
Abstract
Background
The simple and caged xanthones from Clusiaceae showed significant antineoplastic activity. This study aims to identify structural diverse xanthones and search for novel antitumor natural products from this family plants.
Methods
The structures of new compounds 1a and 1b were elucidated mainly through comprehensive NMR and MS spectroscopic data, and their absolute configurations were determined by the comparison of the experimental and calculated electronic circular dichroism.
Results
A pair of new xanthone enantiomers, (+)- and (−)-cracochinxanthone A (1a and 1b), along with thirty known analogues (2–31), were isolated from extracts of the stems and leaves of C. cochinchinense. Preliminary biological assay of some isolates against HL-60, PC-3, and MDA-MB-231 cancer cell lines.
Conclusion
Some isolated xanthones exhibited high sensitivity against three human malignant cell lines and the structure–activity relationship study showed that the prenyl and geranyl units may play an important role in antitumor activity.
Similar content being viewed by others

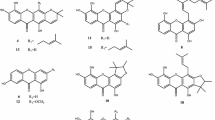
Background
Cratoxylum cochinchinense Blume (Clusiaceae) is a deciduous shrub tree growing abundantly in southeast Asian countries [1]. The leaves, stems, barks, roots and latex of C. cochinchinense have been used as traditional Chinese medicine for the treatment of various diseases such as jaundice, edema, cough, itch, fever, diarrhea, hoarseness, diuretic, flu, colic, ulcer and dental problems and so on [2,3,4]. In addition, the young leaves have been used as an herbal substitute for tea and the immature fruit as a spice for cooking [5]. The simple and caged xanthones with significant antineoplastic activity have been reported from previous phytochemical investigations [6,7,8,9,10,11,12]. Aiming to identify structural diverse xanthones and search for novel antitumor natural products from the Clusiaceae [13,14,15,16,17,18], we continued our studies on the petroleum ether-soluble and dichloromethane-soluble portions of the stems and leaves of C. cochinchinense which exhibited moderate cytotoxicity against human myeloid leukemia (HL-60), human prostate cancer (PC-3) and human breast carcinoma (MDA-MB-231) cell lines with IC50 values of 7.59, 21.49, 19.63 and 7.86, 32.48, 30.40 μg/ml, respectively. A pair of new enantiomers of xanthones, (+)- and (−)-cracochinxanthone A (1a and 1b), as well as thirty known analogues (2–31) were obtained (Fig. 1). In the present paper, the isolation and structure elucidation of new enantiomers of 1a and 1b, as well as the biological evaluation of some selected xanthones are presented.

Chemical structures of xanthones 1–31
Materials and methods
Information of experimental design and resources
The Minimum Standards of Reporting Checklist contains details of the experimental design, and statistics, and resources used in this study (Additional file 1).
General experimental procedures
1H NMR, 13C NMR, HSQC, and HMBC were recorded on the Bruker-ARX-400 and Bruker-AV-600 NMR with tetramethylsilane (TMS) as internal standard. HRESIMS spectra were measured on a Bruker micrOTOF-Q mass spectrometer. Optical rotations were measured by the JASCO P-2000 polarimeter. UV spectra were recorded on a Shimadzu UV-2201 spectrometer. ECD spectra were measured on the BioLogic MOS 450 AF/CD at room temperature. Multimode Reader were used by a Varioskan Flash. The semi-preparative HPLC was a Shimadzu SPD-20A series equipped with an YMC C18 column (250 × 20 mm, 5 μm, 2 mL/min). Chiral HPLC was a CHIRALPAK IB (250 × 4.6 mm) from Daicel Chiral Technologies Co., Ltd., China. Column chromatography (CC) was conducted on silica gel (100–200 and 200–300 mesh) and preparative and analytical TLC was performed on precoated GF254 plates (Qingdao Haiyang Chemical Co., Ltd., China), octadecyl silane (ODS) (50 µm, YMC Co., Ltd., Kyoto, Japan) and Sephadex LH-20 (GE Healthcare, Uppsala, Sweden). All the organic solvents were purchased from Yuwang and Laibo Chemicals Industries, Ltd., China.
Plant material
Stems and leaves of Cratoxylum cochinchinense were collected in December 2016, at Mengla County, Xishuangbanna Autonomous Prefecture, People’s Republic of China, and were identified by Zhi Na (Kunming Institute of Botany, Chinese Academy of Sciences). The voucher specimen (HNMJY-2016) was deposited in the Department of Natural Products Chemistry, Shenyang Pharmaceutical University, Shenyang, China.
Extraction and isolation
The smashed leaves and stems of C. cochinchinense (10 kg) were macerated with 80% aqueous acetone at room temperature (3 × 80 L, 3 days each time). The combined extracts was suspended in water, and successively partitioned to produce petroleum ether (PE), dichloromethane (CH2Cl2), ethyl acetate (EtOAc), n-butyl alcohol (n-BuOH) and water (H2O) fractions. The CH2Cl2 extract (140 g) was fractionated on a silica gel CC and eluted with a PE/EtOAc gradient (100:0, 100:1, 100:3, 100:5, 100:7, 100:10, 100:20, 100:30, 100:50, 0:100) to give ten fractions (Fr. A–J). Fraction D was purified by ODS CC with a stepwise gradient elution using MeOH/H2O to afford 23 (154.8 mg), 29 (8.2 mg), 22 (5.8 mg), 26 (8.7 mg) and yield two subfractions D3 and D4. Fr. D3 was subsequently refined over Sephadex LH-20 (MeOH), followed by semi-preparative HPLC using 77% MeOH in H2O as a mobile phase to get 19 (15.8 mg, tR = 63.5 min) and 1 (3.2 mg, tR = 93.3 min). Then, 1 was separated by chiral HPLC eluting with n-hexane: isopropanol (90:10) to yield 1a (0.92 mg, tR = 15.4 min) and 1b (1.1 mg, tR = 18.4 min). Fr. D4 was also chromatographed on Sephadex LH-20 (MeOH) and semi-preparative HPLC (64% MeOH in H2O) to produce 6 (4.5 mg, tR = 38.4 min) and 9 (6.5 mg, tR = 40.3 min). Fr. F was fractionated by ODS CC (MeOH/H2O) to give 27 (7.5 mg), 18 (5.3 mg), 20 (7.9 mg) and three major subfractions F2, F5 and F8. Fr. F2 was successively partitioned by a Sephadex LH-20 column (MeOH) to provide the key subfraction F2.2. Fr. F2.2 was further processed via semi-preparative HPLC using 56% aqueous MeOH as the mobile phase to afford 3 (10.2 mg, tR = 28.0 min) and 4 (7.8 mg, tR = 32.2 min). Fr. F5 was recrystallized with methanol to yield 19 (50.1 mg). Fr. F8 was loaded onto semi-preparative HPLC using 82% aqueous MeOH to gain 24 (6.9 mg, tR = 87.5 min), 25 (10.2 mg, tR = 92.5 min) and 21 (15.2 mg, tR = 121.5 min). Fr. H was subjected to ODS CC, which afford 15 (6.9 mg) through further recrystallization and subfractions H3 and H5. Fr. H3 and Fr. H5 were applied to Sephadex LH-20 column and eluted with MeOH to obtain 7 (5.4 mg) and 8 (7.5 mg), respectively. Fr. I was subjected to ODS CC to furnish 13 (5.2 mg), 14 (2.8 mg), and subfraction I4. Fr. I4 followed by Sephadex LH-20 CC to afford 28 (11.6 mg). Fr. J was rechromatographed over silica gel CC, affording 12 (4.6 mg), 11 (3.2 mg) and 10 (13.4 mg).
The PE extract (69 g) was chromatographed on a silica gel CC and eluted stepwise with a PE/EtOAc gradient system (100:1, 100:3, 100:7, 100:15, 100:50, 100:100, 0:100) to afford the major fractions A′-G′. Fr. C′ was subjected to separation over ODS CC to yield 30 (8.8 mg) and subfraction C′8. Fr. C′8 was further purified over a silica gel CC and followed by semi-preparative HPLC with 90% aqueous MeOH as mobile phase under isocratic condition to furnish 31 (10.7 mg, tR = 25.5 min). Fr. D′ was separated via ODS CC to provide 2 (15.5 mg), which was crystallized from the 65% MeOH/H2O solution, and to give subfraction D′5. Fr. D′5 was chromatographed over Sephadex LH-20 eluting with MeOH to give 17 (6.7 mg). Fr. E′ was initially subjected to ODS CC to yield subfraction E′3 and E′8. Fr. E′3 was further purified by semi-preparative HPLC eluted with 60% MeOH/H2O to give 16 (9.3 mg, tR = 35.8 min). Fr. E′8 was again subjected to ODS CC to obtain 5 (3.1 mg).
Cracochinxanthone A (1): yellow needle crystal; UV (MeOH) λmax (log ε) 319 (3.86), 268 (4.23), 235 (4.21) nm; 1H, 13C NMR and HMBC data see Table 1; HRESIMS m/z 379.1541 [M + H]+ (calcd for C23H23O5, 379.1540).
(+) Cracochinxanthone A (1a). Yellow needles; \(\left[ \alpha \right]_{\text{D}}^{25}\) + 10.0 (c 0.06 MeOH); ECD (MeOH 0.58) λmax (Δε) 241 (+ 3.65), 270 (− 4.22), 317 (− 1.91) nm.
(−) Cracochinxanthone A (1b). Yellow needles; \(\left[ \alpha \right]_{\text{D}}^{25}\) − 11.3 (c 0.07 MeOH); ECD (MeOH 0.70) λmax (Δε) 242 (− 4.06), 273 (+ 3.72), 316 (+ 1.45) nm.
Anticancer assay in vitro
The antiproliferative activities of some selected compounds against the HL-60, PC-3, and MDA-MB-231 cancer cell lines were evaluated. 5-Fluorouracil (5-FU) was used as a positive control. Detailed methodology for the cell growth inhibition test has been described in a previous report [19]. The IC50 values were calculated by SPSS 16.0 software and results were repeated three times that were expressed as mean ± SD.
Results
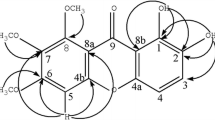
Cracochinxanthone A (1) was obtained as a yellow needle, and its molecular formula was determined as C23H22O5 with 13° of unsaturation from the HRESIMS data of [M + H]+ ion at m/z 379.1541 (calcd for C23H23O5, 379.1540). The UV bands observed at λmax 319, 268 and 235 nm suggested a xanthone skeleton [13]. The 1H NMR data showed signals for a hydrogen bond hydroxy proton at δH 13.35 (1H, s, OH-1), a free phenolic hydroxy proton at δH 9.95 (1H, s, OH-7), a set of ABX coupling system aromatic protons at δH 7.40 (1H, d, J = 3.0 Hz, H-8), 7.46 (1H, d, J = 9.0 Hz, H-5) and 7.28 (1H, dd, J = 9.0, 3.0 Hz, H-6), along with the typical signals of a 3-methylbut-2-enyl (prenyl) moiety at δH 3.43 (2H, d, J = 7.2 Hz, H-1″), 5.19 (1H, t, J = 7.2 Hz, H-2″), 1.82 (3H, s, CH3-4″) and 1.62 (3H, s, CH3-5″). The remaining proton signals were assigned to a dihydrofuran ring with an isopropenyl group at δH 2.95 (1H, dd, J = 14.7, 3.1 Hz, Ha-1′), 2.82 (1H, dd, J = 14.7, 8.0 Hz, Hb-1′), 4.24 (1H, dd, J = 8.0, 2.3 Hz, H-2′), 4.89 and 4.75 (each 1H, s, Ha-4′, Hb-4′), 1.76 (3H, s, CH3-5′), and the corresponding carbon signals at δC 147.1 (C-3′), 110.0 (C-4′), 74.8 (C-2′), 28.9 (C-1′) and 18.1 (C-5′) were assigned through HSQC correlations [20]. The 13C NMR displayed 23 carbon resonances including one conjugated carbonyl carbon, sixteen aromatic/olefinic carbons, three methyl, two methylene and one oxygenated methine (Table 1). The dihydrofuran ring with an isopropenyl group was fused with xanthone skeleton at position C-2 and C-3, based on the HMBC correlations from Ha-1′ (δH 2.95) and Hb-1′ (2.82) to C-1 (δC 158.0), C-2 (108.0) and C-3 (163.5), as well as from H-2′ (δH 4.24) to C-2. The cross peaks between H-1″ (δH 3.43) and C-3, C-4a (δC 152.9) and C-4 (106.3) confirmed the location of the prenyl group at C-4. The correlations of H-5 (δH 7.46) with C-7 (δC 153.8), C-8a (120.1) and C-10a (149.0), H-8 (δH 7.40) with C-6 (124.4), C-9 (δC 179.9) and C-10a, H-6 (δH 7.28) with C-8 (δC 107.9) and C-10a, and OH-7 (δ 9.95) with C-6, C-7 and C-8 indicated that the free phenolic hydroxy located at C-7 (Fig. 2). Based on these results, the structure of 1 was assigned to a new compound, namely cracochinxanthone A.

Key HMBC correlations of 1
Cracochinxanthone A might be a racemic mixture due to the smooth ECD curve as well as close to zero optical rotation. Subsequent chiral HPLC separation of 1 gave the corresponding enantiomers 1a and 1b possessing the opposite ECD curves. Their experimental ECD spectra matched well with the calculated ones for R and S, respectively, thus, explicitly assigning the absolute configurations of 1a and 1b (Fig. 3). And the optical rotations of 1a and 1b were + 10.0 (c 0.06 MeOH) and − 11.3 (c 0.07 MeOH), respectively. Therefore, the structures of 1a and 1b were named as (+) and (−)-cracochinxanthone A.

Experimental and calculated ECD spectra of 1a and 1b
By comparison with those data from the literatures, the known analogues were identified as cochinchinoxanthone (2) [21], 1,4,7-trihydroxy-8-methoxyxanthone (3) [22], gentisein (4) [23], 1,6-dihydroxy-2,5,8-trimethoxyxanthone (5) [23], 1,7-dihydroxyxanthone (6) [24], 1,7-dihydroxy-4-methoxyxanthone (7) [25], 1,7-dihydroxy-3,6-dimethoxyxanthone (8) [26], 1,7-dihydroxy-8-methoxyxanthone (9) [27], 1,5,6-trihydroxy-7-methoxyxanthone (10) [28], 1,4,7-trihydroxyxanthone (11) [29], 1,5,6-trihydroxy-3,7-dimethoxyxanthone (12) [30], 1,3,5,6-tetrahydroxyxanthone (13) [31], 1,3,6,7-tetrahydroxyxanthone (14) [32], 1,3,6-trihydroxy-7-methoxyxanthone (15) [29], cratoxanthone C (16) [33], 1,2,4-trimethoxy-3,8-dimethoxyxanthone (17) [34], 1,3,7-trihydroxy-2-(3-methylbut-2-enyl)-xanthone (18) [35], dulcisxanthone B (19) [20], cudratricusxanthone E (20) [36], γ-mangostin (21) [37], 1,3,7-trihydroxy-2,4-diisoprenylxanthone (22) [38], cochinchinone A (23) [33], cochinchinone B (24) [33], pruniflorone Q (25) [39], 1,3,5-trihydroxy-6′,6′-dimethyl-2H-pyrano(2′,3:6,7)xanthone (26) [30], pruniflorone N (27) [40], xanthone V1 (28) [41], osajaxanthone (29) [42], cochinchinone I (30) [43], 1,7-dihydroxy-4-(3,7-dimethylocta-2,6-dienyl)-5′-(1-hydroxy-1-methylethyl)-4′,5′-dihydrofuro[2′,3′:3,2]-xanthone (31) [44].
Discussions
The antiproliferative activities of some xanthones were evaluated against HL-60, PC-3, and MDA-MB-231 cancer cell lines (Table 2). The isolates 1a, 1b, 3, 4, 6, 10, 12, 19–25, 27 and 28 displayed antiproliferative effect against HL-60 cells with IC50 values ranging from 1.00 to 19.78 μM, especially 23 bearing one prenyl and one geranyl groups with an IC50 value of 1.0 μM and 27 possessing a pyran ring with 1-hydroxy-4,4-dimethyl with an IC50 value of 1.89 μM. Compounds 19–25, 27 and 28 exhibited potent inhibitory activity against PC-3 cells with IC50 values ranging from 11.77 to 27.11 μM and compounds 1b, 19–25 and 28 displayed significant cytotoxicity against MDA-MB-231 cells with IC50 values ranging from 7.94 to 18.46 μM, respectively. It is worth to mention that compounds 23–25 possessing one prenyl and one geranyl and 19–22 with two prenyl groups showed high sensitivity against three human cancer cell lines than others without prenyl unit.
Conclusions
A pair of new racemic mixture of xanthones, (+)-cracochinxanthone A (1a) and (−)-cracochinxanthone A (1b), along with 30 known analogues (2–31) were isolated from the stems and leaves of C. cochinchinense. The antiproliferative activities of some selected compounds against human HL-60, PC-3, and MDA-MB-231 cancer cell lines were screened by the trypan blue and MTT methods. The polyprenylated or geranylated xanthones exhibited potent cytotoxicity against three human malignant cell lines, which could be further developed as potential lead compounds in the design for the treatment of cancer.
Abbreviations
- HL-60:
-
human myeloid leukemia cell line
- PC-3:
-
human prostate cancer cell line
- MDA-MB-231:
-
human breast carcinoma cell line
- ODS:
-
octadecyl silane
- CC:
-
column chromatography
- TMS:
-
tetramethylsilane
- PE:
-
petroleum ether
- CH2Cl2 :
-
dichloromethane
- EtOAc:
-
ethyl acetate
- n-BuOH:
-
n-butyl alcohol
- H2O:
-
water
- ECD:
-
electronic circular dichroic spectroscopy
- HPLC:
-
high performance liquid chromatography
- IC50 :
-
the concentration of drug required to inhibit cell growth by 50% compared with untreated control
References
Rattanaburi S, Daus M, Watanapokasin R, Mahabusarakam W. A new bisanthraquinone and cytotoxic xanthones from Cratoxylum cochinchinense. Nat Prod Res. 2014;28:606–10.
Ito C, Matsui T, Niimi A, Tan HTW, Itoigawa M. Four new xanthones from Cratoxylum cochinchinense and their in vitro antiproliferative effects. Planta Med. 2017;83:812–8.
Yu HY, Jin SL, Zhang X, Liu Y, Ou YF, Wang NL, Yao XS. Two new benzophenone glucosides from Cratoxylon cochinchinensis. Chin Chem Lett. 2009;20:459–61.
Laphookhieo S, Maneerat W, Buatip T, Syers JK. New xanthones from Cratoxylum cochinchinense. Can J Chem. 2008;86:757–60.
Dai DN, Thang TD, Ogunwande IA. Volatile constituents of the leaf oil of Cratoxylum cochinchinense from Vietnam. Chem Nat Compd. 2014;50(1):158–60.
Ren YL, Matthew S, Lantvit DD, Ninh TN, Chai H, James R, Fuch JR, Soejarto DD, de Blanco EJC, Swanson SM, Kinghorn AD. Cytotoxic and NF-κB inhibitory constituents of the stems of Cratoxylum cochinchinense and their semisynthetic analogues. J Nat Prod. 2011;74:1117–25.
Li ZP, Lee HH, Uddin Z, Song YH, Park KH. Caged xanthones displaying protein tyrosine phosphatase 1B (PTP1B) inhibition from Cratoxylum cochinchinense. Bioorg Chem. 2018;78:39–45.
Boonnak N, Karalai C, Chantrapromma S, Ponglimanont C, Fun HK, Kanjana-Opas A, Chantrapromma K, Kato S. Anti-pseudomonas aeruginosa xanthones from the resin and green fruits of Cratoxylum cochinchinense. Tetrahedron. 2009;65:3003–13.
Mahabusarakam W, Rattanaburi S, Phongpaichit S, Kanjana-Opas A. Antibacterial and cytotoxic xanthones from Cratoxylum cochinchinense. Phytochem Lett. 2008;1:211–4.
Duan YH, Dai Y, Wang GH, Chen LY, Chen HF, Zeng DQ, Li YL, Yao XS. Bioactive prenylated xanthones from the stems of Cratoxylum cochinchinense. J Asian Nat Prod Res. 2015;17(5):519–31.
Udomchotphruet S, Phuwapraisirisan P, Sichaem J, Tip-Pyang S. Xanthones from the stems of Cratoxylum cochinchinense. Phytochemistry. 2012;73:148–51.
Nguyen HD, Trinh BTD, Nguyen NK, Dang SV, Pham HD, Nguyen LHD. Xanthones from the twigs of Cratoxylum cochinchinense. Phytochem Lett. 2011;4:48–51.
Jia CC, Xue JJ, Gong C, Li XY, Li ZL, Hua HM. Chiral resolution and anticancer effect of xanthones from Garcinia paucinervis. Fitoterapia. 2018;127:220–5.
Tian DS, Yi P, Xia L, Xiao X, Fan YM, Gu W, Huang LJ, Ben-David Y, Di YT, Yuan CM, Hao XJ. Garmultins A-G, biogenetically related polycyclic acylphloroglucinols from Garcinia multiflora. Org Lett. 2016;18:5904–7.
Niu SL, Li DH, Li XY, Wang YT, Li SG, Bai J, Pei YH, Jing YK, Li ZL, Hua HM. Bioassay- and chemistry-guided isolation of scalemic caged prenylxanthones from the leaves of Garcinia bracteata. J Nat Prod. 2018;81(4):749–57.
Wang LL, Li ZL, Song DD, Sun L, Pei YH, Jing YK, Hua HM. Two novel triterpenoids with antiproliferative and apoptotic activities in human leukemia cells isolated from the resin of Garcinia hanburyi. Planta Med. 2008;74:1735–40.
Jing Y, Jiang C, Ji F, Hua HM, Li ZL. Chemical constituents from the stem barks of Garcinia multiflora. J Asian Nat Prod Res. 2013;15:1152–7.
Ji F, Li ZL, Liu GF, Niu SL, Zhao N, Liu XQ, Hua HM. Xanthones with antiproliferative effects on prostate cancer cells from the stem bark of Garcinia xanthochymus. Nat Prod Commun. 2012;7:53–6.
Xu ST, Yao H, Hu M, Li DH, Zhu ZY, Xie WJ, Yao HQ, Wu L, Chen ZS, Xu JY. 6,7-Seco-ent-kauranoids derived from oridonin as potential anticancer agents. J Nat Prod. 2017;80:2391–8.
Seo EK, Kim NC, Wani MC, Wall ME, Navarro HA, Burgess JP, Kawanishi K, Kardono LBS, Riswan S, Rose WC, Fairchild CR, Farnsworth NR, Douglas A. Cytotoxic prenylated xanthones and the unusual compounds anthraquinobenzophenones from Cratoxylum sumatranum. J Nat Prod. 2002;65(3):299–305.
Phuwapraisirisan P, Udomchotphruet S, Surapinit S, Tip-Pyang S. Antioxidant xanthones from Cratoxylum cochinchinense. Nat Prod Res. 2006;20(14):1332–7.
Linuma M, Tosa H, Ito T, Tanaka T, Madulid DA. Two xanthones from roots of Cratoxylum formosanum. Phytochemistry. 1996;42(4):1195–8.
Na Z, Hu HB, Xu YK. Cytotoxic caged xanthones from the fruits of Garcinia bracteata. Chem Nat Comp. 2013;49(3):505–6.
Yang XD, Xu LZ, Yang SL. Xanthones from the stems of Securidaca inappendiculata. Phytochemistry. 2001;58(8):1245–9.
Marston A, Hamburger M, Sordat-Diserens I, Msonthi JD, Hostettmann K. Xanthones from Polygala nyikensis. Phytochemistry. 1993;33:809–12.
Ramdani ED, Marlupi UD, Sinambela J, Tjandrawinata RR. Isolation and identification of compounds from Phaleria macrocarpa (Scheff.) boerl fruit extract. Asian Pac J Trop Med. 2017;7:300–5.
Kijjoa A, Jose M, Gonzalez TG, Pinto MMM, Damas AM, Mondranondra IO, Silva AMS, Herz W. Xanthones from Cratoxylum maingayi. Phytochemistry. 1998;49(7):2159–62.
Iinuma M, Tosa H, Ito T, Tanaka T, Aqil M. Two prenylated anthrones in Harungana madagascariensis. Phytochemistry. 1995;40(1):267–70.
Duan YH, Da Y, Wang GH, Zhang X, Chen HF, Chen JB, Yao XS, Zhang XK. Bioactive xanthones from the stems of Cratoxylum formosum ssp. Pruniflorum. J Nat Prod. 2010;73(7):1283–7.
Shen J, Yang JS. Two new xanthones from the stems of Garcinia cowa. Chem Pharm Bull. 2006;54(1):126–8.
Tantapakul C, Maneerat W, Sripisut T, Ritthiwigrom T, Andersen RJ, Cheng P, Cheenpracha S, Raksat A, Laphookhieo S. New benzophenones and xanthones from Cratoxylum sumatranum ssp. neriifolium and their antibacterial and antioxidant activities. J Agric Food Chem. 2016;64:8755–62.
Gao S, Ye KH, Zhang Y, Zhou GX. Chemical constituents from barks of Pterocarya stenoptera. Chin Tradit Herbal Drugs. 2013;44(7):803–7.
Mahabusarakam W, Nuangnaowarat W, Taylor WC. Xanthone derivatives from Cratoxylum cochinchinense roots. Phytochemistry. 2006;67(5):470–4.
Peres V, Nagem TJ, de Oliveira FF. Tetraoxygenated naturally occurring xanthones. Phytochemistry. 2000;55(7):683–710.
Cortez DAG, Young MCM, Marston A, Wolfender JL, Hostettmann K. Xanthones, triterpenes and a biphenyl from Kielmeyera coriacea. Phytochemistry. 1998;47(7):1367–74.
Zou YS, Hou AJ, Zhu GF, Chen YF, Sun HD, Zhao QS. Cytotoxic isoprenylated xanthones from Cudrania tricuspidata. Bioorgan Med Chem. 2004;12:1947–53.
Xu Z, Huang L, Chen XH, Zhu XF, Qian XJ, Feng GK, Lan WJ, Li HJ. Cytotoxic prenylated xanthones from the pericarps of Garcinia mangostana. Molecules. 2014;19:1820–7.
Iinuma M, Tosa H, Tanaka T, Riswan S. Three new xanthones from the bark of Garcinia dioica. Chem Pharm Bull. 1996;44(1):232–4.
Raksat A, Sripisut T, Maneerat W. Bioactive xanthones from Cratoxylum cochinchinense. Nat Prod Commun. 2015;10(11):1969–72.
Boonnak N, Khamthip A, Karalai C, Chantrapromma S, Ponglimanont C, Kanjana-Opas A, Tewtrakul S, Chantrapromma K, Fun HK, Kato S. Nitric oxide inhibitory activity of xanthones from the green fruits of Cratoxylum formosum ssp. pruniflorum. Aust J Chem. 2010;63(11):1550–6.
Wabo HK, Kouam SF, Krohn K, Hussain H, Tala MF, Tane P, Ree T, Hu QX, Schulze B. Prenylated anthraquinones and other constituents from the seeds of Vismia laurentii. Chem Pharm Bull. 2007;55(11):1640–2.
Huang ZJ, Yang RY, Guo ZY, She ZG, Lin YC. A new xanthone derivative from mangrove endophytic fungus No. ZSU-H16. Chem Nat Compd. 2010;46(3):348–51.
Boonnak N, Karalai C, Chantrapromma S, Ponglimanont C, Kanjana-Opas A, Chantrapromma K, Kato S. Chromene and prenylated xanthones from the roots of Cratoxylum formosum ssp. pruniflorum. Chem Pharm Bull. 2010;58(3):386–9.
Yao XS, Zhang XK, Dai Y, Wang GH, Zhang X, Chen HF, Duan YH, Chen JB, Jin SL. Method for preparing xanthone derivative medicinal composition, and its application to preparing TR3 receptor inducer. Chinese Patent 101889998.
Authors’ contributions
CCJ designed the experiment. CCJ and JP performed the extraction and isolation experiment. CCJ and CG intepreted the data. HC performed the cell experiment. DHL and ZLL provided advices on study design and technical support. HMH in-charged and supervised the project. CCJ was major contributor in writing the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data for this study are included in this published article and its Additional file 2.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This project was funded by the National Natural Science Foundation of China (31570350), and the Key laboratory basic research projects of Department of Education in Liaoning Province (LZ2014044).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1.
Minimum standards of reporting checklist.
Additional file 2.
Supporting information.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jia, C., Gong, C., Chen, H. et al. A pair of new enantiomers of xanthones from the stems and leaves of Cratoxylum cochinchinense. Chin Med 14, 14 (2019). https://doi.org/10.1186/s13020-019-0235-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13020-019-0235-z