syndromic hearing loss without retinal a phenotype [9, 26, 27]. In contrast, mutations in CNGA1 have only been linked to RP so far [28]. Since patient USHsrf40 didn’t have a vestibular problem, a phenotype observed in both USH I and III type patients, it is possible that the two mutations in MYO7A in USHsrf40 only lead to deafness while mutations in CGNA1 are the underlying cause of the RP phenotype. Supporting this idea, mutations in MYO7A identified in this patient were indeed relatively weak. In particular, one of the missense variants,MYO7A:c.4360 G > A:p.V1454I, was predicted to be neutral by all functional prediction tools used except CADD [29] (Additional file 1: Table S6). Given that the mutation affects a highly conserved amino acid, was absent from all control databases and segregates with the disease, this variant is was likely to be mildly pathogenic.
No mutations were identified in CIB2, ABHD12 and HARS
Three of the known USH disease genes, CIB2, ABHD12 and HARS, were not included in the capture panel. To achieve a comprehensive screening, we performed whole exome sequencing (WES) on patients who were negative for mutations in known USH disease genes after the target capture sequencing. No mutations were identified in these three USH genes that were not included in our panel, indicating that mutations in these three genes are not major causes of USH in Chinese patients.
USH2A mutation severity determines patient phenotype
We identified 40 distinct USH2A alleles in this study. Previous studies from multiple groups, including ours, have already shown that mutations in USH2A can lead to either USH II or non-syndromic RP [10, 29]. We compared the USH2A alleles from 32 USH II patients identified in this paper with a collection of 38 RP patients whose disease was caused by USH2A mutations ([10] and our unpublished data). The number of obviously null alleles (including nonsense mutations, splicing mutations, and frameshift mutations) carried by each patient was counted. As shown in Fig. 4, the vast majority of USH II patients carry at least one null allele (29/32). Specifically, 17 patients carry two null alleles and 12 USH II patients carry one null allele. In contrast, among the 38 RP patients, only 2 carry two null alleles and 12 carry one null allele. Therefore, mutations carried by USH II patients tend to be more severe than those found in RP patients (Fisher’s exact test p-value < 0.0001). Indeed, patients with two severe mutations in USH2A were predominantly USH II patients (53 % USH II vs 5 % RP) while patients with two missense mutations were largely RP patients (9 % USH II vs 63 % RP). Further supporting our observations, the vast majority of USH2A alleles identified from another published USH II patient cohort are null alleles (Fig. 4) [30]. It is likely that severe disruption of USH2A causes both hearing and RP phenotypes in most cases, while milder disruptions to USH2A only cause RP except in patients with a background or environment predisposed to hearing loss.

USH patients are highly enriched in patients with two severe alleles. Patients with USH2A mutations were classified based on number of severe alleles (frameshift mutations, splicing site mutations and nonsense mutations). Enrichment of patients with two severe mutations is significant (Fisher exact test, p-value < 0.0001) in two independent USH patients cohorts (USH patients in this study [30]) compared to that of RP patients
Discussion
In this report, we comprehensively screened 67 unrelated USH families for disease causing mutations. This is the largest Chinese USH cohort molecularly tested to date. All known USH disease genes as well and other known retinal disease genes were screened for mutations using a combination of panel capture and whole exome sequencing, representing the first NGS based comprehensive molecular characterization of a large Usher patient cohort. This approach allowed us to obtain accurate estimates of mutation frequency in known USH disease genes in the Chinese population. Our results reveal a similar but distinct mutation spectrum in Chinese USH patients compared to European patients.
In our cohort, causal mutations were identified in 47 families (49 patients), achieving an overall solving rate of 70 %. This is similar but slightly lower than that of previous studies on patients of European descent, which used sanger sequencing of 9 genes to obtain diagnosis rates of 72 and 85 % [12, 30]. In our patient cohort, USH2A and MYO7A are the most frequently mutated genes, accounting for 46 and 12 % patients, respectively. This rate is similar to the 55 and 14 %, respectively, observed in a previous study that Sanger sequenced 9 USH genes in 172 ethnically heterogeneous UK patients who are primarily Caucasian [30]. In addition to these two most frequently mutated genes, mutations in CDH23, PCDH15, USH1C, USH1G, GPR98, DBNF31, CLRN1, CIB2, ABHD12 and HARS have been reported to cause USH syndrome. However, these 10 genes only each account for a small percent of the patients and together account for no more than 20 % [1, 12, 30]. Until now, patients with mutations in these less frequently mutated genes have not been reported for Chinese USH patients. In this cohort, mutations in these genes together account for about 11.4 % patients. Significantly, we identify the first Chinese patient(s) with mutations inCLRN1, DFNB31, GPR98 and PCDH15.
In contrast to the similarity in frequency of mutations in known USH disease genes between ethnic groups, many alleles identified in the Chinese patient cohort are absent from other ethnic groups. For example, in our study, a total of 40 alleles have been identified in USH2A with 67.5 % being novel alleles. This is striking as a large number of mutations (>200) have already been reported for USH2A with much lower rate (40 % and 48 %) of novel causal alleles identified in studies screened USH genes in European patients [12, 30]. Since the vast majority of reported alleles come from studies of patients of European descent, the allele spectrum in our Chinese patients differs from that present in current allele databases. This idea is further supported by the observation that, for the 13 known mutations in USH2A identified in this study, only 8 alleles were been previously reported in Caucasians, while the other 5 were exclusively reported in Chinese or Japanese patients [16, 18, 31]. Furthermore, strong founder effect has been observed in both ethnic groups. For example, USH2A:c.2299delG, which is the most prevalent European mutation and accounts at approximately 30 % of all European USH2A [19], was not detected in our patients despite being thoroughly tested. In contrast, the splice site mutation USH2A:c.8559-2A > G, which has been previously identified solely in Chinese and Japanese patients, is the most frequent mutation in our patient cohort and was observed in 11 patients.
Our study clearly demonstrates that the molecular basis of USH syndrome is highly heterogeneous in several ways. First, although founder mutations have been identified, the vast majority of the alleles are rare and each only appears in a small fraction of patients. Indeed, 77 % of the alleles identified in our study are novel. The large diversity of mutations in USH genes has also been noted in other Middle Eastern populations [32]. With this in mind, we expect a high rate of novel mutations in the Chinese population. Second, multiple genes have been associated with USH syndrome. To date, 15 USH associated genes have been identified. In our study, mutations in 6 known USH disease genes were found. Third, mutations in the same gene can lead to different clinical phenotypes. For example, CLRN1 mutations have been associated with USH III [23] while in our study, 3 patients with CLRN1 mutations exhibit USH I or USH II. Our study shows CLRN1 causes a broad spectrum of hearing and retinal phenotypes. Finally, the same clinical phenotype can be caused by mutations in multiple genes. For example, one patient in our cohort, USHsrf40, carries mutations in both MYO7A (c.4951G > A and c.4360G > A) and CNGA (c.265delC and c.479C > T) that result in hearing and vision impairment respectively; therefore this patient does not have canonical USH syndrome. Given this heterogeneity, it is important to combine a patient’s clinical information with their molecular diagnosis in order to provide patients with better prognoses and to help match management and treatment strategies with the patient disease.
Several attempts to establish genotype and phenotype correlation in USH2A have been reported [33, 34]. None of these studies identified apparent genotype/phenotype correlations. Similar to previous reports, no apparent genotype/phenotype correlations were observed when we cross referenced the patient’s alleles and their clinical phenotypes. Interestingly, a strong correlation between he genotype and phenotype was observed when we compared alleles obtained from our USH cohort and our own and previously reported RP cohorts. Our study suggests that more severe loss-of-function mutations in USH2A lead to syndromic retinopathy. By determining the severity of various USH mutations we could predict the disease that babies or fetuses with a given genotype are likely to develop.
In our patient cohort we observed an enrichment of mono-allelic mutations genetically undiagnosed patients, particularly in USH2A. For patients where bi-allelic mutations could not be found, mono-allelic USH2A nonsense mutations, frameshift and splicing site mutations occurred in 10 out of 54 USH II patients (20 %) (Additional file 1: Table S4). This is significantly higher than what is observed in controls, which typically have a frequency of less than 1 % (internal unpublished data). This data suggests that a significant portion of USH2A mutations might be missed by exome capture sequencing. Given that the entire coding region of the USH2A was well covered by our design, it is likely that the missing alleles are either in non-coding regions or are structural rearrangements, such as deletions or inversions that affect USH2A protein production. Indeed, a recent study of theUSH2A locus reveals 35 % of USH2A monoallelic cases can be solved by screening for duplications, deletions and deep intronic mutations [35]. Thus, much of the missing heritability in USH could be achieved as follows. First, we could discover mutations not previously annotated by improving functional prediction software. An example of this is a recent study of ABCA4 that demonstrated the effect of synonymous mutations and splice site modification mutations as a major cause of Stargardt’s disease [36]. Second, we can use a compensatory method, such as comparative genomic hybridization, to detect duplications and deletions. Third, sequencing gene promoters and other regulatory regions will allow for the identification of pathogenic regulatory mutations. The lack of reliable prediction tools and high throughput experimental assays are the main bottlenecks in identifying these types of mutations.
In summary, we report the first NGS-based comprehensive molecular survey of a large Chinese USH patient cohort. Our results suggest that up to 90 % of USH patients are due to mutations in known USH disease genes when including patients with monoallelic mutations in USH2A. By combining molecular diagnosis and patient clinical information, a more accurate diagnosis, prognoses and personalized treatment of individual USH patients can be achieved.
Conclusions
Our study provides the first comprehensive characterization of a large collection of Chinese USH patients. Up to 90 % of USH patients are due to mutations in known USH disease genes. By combing NGS-based molecular diagnosis and patient clinical information, a more accurate diagnosis, prognosis and personalized treatment of USH patients can be achieved.
Abbreviations
- USH:
-
USH syndrome
- ERG:
-
Electroretinograms
- OCT:
-
Optical coherence tomography
- NGS:
-
Next generation sequencing
- RP:
-
Retinitis pigmentosa
- BCVA:
-
Best-corrected visual acuity
References
Millan JM, Aller E, Jaijo T, Blanco-Kelly F, Gimenez-Pardo A, Ayuso C. An update on the genetics of usher syndrome. J Ophthalmol. 2011;2011:417217.
Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, Nandamuri SP, et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1 J and nonsyndromic deafness DFNB48. Nat Genet. 2012;44(11):1265–71.
Puffenberger EG, Jinks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One. 2012;7(1);e28936.
Eisenberger T, Slim R, Mansour A, Nauck M, Nurnberg G, Nurnberg P, et al. Targeted next-generation sequencing identifies a homozygous nonsense mutation in ABHD12, the gene underlying PHARC, in a family clinically diagnosed with Usher syndrome type 3. Orphanet J Rare Dis. 2012;7:59.
Zou J, Zheng T, Ren C, Askew C, Liu XP, Pan B, et al. Deletion of PDZD7 disrupts the Usher syndrome type 2 protein complex in cochlear hair cells and causes hearing loss in mice. Hum Mol Genet. 2014;23:2374–90.
Schultz JM, Bhatti R, Madeo AC, Turriff A, Muskett JA, Zalewski CK, et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J Med Genet. 2011;48(11):767–75.
Tlili A, Charfedine I, Lahmar I, Benzina Z, Mohamed BA, Weil D, et al. Identification of a novel frameshift mutation in the DFNB31/WHRN gene in a Tunisian consanguineous family with hereditary non-syndromic recessive hearing loss. Hum Mutat. 2005;25(5):503.
Ouyang XM, Xia XJ, Verpy E, Du LL, Pandya A, Petit C, et al. Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Hum Genet. 2002;111(1):26–30.
Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, et al. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet. 1997;16(2):188–90.
Wang F, Wang H, Tuan HF, Nguyen DH, Sun V, Keser V, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133(3):331–45.
Wang X, Wang H, Sun V, Tuan HF, Keser V, Wang K, et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50(10):674–88.
Bonnet C, Grati M, Marlin S, Levilliers J, Hardelin JP, Parodi M, et al. Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J Rare Dis. 2011;6:21.
Liu F, Li P, Liu Y, Li W, Wong F, Du R, et al. Novel compound heterozygous mutations in MYO7A in a Chinese family with Usher syndrome type 1. Mol Vis. 2013;19:695–701.
Wei X, Sun Y, Xie J, Shi Q, Qu N, Yang G, et al. Next-generation sequencing identifies a novel compound heterozygous mutation in MYO7A in a Chinese patient with Usher Syndrome 1B. Clin Chim Acta. 2012;413(23–24):1866–71.
Xu W, Dai H, Lu T, Zhang X, Dong B, Li Y. Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II. Mol Vis. 2011;17:1537–52.
Dai H, Zhang X, Zhao X, Deng T, Dong B, Wang J, et al. Identification of five novel mutations in the long isoform of the USH2A gene in Chinese families with Usher syndrome type II. Mol Vis. 2008;14:2067–75.
Huang XF, Xiang P, Chen J, Xing DJ, Huang N, Min Q, et al. Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PLoS One. 2013;8(5);e63832.
Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Usami S, Mizuta K, et al. Novel USH2A mutations in Japanese Usher syndrome type 2 patients: marked differences in the mutation spectrum between the Japanese and other populations. J Hum Genet. 2011;56(7):484–90.
Aller E, Najera C, Millan JM, Oltra JS, Perez-Garrigues H, Vilela C, et al. Genetic analysis of 2299delG and C759F mutations (USH2A) in patients with visual and/or auditory impairments. Eur J Hum Genet. 2004;12(5):407–10.
Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, et al. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am J Med Genet. 1994;50(1):32–8.
Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012;44(9):1035–39.
Baux D, Faugere V, Larrieu L, Le Guedard-Mereuze S, Hamroun D, Beroud C, et al. UMD-USHbases: a comprehensive set of databases to record and analyse pathogenic mutations and unclassified variants in seven Usher syndrome causing genes. Hum Mutat. 2008;29(8):e76–87.
Akoury E, El Zir E, Mansour A, Megarbane A, Majewski J, Slim R. A novel 5-bp deletion in Clarin 1 in a family with Usher syndrome. Ophthalmic Genet. 2011;32(4):245–9.
Abd El-Aziz MM, O'Driscoll CA, Kaye RS, Barragan I, El-Ashry MF, Borrego S, et al. Identification of novel mutations in the ortholog of Drosophila eyes shut gene (EYS) causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2010;51 (8):4266-72.
Guilford P, Ayadi H, Blanchard S, Chaib H, Le Paslier D, Weissenbach J, et al. A human gene responsible for neurosensory, non-syndromic recessive deafness is a candidate homologue of the mouse sh-1 gene. Hum Mol Genet. 1994;3(6):989-93.
Liu XZ, Hope C, Walsh J, Newton V, Ke XM, Liang CY, et al. Mutations in the myosin VIIA gene cause a wide phenotypic spectrum, including atypical Usher syndrome. Am J Hum Genet. 1998;63(3):909-12.
Dryja TP, Finn JT, Peng YW, McGee TL, Berson EL, Yau KW. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci U S A. 1995;92(22):10177-81.
Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000;66(6):1975-78.
Le Quesne Stabej P, Saihan Z, Rangesh N, Steele-Stallard HB, Ambrose J, Coffey A, et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J Med Genet. 2012;49(1):27-36.
Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Mizuta K, Mineta H, et al. Identification of 11 novel mutations in USH2A among Japanese patients with Usher syndrome type 2. Clin Genet. 2009;76(4):383-91.
Reddy R, Fahiminiya S, El Zir E, Mansour A, Megarbane A, Majewski J, et al. Molecular genetics of the Usher syndrome in Lebanon: identification of 11 novel protein truncating mutations by whole exome sequencing. PLoS One. 2014;9(9):e107326.
Joensuu T, Hamalainen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet. 2001;69(4):673–84.
Bernal S, Meda C, Solans T, Ayuso C, Garcia-Sandoval B, Valverde D, et al. Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype--phenotype correlation. Clin Genet. 2005;68(3):204–14.
Sadeghi AM, Cohn ES, Kimberling WJ, Halvarsson G, Moller C. Expressivity of hearing loss in cases with Usher syndrome type IIA. Int J Audiol. 2013;52(12):832–7.
Steele-Stallard HB, Le Quesne Stabej P, Lenassi E, Luxon LM, Claustres M, Roux AF, et al. Screening for duplications, deletions and a common intronic mutation detects 35 % of second mutations in patients with USH2A monoallelic mutations on Sanger sequencing. Orphanet J Rare Dis. 2013;8:122.
Braun TA, Mullins RF, Wagner AH, Andorf JL, Johnston RM, Bakall BB, et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet. 2013;22(25):5136–45.
Cremers FP, Kimberling WJ, Kulm M, de Brouwer AP, van Wijk E, te Brinke H, et al. Development of a genotyping microarray for Usher syndrome. J Med Genet. 2007;44(2):153–60.
Bharadwaj AK, Kasztejna JP, Huq S, Berson EL, and Dryja TP. Evaluation of the myosin VIIA gene and visual function in patients with Usher syndrome type I. Exp Eye Res. 2000;71(2):173–81.
Dreyer B, Brox V, Tranebjaerg L, Rosenberg T, Sadeghi AM, Moller C, et al. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat. 2008;29(3):451.
Dreyer B, Tranebjaerg L, Rosenberg T, Weston MD, Kimberling WJ, Nilssen O. Identification of novel USH2A mutations: implications for the structure of USH2A protein. Eur J Hum Genet. 2000;8(7):500–6.
Leijendeckers JM, Pennings RJ, Snik AF, Bosman AJ, Cremers CW. Audiometric characteristics of USH2a patients. Audiol Neurootol. 2009;14(4):223–31.
Ebermann I, Koenekoop RK, Lopez I, Bou-Khzam L, Pigeon R, and Bolz HJ. An USH2A founder mutation is the major cause of Usher syndrome type 2 in Canadians of French origin and confirms common roots of Quebecois and Acadians. Eur J Hum Genet. 2009;17(1):80–4.
Liu X, Tang Z, Li C, Yang K, Gan G, Zhang Z, et al. Novel USH2A compound heterozygous mutations cause RP/USH2 in a Chinese family. Mol Vis. 2010;16:454–61.
Acknowledgements
We gratefully acknowledge all participating patients and their family members. We thank Zhongqi Ge and Natasha Lie for proof reading the manuscript. This work is supported by grants from the Retina Research Foundation, Foundation Fighting Blindness (BR-GE-0613-0618-BCM), and the National Eye Institute (R01EY022356 and R01EY018571) to R.C., the Foundation Fighting Blindness USA (CD-CL-0808-0470-PUMCH and CD-CL-0214-0631-PUMCH), the Ministry of Science and Technology of the People’s Republic of China (Grant No.:2010DFB33430), National Natural Science Foundation of China (81470669) and Beijing Natural Science Foundation (7152116) to R.S.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Please see sample text in the instructions for authors. LJ had full access to all the data in the study and takes responsibility for the integrity of the data the accuracy of the data analysis. LJ and XL contributed equally to this work. Study concept and design: LJ, XL, RC and RS. Acquisition, analysis, or interpretation of data: All authors. Critical revision of the manuscript for important intellectual content: LJ, XL, RC and RS. Obtain funding: RC and RS. Administrative, technical, or material support: All authors. All authors read and approved the final manuscript.
Lichun Jiang and Xiaofang Liang contributed equally to this work.
Additional files
Additional file 1: Table S1.
Genes in the panel design. Table S2. Summary of coverage for coding exons and flanking regions (+ 2bp). Table S3. Clinical information of patients. Table S4. High confidence monoallelic mutations in USH2A genes*. Table S5. Rare biallelic variants in other eye disease genes*. Table S6. In-silico function prediction of nonsynonymous variants (DOCX 80 kb)
Additional file 2: Figure S1.
Both Novel missense alleles identified in MYO7A genes are conserved between human, zebrafish and Drosophila melanogaster. (PPTX 676 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jiang, L., Liang, X., Li, Y. et al. Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: high rate of ethnicity specific mutations in Chinese USH patients. Orphanet J Rare Dis 10, 110 (2015). https://doi.org/10.1186/s13023-015-0329-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-015-0329-3