
Abstract
Background
HIV-1 protease (PR) activation is triggered by Gag-Pol dimerization. Premature PR activation results in reduced virion yields due to enhanced Gag cleavage. A p6* transframe peptide located directly upstream of protease is believed to play a modulating role in PR activation. Previous reports indicate that the C-terminal p6* tetra-peptide prevents premature PR activation triggered by a leucine zipper (LZ) dimerization motif inserted in the deleted p6* region. To clarify the involvement of C-terminal p6* residues in mitigating enhanced LZ-incurred Gag processing, we engineered constructs containing C-terminal p6* residue substitutions with and without a mutation blocking the p6*/PR cleavage site, and created other Gag or p6* domain-removing constructs. The capabilities of these constructs to mediate virus maturation were assessed by Western blotting and single-cycle infection assays.
Results
p6*-PR cleavage blocking did not significantly reduce the LZ enhancement effect on Gag cleavage when only four amino acid residues were present between the p6* and PR. This suggests that the potent LZ dimerization motif may enhance PR activation by facilitating PR dimer formation, and that PR precursors may trigger sufficient enzymatic activity without breaking off from the PR N-terminus. Enhanced LZ-induced activation of PR embedded in Gag-Pol was found to be independent of the Gag assembly domain. In contrast, the LZ enhancement effect was markedly reduced when six amino acids were present at the p6*-PR junction, in part due to impaired PR maturation by substitution mutations. We also observed that a proline substitution at the P3 position eliminated the ability of p6*-deleted Gag-Pol to mediate virus maturation, thus emphasizing the importance of C-terminal p6* residues to modulating PR activation.
Conclusions
The ability of HIV-1 C-terminal p6* amino acid residues to modulate PR activation contributes, at least in part, to their ability to counteract enhanced Gag cleavage induced by a leucine zipper substituted for a deleted p6*. Changes in C-terminal p6* residues between LZ and PR may affect PR-mediated virus maturation, thus providing a possible method for assessing HIV-1 protease precursor activation in the context of virus assembly.
Similar content being viewed by others


Background
The HIV-1 retrovirus contains three major genes (gag, pol and env) and several accessory genes [1]. HIV-1 pol encodes viral enzymes such as protease (PR), reverse transcriptase (RT) and integrase (IN), while gag encodes viral structural proteins. Both Pol and Gag are translated from the same mRNA template. Pol is translated as a Gag-Pol fusion protein associated with a ribosome shift during Gag translation that occurs at a 5% frequency, leading to a Gag-Pol versus Gag expression ratio of approximately 1:20 [2]. Pr160gag-pol and Pr55gag are transported to plasma membranes, where Pr55gag molecules assemble into virus particles [3]; Pr160gag-pol is incorporated into these particles via Pr55gag interaction [4,5,6,7]. During or after virus budding, activated PR auto-cleaves from Gag-Pol and mediates virus maturation through the proteolytic processing of Pr55gag and Pr160gag-pol [8]. Pr55gag cleavage yields four main products: matrix (p17; MA), capsid (CA; p24), nucleocapsid (NC; p7) and C-terminal p6 [9]. Two spacer peptides—SP1 (or p2) and SP2 (or p1)—respectively separate NC from CA and p6. Pr160gag-pol cleavage generates RT and IN in addition to the Gag proteins MA, CA and NC. PR-mediated virus maturation is necessary for viral infectivity acquisition [10, 11].
It is generally believed that Gag-Pol dimerization triggers PR activation [3]. In agreement with this assumption, mutations upstream or downstream of PR may significantly reduce PR-mediated Gag cleavage efficiency due to inadequate Gag-Pol dimerization [12,13,14,15,16]. In contrast, the promotion of PR activation as a result of enhanced Gag-Pol dimerization or Gag-PR dimer interaction likely triggers premature or enhanced Pr55gag cleavage, resulting in markedly reduced virus production. [17,18,19]. Accordingly, preventing premature PR activation is central to virus assembly.
Within Gag-Pol, truncated p1-p6gag is replaced with a transframe region referred to as p6* or p6pol. Located directly adjacent to the PR N-terminus, p6* has been described as playing a role in modulating PR activation even though it lacks a specific structure. Mature HIV-1 PR has a dimeric form, and the removal of p6* from PR precursor is essential for PR to be fully functional [20,21,22,23]. Mutations that block p6*-PR cleavage markedly impede PR-mediated virus maturation, implying a suppressive effect of p6* on PR activation [22, 23]. Molecular models suggest that p6* may prevent early PR maturation by inducing instability in the folded PR dimer structure [24,25,26,27]. However, virus assembly-associated evidence in support of this assumption is limited, since deletion analysis of p6* function has the potential to compromise virus assembly due to an overlap of p6* with the p6gag budding domain.
To investigate p6* function without affecting the p6gag coding region, we engineered an HIV-1 virus-producing vector by placing the pol coding sequence at the PR-inactivated C-terminus. This construct, designated Dp6*PR, was capable of assembling and processing virus particles in a manner similar to that of wild-type (wt). Replacement of p6* with a leucine-zipper (LZ) dimerization domain has been shown to eliminate virus production as a result of enhanced Gag cleavage, but as few as four C-terminal p6 residues remaining between LZ and PR significantly counteract the LZ enhancement effect [28]. These observations provide supporting evidence that p6* may contribute to the prevention of premature PR activation, but it remains unknown whether a correlation exists between the ability of C-terminal p6* residues to counteract LZ enhancement and their ability to modulate PR maturation. Further, it is unknown whether specific amino acids must be present between LZ and PR in order to counteract the LZ enhancement effect.
To study these questions, we engineered multiple constructs to further analyze the role of C-terminal p6* residues in counteracting LZ enhancement and modulating PR activation. Our results indicate that an HIV-1 PR precursor containing a leucine zipper (LZ) motif linked at the N-terminus eliminated virus particle production associated with enhanced Gag cleavage, suggesting that the HIV-1 PR precursor is capable of exhibiting enhanced enzymatic activity. C-terminal p6* residue substitutions can subvert the Gag cleavage enhancement effect induced by a LZ substitution for p6*, likely the result of interference with PR maturation. While p6*-deleted Gag-Pol (∆p6*fs) containing the last two remaining C-terminal p6* residues was still capable of producing infectious virions following co-expression with Pr55gag, a single amino acid residue change at the deleted p6* region completely removed the ability of ∆p6*fs to mediate virus maturation. Our results confirm the importance of C-terminal p6* residues for the spatiotemporal modulation of PR activity, and provide a virus assembly system for studying HIV-1 protease precursor activation by manipulating linker residues between fused peptides and PR.
Methods
Plasmid construction
The parental HIV-1 proviral sequence in this study is HXB2 [29]. The HIV-1 proviral plasmid HIVgpt is considered the backbone of all expression constructs [30]. The constructs used in this research were mostly derived from Dp6*PR, DPR, DWzPR, DWz/PR and DWz//PR. As described previously [31], Dp6*PR contains p6* domain between an inactivated and an active PR. DPR contains BamHI-linked duplicate PR pairs, with the proximal PR was inactivated. DWzPR, DWz/PR and DWz//PR contain leucine zipper replacements of p6* with two, four and six C-terminal p6* residues remaining in the p6*/PR junction, respectively [28]. PSHL and PIDL substitutions for the four C-terminal p6* residues in DWz/PR yielded DWz/PSHL/PR and DWz/PIDL/PR [28]. Primers used for engineering the designated mutations were listed in Table 1.
V/P, as described previously was created by changing amino acid residues at p6*-PR cleavage site from Phe/Pro into Val/Pro [32]. BamHI-containing forward primers were used to amplify the p6* C-terminal coding fragment containing the desired mutation DWzPRV/P, DWz/PRV/P, DWz//PRV/P or DWz/PANF/PR (Table 1). V/P or HIVgpt served as a template and the reverse primer (nt.3116-90) sequence was 5′-TACATACAAATCATCCATGTTATTGATA-3′. Amplified fragments were digested with BamHI and EcoRV, and subcloned into pBRClaSal/DPR [28]. BamHI-flanking leucine zipper coding fragments derived from PRWzPR [28] were inserted into each pBRClaSal/DPR recombinant, yielding DWzPRV/P, DWz/PRV/P, DWz//PRV/P and DWz/PANF/PR, respectively.
p6*PSHL, p6*PIDL and p6*PANF were constructed by megaprimer PCR method [33] using a forward primer containing the desired mutation (Table 1) and a reverse primer 5′-GGTACAGTCTCAATAGGGCTAATG-3. HIVgpt serves as a template. The amplified fragments were digested with ApaI and BclI, and subcloned into a plasmid cassette pBRCla-Sal that contains HIV-1 coding sequence (from ClaI-nt.831 to SalI-nt.5786). Each mutation-containing pBRCla-Sal cassette was then digested with SpeI and SalI, and ligated into HIVgpt, yielding p6*PSHL, p6*PIDL and p6*PANF.
p6*PSHL, p6*PIDL and p6*PANF were digested with BglII and EcoRV and ligated into DPR digested with BamHI and EcoRV, yielding Dp6*PSHL, Dp6*PIDL/PR and Dp6*PANF/PR, respectively.
GPfs has the Gag and Pol in the same reading frame due to a deletion of the frame shift signal [34]. DWzPR and DWz/PR were digested with BclI. The BclI-flanking fragment containing WzPR and Wz/PR mutations were ligated into PR-inactivated GPfs (fsd) digested with BclI, yielding fsdWzPR and fsdWz/PR. Recombination of fsdWzPR and a Gag-deleted fsd [34] generated construct fsdWzPR∆Gag.
Cell culture and transfection
293T and HeLa cells were maintained in DMEM supplemented with 10% fetal calf serum. Confluent 293T cells were trypsinized, split 1:10 and seeded onto 10-cm plates 18–24 h before transfection. For each construct, 293T cells were transfected with 20 μg of plasmid DNA by the calcium phosphate precipitation method, with the addition of 50 μM chloroquine to enhance transfection efficiency. Culture media and cells were harvested for protein analysis at 48–72 h post-transfection. When pGAG was co-transfected with the Gag-Pol expression constructs at a DNA ratio of 1:1 or 10 to 1, 10 or 15 μg of pGAG were used with the addition of pBlueScript plasmid DNA to a final quantity of 20 μg DNA. The cells and media were harvested for protein analysis 48–72 h post-transfection.
Single-cycle infection assays
293T cells were either co-transfected with 10 μg wt or each of the mutant HIVgpt plus 5 μg of the VSV-G protein expression plasmid pHCMV-G [35], or co-transfected with 1 μg of the GPfs or the p6*-deleted GPfs plasmid with 10 μg pGAG plus 5 μg pHCMV-G. At 48 h after transfection, virus-containing supernatants were collected, filtered, diluted, and used to infect HeLa cells. Aliquots of the same filtered supernatants and cell samples were prepared and subjected to Western blot. Adsorption of virions is allowed to proceed in the presence of 4 μg/ml polybrene. Twenty-four hours after infection, cells were trypsinized, split into dishes, and refed with medium containing drug selection cocktail [36]. Selected drug resistant colonies were fixed and stained with 50% methanol containing 0.5% methylene blue. Numbers of drug-resistant colonies were converted into titers (cfu/ml). Infectivity was expressed as the ratio of the mutant titer to the titer of wt, and normalized to Gag protein levels in parallel experiments.
Western immunoblot analysis
Culture media from transfected 293T cells were filtered through 0.45-µm pore-size and then centrifuged through 2 ml 20% sucrose in TSE (10 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA) containing 0.1 mM phenylmethylsulfonyl fluoride (PMSF) at 4 °C for 40 min at 274,000 × g. Viral pellets and cell lysates mixed with sample buffer were then subjected to SDS-10% PAGE or 4–12% Bis–Tris gradient gels (NuPage Bis–Tris Mini Gels; Thermo Fisher Scientific) followed by immunoblotting analysis as previously described [37]. HIV-1 Gag proteins were probed with an anti-p24gag monoclonal antibody (mouse hybridoma clone 183-H12-5C) from ascites. For HIV-1 RT detection, the primary antibody was rabbit antiserum or a mouse anti-RT monoclonal antibody [38, 39]. Cellular β-actin was detected using a mouse anti-β-actin monoclonal antibody (Sigma). The secondary antibody was either a sheep anti-mouse or a donkey anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (Jackson ImmunoResearch). An enhanced chemiluminescence (ECL) detection system (SuperSignal West Pico Chemiluminescent Substrate; Thermo Fisher Scientific) was used to detect membrane-bound proteins.
Statistical analysis
Differences between control (wt) and experimental (mutant) groups were assessed using Student’s t-tests. Data are expressed as mean ± standard deviation. Significance was defined as *p < 0.05, **p < 0.01, ***p < 0.001.
Results
p6*-PR cleavage blocking does not significantly mitigate leucine zipper-induced Gag cleavage enhancement
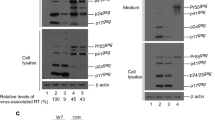
In a previous study we reported that an HIV-1 mutant (DWzPR) containing a LZ dimerization motif adjacent to and upstream of PR was not capable of producing virions due to the strong enhancement of Gag cleavage [28]. DWzPR is derived from Dp6*PR by replacing a deleted p6* with LZ, but retaining the last two C-terminal p6* residues at the LZ/PR junction (Fig. 1a). We observed that this LZ replacement of p6* led to the elimination of virus assembly, likely due to premature PR activation triggered by the LZ. Based on our prior finding that the DWzPR virus assembly defect is PR activity-dependent, and since p6*-PR cleavage is required for fully active PR, we postulated that blocking p6*-PR cleavage within the LZ/PR junction might help restore DWzPR-associated virus production by reducing PR activity. To test this possibility, we substituted Val for the last C-terminal p6* residue (Phe) at the LZ/PR junction of DWzPR and designated the resulting construct as DWzPRV/P (Fig. 1a; note that p6*-PR cleavage site residues were changed from F/P to V/P). Our data indicate that DWzPR virus yields were still hardly detected following the Val substitution unless it was accompanied by treatment with an HIV-1 PR inhibitor (Fig. 1b middle panel, lane 4 vs. lane 5). These results suggest that (a) the blocking of p6*-PR cleavage exerted no major impacts on LZ-induced Gag cleavage enhancement, and (b) a HIV-1 PR precursor containing a LZ motif fused at the N-terminus exhibited enhanced enzymatic activity.

Effects of C-terminal HIV-1 p6* residue substitutions on virus assembly and processing. a Schematic representations of HIV-1 Gag and Gag-Pol expression constructs. Indicated are the HIV-1 Gag protein domains MA (matrix), CA (capsid), NC (nucleocapsid), p6, pol-encoded p6*, PR, RT and IN. “X” denotes a PR-inactivated mutation. Arrows indicate PR cleavage sites. Underlined “V” indicates a Val residue substitution for the final C-terminal p6* residue Phe. Striped (Wz) box denotes wild-type (wt) leucine zipper (LZ). Remaining C-terminal p6* residues are in boldface. Altered or additional residues are in italics. b Blocking p6*-PR cleavage is insufficient for mitigating the enhanced Gag cleavage incurred by an LZ replacement for a deleted p6* domain. 293T cells were transfected with designated constructs. At 4 h post-transfection, equal amounts of cells were plated on two dishes and either left untreated or treated with saquinavir (a HIV-1 protease inhibitor) at a concentration of 5 μM. Supernatants and cells were collected 48 h post-transfection, prepared, and subjected to Western immunoblotting. c Blocking p6*-PR cleavage (V/P mutation) disrupted the function of a C-terminal p6* tetra-peptide for modulating PR activation. 293T cells were transfected with designated constructs. Culture supernatants and cells were collected and subjected to Western immunoblotting at 48–72 h post-transfection. d Relative virus assembly efficiencies of HIV-1 mutants. Gag proteins from medium or cell samples were quantified by scanning mutant and wt p24gag-associated band densities from immunoblots. Ratios of total Gag protein levels in medium to those in cells were determined for each construct and compared with wt release levels; release ratios for each mutant were divided by wt ratios in parallel experiments. Error bars indicate standard deviation. *p < 0.05; **p < 0.01. e Relative virus particle processing efficiency data for HIV-1 mutants. Virus-associated Pr55gag and p24gag levels were quantified by scanning immunoblot band densities. Ratios of p24gag to p55gag were determined for each mutant and normalized to those of the wt in parallel experiments. Bars indicate standard deviations. *p < 0.05; **p < 0.01
As a control, Val substitution for Phe (referred to as a V/P mutation) significantly reduced virus processing efficiency when tested in a wild-type (wt) HIV-1 Gag/Gag-Pol expression vector (Fig. 1c, middle panel, lanes 2 and 6). Constructs with either four or six remaining C-terminal p6* residues at the LZ/PR junction (DWz/PR or DWz//PR) exhibited particle assembly and processing profiles similar to that of the wt (Fig. 1c middle panel, lanes 4 and 5). While the V/P mutation exerted no major impacts on wt virus production, it significantly reduced DWz/PR and DWz//PR virus yields in addition to impairing virus maturation (Fig. 1c middle panel lanes 4–5 vs. 8–9 and panels d and e). The capacity of the C-terminal p6* tetra-peptide to counteract the LZ enhancement effect and modulate PR activation was subject to weakening by the V/P mutation. This may account, at least in part, for the decreased virus assembly and processing efficiency of DWz/PRV/P and DWz//PRV/P (Fig. 1c middle panel, lanes 8 and 9).
Specific C-terminal p6* residues are required to modulate PR maturation
Our results support the proposal that the C-terminal p6* tetra-peptide plays a central role in mitigating PR maturation, likely due to the absence of an intact C-terminal p6* tetra-peptide, which lets LZ dictate the PR maturation process and trigger premature PR activation. To test this hypothesis, we inserted substitution mutations at the C-terminal p6* tetra-peptide without affecting the p6gag amino acid residues. Given our observation of DWzPR exhibiting Gag cleavage enhancement, the last two C-terminal p6* residues (NF) remaining at the LZ/PR junction might be required for PR activation. We therefore engineered a DWz/PANF/PR construct by replacing SF with PA (Fig. 2a). Both the DWz/PSHL/PR and DWz/PIDL/PR constructs contain substitutions for all four C-terminal p6* residues. We found that DWz/PANF/PR displayed a virus assembly and processing profile similar to that of DWz/PR (Fig. 2b, lane 4 vs. lane 1), but evidence from statistical analyses suggest that its virus particle processing was not as efficient as that of DWz/PR (Fig. 2b, d). Whereas DWz/PIDL/PR exhibited readily detected virus-associated p24gag and mature PR, DWz/PSHL/PR had virus-associated Gag or PR mostly present in unprocessed or incompletely processed precursor forms (Fig. 2b, lane 3 vs. lane 2). Similar effects on virus processing were observed when PSHL, PIDL or PANF mutations were cloned into Dp6*PR (Fig. 2c). Western blot data indicate a strong correlation between the virus processing efficiencies of the mutants and their virus-associated mature PR levels. Although Dp6*PANF/PR and DWz/PANF/PR both exhibited relatively low processing efficiency, no statistical significance was noted when compared with their Dp6*PR and DWZ/PR prototypes (Fig. 2d).

Effects of C-terminal p6* amino acid substitutions on protease maturation and virus processing. a Schematic representations of HIV-1 Gag and Gag-Pol expression constructs. HIV-1 Gag protein domains and pol-encoded proteins and the leucine zipper (LZ) motif (striped box, Wz) are indicated as described in the Fig. 1 caption. Also indicated are amino acid residues in the junction area. Two or four C-terminal p6* residues remaining at the LZ/PR junction are shown in boldface. Amino acid changes at the C-terminal p6* tetra-peptide are underlined. Altered or additional residues are in italics. b, c 293T cells were transfected with designated constructs. Culture supernatants and cells were collected at 48–72 h post-transfection. To detect PR-associated products, aliquots of supernatant samples were separated by 4–12% Bis–Tris gradient gels. Membrane-bound proteins were initially probed with anti-PR serum prior to stripping and probing with anti-RT serum, followed by probing with an anti-p24CA monoclonal antibody. Molecular weight size markers (in kDa) are indicated on right side (middle panels). d Relative virus particle processing efficiency of HIV-1 mutants. Virus-associated Pr55gag and p24gag levels were quantified by scanning immunoblot band densities. Ratios of p24gag to p55gag were determined for each mutant and normalized to those of the wt in parallel experiments. Bars indicate standard deviations. *p < 0.05; **p < 0.01; ***p < 0.001
To confirm our conclusions, we tested PIDL, PSHL and PANF substitution mutations in a wt HIVgpt backbone (Fig. 3a). Results indicate that neither PIDL nor PANF mutations significantly affected virus infectivity in single cycle infection assays, although both mutations reduced virus processing efficiency (Fig. 3b–d). The data also show that PANF exerted a weaker impact on virus processing and infectivity compared to PIDL. In contrast, the PSHL mutation significantly impaired both virus processing and infectivity (Fig. 3c, d). Combined, these results suggest that specific C-terminal p6* residues, especially the last two, are essential for modulating PR activation.

Effects of C-terminal p6* tetra-peptide mutations on virus processing and infectivity. a Schematic representations of HIV-1 Gag and Gag-Pol expression constructs. HIV-1 Gag protein domains and pol-encoded proteins are indicated as described in the Fig. 1 caption. Native C-terminal p6* residues are shown in boldface. Altered amino acid residues are underlined. b 293T cells were transfected with designated constructs. Culture supernatants and cells were collected 48–72 h post-transfection and subjected to Western immunoblotting. c Relative virus particle processing efficiency of HIV-1 mutants. Virus-associated Pr55gag and p24gag levels were quantified by scanning immunoblot band densities. Ratios of p24gag to p55gag were determined for each mutant and normalized to those of the wt in parallel experiments. Bars indicate standard deviations. *p < 0.05; **p < 0.01; ***p < 0.001. d Infectivity of HIV-1 mutants. 293T cells were co-transfected with one of the designated constructs plus a VSV-G expression vector. At 48 h post-transfection, supernatants were collected, filtered, and used to infect HeLa cells. Infection and selection of drug-resistant colonies was performed as described in Methods. Infectivity for each mutant was determined as the ratio of mutant titers to wt titers, normalized to Gag protein levels in parallel experiments. ***p < 0.001
A single amino acid residue change eliminated the ability of p6*-deleted Gag-Pol to mediate virus maturation
The above conclusions agree with our past observations of HIV-1 Gag-Pol (∆p6*fs) with most p6* deleted, but with the final two C-terminal p6* residues still capable of mediating virus maturation, although less efficiently than wt Gag-Pol [40]. To determine if additional changes at C-terminal p6* residues affect the ability of ∆p6*fs to mediate Gag processing, we engineered p6*-deleted constructs with two, four or six C-terminal p6* residues remaining at the p6*/PR junction (respectively designated ∆p6*fsPR, ∆p6*fs/PR and ∆p6*fs//PR). All three contained an additional Pro residue insertion in the deleted p6* region due to cloning procedures (Fig. 4a). All of the p6*-deleted Gag-Pol mutants and wild-type GPfs were co-transfected with a Pr55gag expression vector designated pGAG. Unsurprisingly, virus production was almost completely blocked when each construct was co-expressed with equal amounts of pGAG (10 μg each), presumably due to enhanced Gag cleavage from over-expressed PR activity. The only exception was ∆p6*fsPR, which produced significant amounts of mostly unprocessed or incompletely processed virus-associated Gag (Fig. 4b middle panel, lane 7). All constructs other than ∆p6*fsPR were capable of producing readily detectable virus-associated p24gag and p66/51RT when co-expressed with pGAG at a plasmid DNA ratio of 1:10 (Fig. 4b, c). In contrast, ∆p6*fsPR co-expression with pGAG consistently yielded virions that mostly contained incomplete or unprocessed Gag and RT-associated Gag-Pol (Fig. 4b, lane 4, and Fig. 4c, lane 6). These data suggest that ∆p6*fsPR is profoundly defective in auto-processing and the in trans processing of virus particles.

Effects of C-terminal p6* residue substitutions on the capability of p6*-deleted Gag-Pol mutants to mediate virus maturation. a Schematic representations of HIV-1 Gag-Pol expression constructs with deletions of most p6* coding sequences. HIV-1 Gag domains, pol-encoded p6*, PR, RT and IN are indicated. All constructs contain a frame shift (fs) mutation forcing gag and pol into the same reading frame. Dashed lines denote deleted p6* regions. Remaining N-terminal and C-terminal p6* residues are indicated in boldface. Altered or foreign residues are in italics. b, c 293T cells were co-transfected with 10 μg of an HIV-1 Pr55gag expression plasmid (pGAG) and 1 or 10 µg (panel b) or 1 µg (panel c) of the designated Gag-Pol expression construct. At 48 h post-transfection, cells and supernatants were collected and analyzed by Western immunoblotting. Membrane-bound proteins were initially probed with anti-RT serum, stripped, and probed again with anti-p17MA and anti-p24CA monoclonal antibodies. Indicated are HIV-1 Gag-Pol, 66/51RT, Pr55gag, p41gag, p24gag and p17gag positions. d A single amino acid change blocked the capability of p6*-deleted Gag-Pol to confer virus infectivity. 293T cells were co-transfected with 10 µg of an HIV-1 Pr55gag expression vector (pGAG) and 1 µg of one of the designated constructs plus 5 µg of a VSV-G expression vector. At 48 h post-transfection, supernatants were collected, filtered, and used to infect HeLa cells. Infectivity for each Gag-Pol construct was determined as the ratio of mutant titers to wt Gag-Pol titers, normalized to virus-associated p24gag protein levels in parallel experiments. **p < 0.01; ***p < 0.001
In another test designed to determine whether the p6*-deleted Gag-Pol mutants mediated virus maturation and produced infectious virions, each Gag-Pol construct was co-expressed with pGAG plus a VSV-G envelope expression plasmid. Culture supernatants were collected for protein analysis and used to infect HeLa cells. Our data indicate that with the exception of ∆p6*fsPR, all p6*-deleted Gag-Pol mutants were capable of producing infectious virions with infectivity levels of approximately 20–40% relative to wt GPfs (Fig. 4d). The third N-terminal PR residue at ∆p6*fsPR (Val instead of Ile) apparently does not account for the virus processing defect, since a Val/Ile polymorphism was found at this position. Further, both ∆p6*fs/PR and ∆p6*fs//PR containing a Val polymorphism were capable of mediating virus particle maturation. ∆p6*fsPR contains an inserted proline adjacent to the last two C-terminal p6* residues. Although ∆p6*fs/PR and ∆p6*fs//PR both contain the same proline insertion at the p6*-deleted region, both were still found to be capable of mediating virus maturation. The four remaining C-terminal p6* residues within ∆p6*fs/PR and ∆p6*fs//PR might prevent the Pro insertion from interfering with PR activation.
Enhanced PR activation due to the LZ replacement of p6* is Gag domain-independent
Given the contribution that Gag makes to PR activation by promoting Gag-Pol dimerization, we hypothesized that Gag removal from DWzPR might impair Gag-Pol dimerization, thereby mitigating the LZ enhancement effect on PR activation and reducing both Gag-Pol auto-cleavage and Gag cleavage efficiency. To test this idea, we engineered GPfs versions of DWzPR with a Gag deletion (fsdWzPR∆Gag) and without one (fsdWzPR) (Fig. 5a). GPfs and a GPfs version of DWz/PR (designated fsdDWz/PR) served as controls. Each construct was co-expressed with D25, a PR-inactivated Gag/Gag-Pol expression plasmid. According to our results, both fsdWzPR and fsdWzPR∆Gag produced barely detectable virus-associated p24gag when co-transfected with D25 at a DNA ratio of 1:10 (Fig. 5b middle panel, lanes 3 and 5). In contrast, virus-associated p24gag was readily detected in fsdWz/PR-plus-D25 co-transfection samples although at a much lower level compared to Pr55gag and p41gag (Fig. 5b, lane 7). Incorporated Gag-Pol deficiency due to premature or enhanced Gag-Pol auto-cleavage might result in insufficient virus processing. The over-expression of fsdWzPR∆Gag and other GPfs mutants led to the complete blocking of virus assembly (Fig. 5b, lanes 4, 6 and 8).

Enhanced Gag-Pol auto-cleavage reduces virus yields and Gag-Pol viral incorporation. a Schematic representations of HIV-1 Gag-Pol expression constructs in a gag-pol frame shift (fs) mutation backbone are as described in the Fig. 4 caption. Striped (Wz) box denotes leucine zipper (LZ). Dashed line indicates deleted Gag coding sequence. “X” denotes a PR-inactivated mutation. Remaining C-terminal p6* residues are in boldface. Altered or additional residues are shown in italics. b, c Leucine zipper-induced Gag-Pol auto-cleavage enhancement leads to reductions in virion yields and Gag-Pol packaging. Indicated amounts of designated plasmids were co-expressed with 15 μg of an HIV-1 protease-defective (D25) Gag/Gag-Pol expression vector (panel b) or co-expressed with pGAG (panel c) at a DNA ratio of 1:10. Culture supernatants and cells were collected at 48–72 h post-transfection and subjected to Western immunoblotting. Indicated are HIV-1 Gag-Pol, 66/51RT, Pr55gag, p41gag and p24gag positions
Virus-associated Gag-Pol molecules detected in medium were likely from D25 (Fig. 5b, upper panel). It is possible that D25 Gag-Pol competes with other Gag-Pol mutants in terms of viral incorporation. There is also the possibility that D25 PR-defective Gag-Pol interferes with the ability of incorporated Gag-Pol mutants to mediate virus particle processing. This may partly explain the relatively lower levels of virus-associated p24gag that we observed in fsdWz/PR co-transfection samples (Fig. 5b middle panel lane 7). To study these possibilities, Gag-Pol mutants were co-expressed with pGAG. Results indicate that virus-associated RT and p24gag were both readily detected in wt GPfs or fsdWz/PR co-transfection samples (Fig. 5c). In contrast, virus-associated RT and p24gag were both barely detectable in fsdWzPR co-transfection samples, likely due to a Gag-Pol incorporating defect (Fig. 5c, lane 3).
Combined, these results suggest that an LZ replacement for p6* leads to enhanced Gag-Pol auto-cleavage and associated Gag-Pol incorporation deficiency, and that Gag removal does not reduce the LZ enhancement effect on PR activation.
Discussion
Even though the removal of p6* from PR precursor is necessary for PR to be fully functional, the enhancement effect of LZ on PR-mediated Gag processing was not significantly compromised by blocking p6*-PR cleavage. Further, we noted that DWz/PRV/P, DWz//PRV/P and other constructs with substitution mutations at the remaining C-terminal p6* tetra-peptide were all capable of producing readily detectable virus-associated Gag, although with processing defects for some of the mutants (Figs. 1, 2). These results suggest that the enhancement of PR activation by LZ may be reduced when as few as six amino acid residues, whether native or foreign, are present between LZ and PR. The hexa-peptide between LZ and PR may serve as a spacer that prevents the potent LZ dimerization motif from facilitating PR dimer formation. Additionally, substitutions at the C-terminal p6* tetra-peptide might interfere with PR maturation, thereby contributing, at least in part, to reduced Gag processing efficiency.
As a result of blocked cleavage at the PR-RT site, HIV-1 PR-RT fusion is capable of mediating virus processing and supporting virus replication [41]. In contrast, p6* appears to be capable of inhibiting PR maturation, and therefore must be removed for PR to be fully active. However, DWzPR virus yields were not significantly restored when p6*-PR cleavage was blocked, suggesting that HIV-1 PR precursors containing a LZ motif fused at the N-terminus may exhibit markedly enhanced enzymatic activity even when free mature PR is not released. Theoretically, PR-associated products with LZ linked at the PR N-terminus may exist in DWzPRV/P transfectant samples due to blocked cleavage at p6*/PR. Since attempts to detect these PR-associated products were unsuccessful, we believe PR may access putative cryptic cleavage sites within the LZ in addition to being self-degrading. Regardless, our findings suggest that PR precursors containing foreign peptides fused at the N-terminus are still capable of being functionally active. Specifically, PR activity may be significantly enhanced when a potent dimerization motif is present at the PR N-terminus.
Although C-terminal p6* substitutions with either PIDL or PANF residues exerted no major impacts on HIVgpt virus infectivity according to single-round infection assays, they did reduce virus processing efficiency (Fig. 3). These data agree with an earlier report that substitutions for C-terminal p6* residues can impair PR maturation, resulting in a virus processing defect [20]. Negative effects of C-terminal p6* tetra-peptide substitution mutations on virus processing become increasingly noticeable when tested with a Dp6*PR backbone (Fig. 2c), presumably due to PR function perturbation by an upstream inactivated PR copy.
According to single-cycle infection assays, ∆p6*fs generated mature infectious virions. In contrast, ∆p6*fsPR (with only one amino acid difference from ∆p6*fs in the linker region) was completely blocked in terms of mediating virus maturation (Fig. 4). ∆p6*fs has a Leu at the P3 position, while ∆p6*fsPR has a foreign Pro insertion at P3. Pro residues were barely detected at the P3 position flanking the PR cleavage site (i.e., the third amino acid residue immediately upstream from the PR substrate cleavage site). Leu was one of several amino acids found at the P3 position [42]. This raises the possibility of P3-Pro disrupting PR maturation, thereby blocking the ability of ∆p6*fsPR to mediate virus processing. Even though they both contain a Pro insertion in the deleted p6* region, ∆p6*fs/PR and ∆p6*fs//PR were still capable of mediating virus maturation, likely due to the containment of an intact C-terminal p6* tetra-peptide SFNF at the p6*/PR junction. This agrees with our observations involving DWz/PR and DWz//PR, both of which contain an intact C-terminal p6* tetra-peptide and are capable of counteracting the LZ enhancement of PR activation.
In addition to containing only four residues between LZ and PR, DWzPR has a Pro at the P3 position, which might contribute to its failure to counteract the LZ enhancement effect on PR-mediated Gag cleavage. C-terminal p6* residues might inhibit PR maturation by destabilizing the structure of the folded PR dimer [43]. The absence of regulated PR dimer structure folding due to mutations at C-terminal p6* residues might allow the potent LZ dimerization motif to facilitate or exert a synergetic effect on Gag-Pol or PR dimer formation via PR dimer stabilization. PR is consequently prematurely activated, resulting in a significant enhancement of Gag processing as was observed for DWzPR and DWzPRV/P.
In conclusion, our findings suggest that an HIV-1 PR precursor containing an N-terminally extended peptide can function efficiently without liberating free mature PR. HIV-1 PR precursor activity might be manipulated via the altering of peptide residues adjacent to the PR N-terminus.
References
Petropoulos C. Retroviral taxonomy, protein structures, sequences, and genetic maps. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. New York: Cold Spring Harbor Laboratory Press; 1997.
Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature. 1988;331:280–3.
Freed EO. HIV-1 assembly, release and maturation. Nat Rev Microbiol. 2015;13:484.
Huang M, Martin MA. Incorporation of Pr160(gag-pol) into virus particles requires the presence of both the major homology region and adjacent C-terminal capsid sequences within the Gag-Pol polyprotein. J Virol. 1997;71:4472–8.
Smith AJ, Srinivasakumar N, Hammarskjöld ML, Rekosh D. Requirements for incorporation of Pr160gag-pol from human immunodeficiency virus type 1 into virus-like particles. J Virol. 1993;67:2266–75.
Srinivasakumar N, Hammarskjöld ML, Rekosh D. Characterization of deletion mutations in the capsid region of human immunodeficiency virus type 1 that affect particle formation and Gag-Pol precursor incorporation. J Virol. 1995;69:6106–14.
Park J, Morrow CD. The nonmyristylated Pr160gag-pol polyprotein of human immunodeficiency virus type 1 interacts with Pr55gag and is incorporated into viruslike particles. J Virol. 1992;66:6304–13.
Lee SK, Potempa M, Swanstrom R. The choreography of HIV-1 proteolytic processing and virion assembly. J Biol Chem. 2012;287:40867–74.
Swanstrom R, Wills JW. Synthesis, assembly, and processing of viral proteins. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. New York: Cold Spring Harbor Laboratory Press; 1997.
Pettit SC, Moody MD, Wehbie RS, Kaplan AH, Nantermet PV, Klein CA, Swanstrom R. The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J Virol. 1994;68:8017–27.
Peng C, Ho BK, Chang TW, Chang NT. Role of human immunodeficiency virus type 1-specific protease in core protein maturation and viral infectivity. J Virol. 1989;63:2550–6.
Liao WH, Wang CT. Characterization of human immunodeficiency virus type 1 Pr160 gag-pol mutants with truncations downstream of the protease domain. Virology. 2004;329:180–8.
Louis JM, Nashed NT, Parris KD, Kimmel AR, Jerina DM. Kinetics and mechanism of autoprocessing of human immunodeficiency virus type 1 protease from an analog of the Gag-Pol polyprotein. Proc Natl Acad Sci USA. 1994;91:7970–4.
Pettit SC, Gulnik S, Everitt L, Kaplan AH. The dimer interfaces of protease and extra-protease domains influence the activation of protease and the specificity of GagPol cleavage. J Virol. 2003;77:366–74.
Quillent C, Borman AM, Paulous S, Dauguet C, Clavel F. Extensive regions of pol are required for efficient human immunodeficiency virus polyprotein processing and particle maturation. Virology. 1996;219:29–36.
Zybarth G, Carter C. Domains upstream of the protease (PR) in human immunodeficiency virus type 1 Gag-Pol influence PR autoprocessing. J Virol. 1995;69:3878–84.
Figueiredo A, Moore KL, Mak J, Sluis-Cremer N, de Bethune MP, Tachedjian G. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLoS Pathog. 2006;2:e119.
Tachedjian G, Orlova M, Sarafianos SG, Arnold E, Goff SP. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc Natl Acad Sci USA. 2001;98:7188–93.
Pan YY, Wang SM, Huang KJ, Chiang CC, Wang CT. Placement of leucine zipper motifs at the carboxyl terminus of HIV-1 protease significantly reduces virion production. PLoS One. 2012;7:e32845.
Ludwig C, Leiherer A, Wagner R. Importance of protease cleavage sites within and flanking human immunodeficiency virus type 1 transframe protein p6* for spatiotemporal regulation of protease activation. J Virol. 2008;82:4573–84.
Partin K, Zybarth G, Ehrlich L, DeCrombrugghe M, Wimmer E, Carter C. Deletion of sequences upstream of the proteinase improves the proteolytic processing of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1991;88:4776–80.
Tessmer U, Krausslich HG. Cleavage of human immunodeficiency virus type 1 proteinase from the N-terminally adjacent p6* protein is essential for efficient Gag polyprotein processing and viral infectivity. J Virol. 1998;72:3459–63.
Paulus C, Ludwig C, Wagner R. Contribution of the Gag-Pol transframe domain p6* and its coding sequence to morphogenesis and replication of human immunodeficiency virus type 1. Virology. 2004;330:271–83.
Chatterjee A, Mridula P, Mishra RK, Mittal R, Hosur RV. Folding regulates autoprocessing of HIV-1 protease precursor. J Biol Chem. 2005;280:11369–78.
Ishima R, Torchia DA, Louis JM. Mutational and structural studies aimed at characterizing the monomer of HIV-1 protease and its precursor. J Biol Chem. 2007;282:17190–9.
Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat Struct Biol. 1999;6:868–75.
Sadiq SK, Noe F, De Fabritiis G. Kinetic characterization of the critical step in HIV-1 protease maturation. Proc Natl Acad Sci USA. 2012;109:20449–54.
Yu F-H, Huang K-J, Wang C-T. C-terminal HIV-1 transframe p6* tetrapeptide blocks enhanced Gag cleavage incurred by leucine zipper replacement of a deleted p6* domain. J Virol. 2017;91:e00103-17.
Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, et al. Complete nucleotide sequence of the AIDS virus. HTLV-III. Nature. 1985;313:277–84.
Page KA, Landau NR, Littman DR. Construction and use of a human immunodeficiency virus vector for analysis of virus infectivity. J Virol. 1990;64:5270–6.
Yu F-H, Chou T-A, Liao W-H, Huang K-J, Wang C-T. Gag-Pol transframe domain p6* is essential for HIV-1 protease-mediated virus maturation. PLoS ONE. 2015;10:e0127974.
Chen S-W, Chiu H-C, Liao W-H, Wang F-D, Chen SSL, Wang C-T. The virus-associated human immunodeficiency virus type 1 Gag-Pol carrying an active protease domain in the matrix region is severely defective both in autoprocessing and in trans processing of gag particles. Virology. 2004;318:534–41.
Boles E, Miosga T. A rapid and highly efficient method for PCR-based site-directed mutagenesis using only one new primer. Curr Genet. 1995;28:197–8.
Chiu H-C, Yao S-Y, Wang C-T. Coding sequences upstream of the human immunodeficiency virus type 1 reverse transcriptase domain in Gag-Pol are not essential for incorporation of the Pr160gag-pol into virus particles. J Virol. 2002;76:3221–31.
Yee JK, Friedmann T, Burns JC. Generation of high-titer pseudotyped retrovirus with very broad host range. Methods in Cell Biology. 1994;43:99–112.
Chen YL, Ts’ai PW, Yang CC, Wang CT. Generation of infectious virus particles by transient co-expression of human immunodeficiency virus type 1 gag mutants. J Gen Virol. 1997;78(Pt 10):2497–501.
Chiang C-C, Wang S-M, Tseng Y-T, Huang K-J, Wang C-T. Mutations at human immunodeficiency virus type 1 reverse transcriptase tryptophan repeat motif attenuate the inhibitory effect of efavirenz on virus production. Virology. 2009;383:261–70.
Ferris AL, Hizi A, Showalter SD, Pichuantes S, Babe L, Craik CS, Hughes SH. Immunologic and proteolytic analysis of HIV-1 reverse transcriptase structure. Virology. 1990;175:456–64.
Hizi A, McGill C, Hughes SH. Expression of soluble, enzymatically active, human immunodeficiency virus reverse transcriptase in Escherichia coli and analysis of mutants. Proc Natl Acad Sci. 1988;85:1218–22.
Chiu H-C, Wang F-D, Chen Y-MA, Wang C-T. Effects of human immunodeficiency virus type 1 transframe protein p6* mutations on viral protease-mediated Gag processing. J Gen Virol. 2006;87:2041–6.
Cherry E, Liang C, Rong L, Quan Y, Inouye P, Li X, Morin N, Kotler M, Wainberg MA. Characterization of human immunodeficiency virus type-1 (HIV-1) particles that express protease-reverse transcriptase fusion proteins11. J Mol Biol. 1998;284:43–56.
Pettit SC, Simsic J, Loeb DD, Everitt L, Hutchison CA, Swanstrom R. Analysis of retroviral protease cleavage sites reveals two types of cleavage sites and the structural requirements of the P1 amino acid. J Biol Chem. 1991;266:14539–47.
Tang C, Louis JM, Aniana A, Suh JY, Clore GM. Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature. 2008;455:693–6.
Authors’ contributions
CTW designed this project and wrote the manuscript. FHY performed the experiment and helped with the data analysis. Both authors read and approved the final manuscript.
Acknowledgements
We thank K.J. Huang and Y.R. Lin for materials and reagents. We also thank the following individuals from the National Institutes of Health AIDS Research and Reference Reagent Program for their assistance in obtaining the following reagents: Stephen Hughes for MAb21 anti-RT monoclonal antibodies and D. Bailey and Mark Page for HIV-1 PR antiserum.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by Grants V104C-056 and V105C-036 from the Taipei Veterans General Hospital and by Taiwan Ministry of Science and Technology Grants 104-2320-B-010-035, 105-2320-B-010-023 and 106-2320-B-010-017-MY2.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yu, FH., Wang, CT. HIV-1 protease with leucine zipper fused at N-terminus exhibits enhanced linker amino acid-dependent activity. Retrovirology 15, 32 (2018). https://doi.org/10.1186/s12977-018-0413-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-018-0413-6