
Abstract
Background
Complex regional pain syndrome (CRPS) is a debilitating condition where trauma to a limb results in devastating persistent pain that is disproportionate to the initial injury. The pathophysiology of CRPS remains unknown; however, accumulating evidence suggests it is an immunoneurological disorder, especially in light of evidence of auto-antibodies in ~ 30% of patients. Despite this, a systematic assessment of all circulating leukocyte populations in CRPS has never been performed.
Methods
We characterised 14 participants as meeting the Budapest clinical criteria for CRPS and assessed their pain ratings and psychological state using a series of questionnaires. Next, we performed immunophenotyping on blood samples from the 14 CRPS participants as well as 14 healthy pain-free controls using mass cytometry. Using a panel of 38 phenotypic and activation markers, we characterised the numbers and intracellular activation status of all major leukocyte populations using manual gating strategies and unsupervised cluster analysis.
Results
We have shown expansion and activation of several distinct populations of central memory T lymphocytes in CRPS. The number of central memory CD8+ T cells was increased 2.15-fold; furthermore, this cell group had increased phosphorylation of NFkB and STAT1 compared to controls. Regarding central memory CD4+ T lymphocytes, the number of Th1 and Treg cells was increased 4.98-fold and 2.18-fold respectively, with increased phosphorylation of NFkB in both populations. We also found decreased numbers of CD1c+ myeloid dendritic cells, although with increased p38 phosphorylation. These changes could indicate dendritic cell tissue trafficking, as well as their involvement in lymphocyte activation.
Conclusions
These findings represent the first mass cytometry immunophenotyping study in any chronic pain state and provide preliminary evidence of an antigen-mediated T lymphocyte response in CRPS. In particular, the presence of increased numbers of long-lived central memory CD4+ and CD8+ T lymphocytes with increased activation of pro-inflammatory signalling pathways may indicate ongoing inflammation and cellular damage in CRPS.
Similar content being viewed by others

Background
Complex regional pain syndrome (CRPS) is a disease that has perplexed clinicians and basic scientists alike, not only for the refractory nature of the condition but also for its protean nature. It can manifest in any given individual, although it is more common in females, and many of its features are difficult to explain on a pathological level. This has led to some clinicians even declaring that the condition is psychosomatic in origin [1, 2]. CRPS clearly presents as a neuropathic pain condition but is mostly singularly unresponsive to oral anti-neuropathic agents (e.g. Gabapentin, Pregabalin). It has classical features that mostly occur after trauma or surgery to a distal limb, and that start in the periphery and ascend to the groin in lower limb cases and to the axilla in upper limb cases, becoming geographically demarcated at these levels. Traditional subtyping of CRPS depends on whether disease onset is associated with a clinically confirmed direct peripheral nerve injury (CRPS type II—previously ‘causalgia’) or without a confirmed nerve injury (CRPS type I—previously ‘reflex sympathetic dystrophy’); however, research indicates that CRPS-I also involves nerve trauma [3,4,5]. Regardless of subtype, the pain is disproportionate to the initiating event and is persistent. It is associated with some degree of swelling of the distal limb, colour change, temperature change, sweating change and motor impairment (weakness, tremor, dystonic posture). A ‘CRPS-not otherwise specified’ label may be applied to those few patients in whom some of these symptoms resolve over time but significant pain remains [6].
Whilst CRPS was initially considered a neurological phenomenon, there is accumulating evidence that it is an immunoneurological disorder [7,8,9]. Early evidence for this can be found as far back as the 1950s in the response to oral corticosteroid administration in early presentations of the condition (< 6 months) [10]. In most cases, acute CRPS resembles peripheral inflammation. The inflammatory response is excessive in CRPS, and this can have pathological effects on the nervous system and lead to perpetuating inflammation and chronic pain [11,12,13]. Multiple individual findings of immune system involvement have been documented. This has included increased expression of pro-inflammatory cytokines in the skin, blood and cerebrospinal fluid of CRPS patients [14,15,16,17,18,19]. There is evidence for local neurogenic inflammation, with elevation of neuropeptides, particularly CGRP and Substance P that mediate oedema and inflammation and may lead to an immune mediator cascade [20,21,22]. Auto-antibody production has also been implicated, especially involving the muscarinic-2 acetylcholine (M2) receptor, and the α1a and β2 adrenoreceptors [8, 23,24,25,26], whilst elevated plasma levels of soluble IL-2 suggest a possible T lymphocyte-mediated response [27]. A recent review has summarised the auto-inflammatory and auto-immune contributions to CRPS (see [28]).
Flow cytometry has been used to investigate the immune function in CRPS patients with mixed results [29,30,31,32]. These studies analysed a small subset of immune cell types and did not investigate intracellular signalling. An alternative approach to assess immune function is via mass cytometry, which uses metal isotopes instead of fluorophores as reporters, increasing the multiplexing capacity and allowing a large array of immune cells and intracellular activation markers to be examined concurrently. Mass cytometry is a powerful tool that is allowing for new insights into cell biology and systems function to be gained [33,34,35,36,37,38]. Therefore, the current study assessed peripheral blood mononuclear cells (PBMCs) and their intracellular activation status using mass cytometry and systematic unsupervised analysis approaches in chronic (> 6 months) CRPS patients and control subjects.
Methods
Participants
Participants were recruited by Genesis Research Services (Broadmeadow, NSW, Australia) between September 2017 and April 2018, and signed informed consent. The study included both male and female participants aged 19–70 years old, who met the Budapest Criteria for clinical CRPS diagnosis [39] (n = 14, Table 1) or were age-matched pain-free controls (n = 14). We chose to investigate the long-standing immune changes in cases of CRPS (> 6 months), rather than acute changes associated with the initial trauma and healing (< 6 months). Exclusion criteria were less than 18 years of age; CRPS symptom onset less than 6 months (CRPS group); any acute pain (control group); any other neurological, psychiatric or pain conditions that could confound the study endpoints; and pregnancy. On the day of blood collection, participants had ceased taking immune modulating medications (e.g. NSAIDs, steroids and opioids) for at least 7 days. The 7-day drug wash-out could still leave residual effects on immune cells, especially lymphocytes; however, it was the longest wash-out practicable.
Pain and psychological profiling
All participants rated pain from 0 to 100 on a visual analogue scale (VAS) and completed 5 questionnaires to assess pain and psychological variables: Short form McGill pain questionnaire (SF-MPQ-2), short form depression, anxiety and stress Scale (DASS21), Pain self-efficacy questionnaire, (PSEQ), Tampa scale for kinesiophobia (TSK) and Pain catastrophizing scale (PCS).
Blood collection protocol
Blood was taken by venepuncture from the unaffected limb antecubital fossa by a trained phlebotomist, and collected into a 5 ml ethylenediaminetetraacetic acid (EDTA) tube and inverted immediately. Blood was immediately transferred to ten 1.5 ml tubes, 0 .5 ml in each, that contained 0 .7 ml of proteomic stabliser buffer (Smart Tube Inc., CA, USA), and mixed by inversion. Samples were then incubated in proteomic stabliser buffer at room temperture for 10 min, before being transferred to a − 80 °C freezer.
Mass cytometry staining
Blood samples in 1.5 ml tubes were taken out of − 80 °C and transferred to the fridge to thaw samples for 30–45 min. Once thawed, 1 ml of Thaw-Lyse buffer 1 (Smart Tube Inc) was added to each tube and thoroughly mixed. Two 1.5 ml tubes from the same participant (total of 1 ml original blood volume) were combined in a 15 ml tube, topped up to 10 ml with Thaw-Lyse buffer 1, and left at room temperature for 10 min. Samples were spun at 600×g at 4 °C, the supernatant was removed and then the cells lysed a second time with 10 ml Thaw-Lyse buffer 1. Samples were then lysed with 10 ml of Lyse Buffer 2 (Smart Tube Inc.). At this point, if the pellet was not completely white (i.e. incomplete red blood cell lysis), an extra lysis step with Lyse Buffer 2 (Smart Tube Inc) was undertaken. Cells were then suspended in fluorescence-activated cell sorting (FACS) buffer (PBS with 5% FCS, 5 mM EDTA and 0.2% sodium azide) and counted with a haemocytometer to calculate total leukocytes per ml of blood.
Next, 2.5 × 106 cells were transferred to a 1.5 ml tube and incubated with 200 μl heparin (100 IU/ml) in FACS buffer for 20 min at room temperature. Heparin reduces non-specific eosinophil staining artefacts in mass cytometry [40]. Cells were then washed with FACS buffer, spun at 500×g for 5 min and the supernatant was removed before being incubated with surface antibodies on ice for 30 min (see Table 1 for antibody manufacturers and concentrations). Note, the concentrations for all antibodies were optimised for use with the proteomic stabliser buffer prior to staining the experimental samples. Cells were washed and incubated with fixation buffer (eBioscience) for 45 min on ice. Cells were then washed twice in permeabilization buffer (eBioscience), before being incubated with 200 μl heparin (100 IU/ml) in permeabilization buffer for 20 min at room temperature. Cells were washed and spun at 800×g for 7 min, and the supernatant was removed before incubation with intracellular antibodies for forkhead box P3 (FoxP3), T-box protein expressed in T lymphocytes (T-bet), Arg-1 and DAP12 for 30 min on ice (Table 2). Cells were then washed once with permeabilization buffer and then with FACS buffer, before incubation with 500 μl ice-cold methanol for 30 min on ice. Cells were then washed in FACS buffer before being incubated with 200 μl heparin (100 IU/ml) in FACS buffer for 20 min at room temperature. Cells were washed and then incubated with antibodies against intracellular signalling markers for 30 min on ice (Table 1). Cells were washed before incubation with an anti-PE antibody for 15 min on ice. This was necessary as we used a pPLCγ2-PE antibody instead of a direct metal conjugated pPLCγ2 antibody. Cells were washed before being incubated overnight in the fridge with 200 μl DNA intercalator (Cell ID, Fluidigm) diluted 1:4000 in paraformaldehyde (PFA).
Running mass cytometry
On the day of sample acquisition, the cells were diluted in 1 ml FACS buffer and washed. The cells were then washed twice in MilliQ water, and then re-suspended at 1 × 106/ml in 1:10 EQ calibration beads (Fluidigm). A minimum number of 500,000 events were then acquired on a mass cytometer (Helios CyTOF, Fluidigm) using CyTOF software (Fluidigm). Following acquisition, all flow cytometry standard (.fcs) files were normalised to bead signal levels using CyTOF software (see [41] for more details).
Manual gating
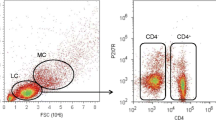
Using FlowJo 10 (OR, USA), normalisation beads were gated out, and then singlets were gated on event length and DNA1. Leukocytes were gated using CD45+ and DNA1. From the CD45+ population, CD66b−CD61b− mononuclear leukocytes were gated, and red blood cell contamination was eliminated by further gating a CD235− population. Major immune cell populations were gated using a manual gating strategy (Fig. 1). All absolute cell numbers of mononuclear cells per ml of blood were back-calculated from the total leukocyte counts made following red blood cell lysis.

A flow diagram representing the manual gating strategy used to isolate major immune cell populations from mass cytometry output. Firstly, quality control gates were performed to remove normalisation beads, isolate singlets and gate CD45+ leukocytes. Next, mononuclear leukocytes (CD66−CD61b−CD235−) were isolated by gating out platelets, granulocytes and erythrocytes. Major lymphocyte and myeloid populations were then isolated using standard phenotypic markers. For example, CD19+ for B lymphocytes, CD3+ for T lymphocytes, CD56+ for NK cells and HLA-DR+ for myeloid cells. Finally, major cell populations were gated further into known subsets
SPADE and FlowSOM
For all data sets, pre-gating to exclude doublets, debris, platelets and red blood cells was performed (top row of Fig. 1). Data was also pre-processed via application of an arcsinh conversion with a standard co-factor of 5. To visualise global changes in intracellular markers in CRPS participants, spanning tree progression of density-normalised events (SPADE) clustering algorithm were run on remaining single mononuclear leukocytes; no additional pre-gating was performed [42]. In brief, the SPADE tree was built with a maximum downsampling population of 50,000 cells and a k means of 100. The locations of clusters representing major leukocyte populations within the SPADE trees were first identified by visualising SPADE trees for all major phenotypic markers, e.g. CD3 for T lymphocytes, CD19 for B lymphocytes, CD14 for classical monocytes. Combinations of phenotypic markers could be used to identify smaller subsets within the SPADE trees, e.g. CD14 and CD16 for inflammatory monocytes. Once these leukocyte populations were identified, SPADE trees displaying intracellular markers could be interpreted.
To analyse the cellular identity and marker expression characteristics of similar cells within a node, phenotypical clustering analysis via a self-organising map (FlowSOM) was performed on single live cells that were pre-gated on CD3+ or HLA-DR+. FlowSOM clustering was conducted via the cytofkit-package (https://github.com/JinmiaoChenLab/cytofkit), which integrates the FlowSOM algorithm from the FlowSOM package [43]. In brief, each set of pre-gated cells were initially organised into 40 clusters and then phenotypically similar nodes were grouped into meta-clusters to allow identification of the cellular identity of each node.
Statistical analysis
Fisher’s exact test was used to compare the proportion of male and female participants between CRPS and control groups (Prism 7, GraphPad Software Inc.). All other data were first analysed for normality using the D’Agostino-Pearson normality test, and then an unpaired Student’s T test (for normally distributed data), or a Mann-Whitney U test (for non-normally distributed data) were used to test for statistically significant differences between control and CRPS groups (Prism 7). For families of analyses, the family-wise error rate was corrected using the Benjamini-Hochberg procedure, at a false discovery rate of q < 0.1. This is the most appropriate and stringent methodology to correct for multiple comparisons and has been used by similar studies [44, 45]. A ‘family of analyses’ was considered to be where a comparable biological measure was made multiple times on the same biological tissue (i.e. the expression of multiple intracellular signalling molecules within a cell population). The data from individual CRPS participants were also analysed using linear regression to determine significant relationships between changes in cell numbers, intracellular signalling and pain or psychological variables.
Results
Participants and clinical findings
All participants in the CRPS group met the Budapest diagnostic criteria for CRPS [39] and were in the chronic phase of disease, having symptoms for at least 6 months. Symptom onset in all 14 participants in the CRPS group occurred following trauma; 7 suffered fractures, 4 had soft tissue injuries and 3 had undergone surgery, and there was an equal proportion of participants with upper and lower limbs affected. CRPS is more common in females than males at a ratio of 3:1, and although the proportion of females in our CRPS group was greater than in the healthy control group, this was not statistically significant (P = 0.13). Severe pain was confirmed in the CRPS group, with a mean pain rating of 69/100 on a visual analogue scale (VAS) and a mean score of 114/220 on the Short-form McGill Pain Questionnaire (SF-MPQ2), both significantly higher than the control group (both U = 0, P < 0.001, Table 3). Psychological state was examined using the Short-form Depression, Anxiety and Stress scale (DASS21). According to the DASS21 severity ratings, the CRPS group had mild depression (U = 34.5, P < 0.01), moderate anxiety (U = 22.5, P < 0.001) and mild stress (U = 1, P < 0.001), all increased compared to the control group, which had normal scores on all ratings. Kinesiophobia, pain catastrophising and reduced pain self-efficacy were also found in the CRPS group, which had higher scores on the Tampa Scale for Kinesiophobia (TSK) and Pain Catastrophising Scale (PCS), and lower scores on the Pain Self-efficacy Questionnaire (PSEQ) compared to the control group (U = 14.5, U = 12.5 and U = 0 respectively, all P < 0.001).
Manual gating results
A manual gating strategy was used to quantify changes in major leukocyte populations that would undergo subsequent validation using unsupervised cluster analyses. In total, we quantified 22 distinct immune subsets in the peripheral blood of CRPS patients, a magnitude more than previous studies. This approach identified higher numbers of total CD3+ (excluding TCRγδ +) T lymphocytes in the CRPS group (t26 = 2.139, P < 0.05, Table 4). Further analysis of major T lymphocyte subsets revealed significantly higher numbers of CD4+CD127loCD25+FoxP3+ Tregs compared to control (t26 = 2.286, p < 0.05). CD4+CD45RO+T-bet+ Th1 cells were twice as numerous in the CRPS group; however, this dataset was not normally distributed and did not reach statistical significance (U = 65, P = 0.14). There were no significant differences in classical and inflammatory monocytes or B, NK and NKT cell numbers. Surprisingly, there were reduced numbers of CD11c+CD1c+CD141− myeloid dendritic cells (mDCs) in the CRPS group (U = 44.5, P < 0.05).
Unsupervised cluster analysis of immune cells and intracellular activation
Unsupervised clustering analyses FlowSOM and SPADE were used to further probe all major adaptive and innate cell populations in CRPS, in particular to confirm significant changes identified with manual gating, but also to investigate more specific cell clusters, as well as their intracellular signalling activation. For the FlowSOM analysis, only clusters with statistically significant differences in cell numbers between control and CRPS are reported below.
CD8+ T lymphocytes
FlowSOM analysis identified significantly higher numbers of central memory CD8+ T lymphocytes (CD3+CD8+CD45RO+CCR7+) in the CRPS group compared to controls (t26 = 2.163, P < 0.05), whereas the number of naïve, effector and effector memory CD8+ cells were stable across both groups (Fig. 2a). Moreover, the central memory CD8+ T lymphocytes cluster contained greater expression of phosphorylated p65 (subunit of NFκB) and STAT1, as well as increased expression of gp130, compared to control (t26 = 2.384, t26 = 2.23 and t26 = 2.436 respectively, all P < 0.05, Fig. 2b). There was no correlation between pain scores (VAS or SF-MPQ2) and central memory CD8+ T lymphocytes within the CRPS group. However, the number of central memory CD8+ T lymphocytes in individuals in the CRPS group positively correlated with the stress scores on the DASS21 (F1,12 = 5.13, r2 = 0.30, P < 0.05, Fig. 2c). That is more central memory CD8+ T lymphocytes are associated with higher perceived stress levels in CRPS.

Increased numbers of central memory CD8+ T lymphocytes with increased pro-inflammatory activation are found in the blood of CRPS participants compared to healthy controls. a A bar graph showing the number of cells in FlowSoM clusters representing the four major CD8+ T lymphocyte populations; naive (Tn, CD3+CD8+CD45RA+CCR7+), effector (Teff, CD3+CD8+CD45RA+CCR7−), effector memory (Tem, CD3+CD8+CD45RO+CCR7−), central memory (Tcm, CD3+CD8+CD45RO+CCR7+). b Intracellular signalling and functional marker expression levels in the FlowSoM cluster representing central memory CD8+ T lymphocytes. All data are presented as group means (± S.E.M.). * represents P < 0.05 in an unpaired Student’s T Test. c Linear correlation between the number of central memory CD8+ T and stress scores on DASS21 in the CRPS group. N = 14 CRPS and N = 14 controls. * represents P < 0.05 in a linear regression analysis
CD4+ T lymphocytes
FlowSOM analysis identified higher numbers of central memory CD4+ T lymphocytes (CD3+CD4+CD45RO+CCR7+) compared to controls (t26 = 2.185, P < 0.05), whereas naïve, effector and effector memory were unchanged (Fig. 3a). Looking at more specific clusters, the number of central memory Th1 lymphocytes (CD3+CD4+CD45RO+CCR7+T-bet+) and central memory Tregs (CD3+CD4+CD45RO+CCR7+CD127loCD25+FoxP3+) were greater in the CRPS group (U = 44, P < 0.05 and t25 = 2.208, P < 0.05 respectively; Fig. 3b). There was no correlation between pain scores (VAS or SF-MPQ2) and any CD4+ T lymphocyte populations within the CRPS group. Unexpectedly, the number of central memory Th1 lymphocytes in individuals in the CRPS group negatively correlates with anxiety scores on DASS21 (F1,12 = 7.7, r2 = 0.39, P < 0.05). Indicating that lower perceived anxiety levels occur in the patients with the greatest number of Th1 lymphocytes.

Increased numbers of central memory CD4+ T lymphocytes with increased pro-inflammatory activation are found in the blood of CRPS participants compared to healthy controls. a A bar graph showing the number of cells in FlowSoM clusters representing the four major CD4+ T lymphocyte populations; naive (Tn, CD3+CD4+CD45RA+CCR7+), effector (Teff, CD3+CD4+CD45RA+CCR7−), effector memory (Tem, CD3+CD4+CD45RO+CCR7−), central memory (Tcm, CD3+CD4+CD45RO+CCR7+). b A bar graph showing the number of cells in FlowSoM clusters representing central memory Th1 lymphocytes (CD3+CD4+CD45RO+CCR7+T-bet+) and Tregs (CD3+CD4+CD45RO+CCR7+CD127loCD25+FoxP3+). Intracellular signalling and functional marker expression levels in FlowSoM clusters representing c Th1 lymphocytes and d Tregs. N = 14 CRPS and N = 14 controls. All data are presented as group means (± S.E.M.). * represents P < 0.05 and ** represents P < 0.01 in an unpaired Student’s T Test, and # represents P < 0.01 in a Mann-Whitney U Test
Several markers of intracellular expression were increased in the CRPS group. Expression of phosphorylated P65 in the central memory Th1 lymphocytes (t26 = 2.817, P < 0.01; Fig. 3c), and expression of phosphorylated p65 and 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 (PLCγ2) in central memory Tregs were increased compared to controls (t25 = 2.746, P < 0.05 and t25 = 3.467, P < 0.01 respectively; Fig. 3d). Further, expression of phospho-p65 in central memory Th1 lymphocytes was negatively correlated with PCS scores (F1,12 = 5.0, r2 = 0.29, P < 0.05), indicating reduced activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) in Th1 cells is associated with greatest pain catastrophising.
SPADE trees are a useful visualisation tool to overlay expression patterns of intracellular markers across major leukocyte populations. They are useful to highlight trends in expression patterns, although unlike FlowSOM clusters, they are not well suited for statistical comparison. In adaptive cell types, SPADE trees reveal a generalised increased in phospho-p65 expression in T lymphocytes (Fig. 4a), a moderate increase in phospho-ERK in some lymphocyte populations (Fig. 4c), and a generalised reduction in phospho-STAT3 expression, most apparent in CD4+ T lymphocytes (Fig. 4h).

Spanning tree progression of density-normalised events (SPADE) trees showing evidence of pro-inflammatory activation in distinct lymphocyte and myeloid cell populations in the blood of CRPS participants relative to healthy controls. The SPADE algorithm was run on a downsampled population of single leukocytes from all CRPS and control participants. SPADE trees were generated showing the fold-change between CRPS and control groups in the expression of phosphorylated (activated), a p65 NFκB, b AKT, c ERK, d MAPKAPK2, e p38, f PLCγ2, g STAT1, h STAT3 and i STAT5. The major cell populations labelled in (a) are representative of all SPADE trees. Note: colour scales vary for each marker
Myeloid cells
FlowSOM analysis of monocytes confirmed manual gating with no significant differences in the number of classical or inflammatory monocytes. This is in contrast to Ritz et al (2011), who previously reported an increase in CD14+CD16+ inflammatory monocytes in CRPS, whereas there were 20% more CD14+CD16+ cells in our CRPS cohort, although this did not reach statistical significance. There was, however, a generalised increase in pAKT expression in all leukocytes, although it was strongest in myeloid cells (Fig. 4b). There was a selective increase in pMAPKAPK2 (Fig. 4d), pp38 (Fig. 4e), pPLCγ2 (Fig. 4f), pSTAT1 (Fig. 4g) and pSTAT5 (Fig. 4i) expression in myeloid cells, relative to lymphocytes, with phosphorylation of p38, STAT1 and STAT5 in particular indicative of pro-inflammatory activation.
Dendritic cells
Unsupervised clustering using FlowSOM confirmed that there were significantly fewer CD1c+ mDCs (HLA-DR+CD14−CD11c+CD1c+CD141−) in CRPS (U = 34, P < 0.01), whilst the other two major dendritic cell populations, CD141+ mDCs and pDCs, were unchanged (Fig. 5a). Intracellular expression patterns within the CD1c+ mDC cluster of the CRPS group showed increased phospho-p38 expression compared to controls (U = 36, P < 0.01; Fig. 5b). There were no changes in intracellular expression in CD141+ DCs and pDCs (data not shown).

Reduced CD1c+ myeloid dendritic cell numbers but with increased p38 activation are found in the blood of CRPS participants compared to healthy controls. a A bar graph showing the number of cells in FlowSoM clusters representing the three major dendritic cell populations; CD1c+ mDCs (HLA-DR+CD14−CD11c+CD1c+CD141−), CD141+ mDCS (HLA-DR+CD14−CD11c+CD141+CD1c−) and plasmacytoid DCS (pDCs, HLA-DR+CD14−CD123+) in CRPS and control participants. b Intracellular signalling and functional marker expression levels in the FlowSoM cluster representing CD1c+ mDCs. N = 14 CRPS and N = 14 controls. All data are presented as group means (± S.E.M.). ## represents P < 0.01 in a Mann-Whitney U Test
We probed the relationship of intracellular signalling in different cell types in individuals in the CRPS group and found that pp38 expression in CD1c+ mDC positively correlated with pSTAT1 expression in central memory CD8+ T lymphocytes (F1,12 = 8.52, r2 = 0.42, P < 0.05, Fig. 6a), and with pp65 expression in central memory Th1 lymphocytes (F1,12 = 4.98, r2 = 0.29; Fig. 6b). That is, activation of p38 in CD1c+ mDC is associated with increased activation markers in T lymphocytes.

Increased pp38 signalling in CD1c+ myeloid dendritic cells correlates with increased signalling in T lymphocytes in the blood of CRPS participants. a Linear correlation between pp38 in CD1c + mDCs and pSTAT1 in central memory CD8+ T lymphocytes. b Linear correlation between pp38 in CD1c + mDCs and pP65 in central memory CD4+ Th1 lymphocytes. * represents P < 0.01 in a linear regression analysis
Discussion
Using a mass cytometric approach, we have identified increased numbers of central memory CD8+ and CD4+ T lymphocytes in 14 participants in the chronic phase of CRPS; moreover, these cells bare the hallmark of pro-inflammatory activation. Further, increased phospho-p38 expression provides evidence of CD1c+ mDCs activation, despite a reduction in their number. These findings provide preliminary evidence of an antigen-mediated T lymphocyte response in CRPS.
CD8+ lymphocytes
Central memory CD8+ T lymphocytes show a selective expansion in CRPS, without changes to naïve and effector subsets. A previous study reported a reduction in total CD8+ T lymphocytes; however, they did not examine the central memory subtype and had a limited medication washout period [30]. Central memory CD8+ T lymphocytes persist in the blood, maintaining a strong proliferative capacity, but with reduced effector functions compared to other CD8+ T lymphocytes [46]. Central memory CD8+ T lymphocyte expansion is favoured following low-affinity antigen presentation via major histocompatibility complex (MHC) class I [47], although the exact antigen in CRPS remains unknown. It could be a damage-associated auto-antigen [48], or it may be related to an infection, given evidence of prior exposure to campylobacter in CRPS patients [49].
Central memory CD8+ T lymphocytes had increased expression of phosphorylated STAT1 and p65 (NF κ B subunit), as well as increased gp130 expression. NFκB is a critical transcription factor in interferon-gamma (IFNγ) production in CD8+ T lymphocytes [50], whilst STAT1 signalling mediated by IFNγ regulates clonal expansion and production of memory CD8+ lymphocytes [51]. It is therefore possible that NFκB-IFN γ-STAT1 signalling may be involved in the expansion of central memory T lymphocytes seen in CRPS. gp130 is a common signal transducing component of functional receptor complexes of the IL-6 family, including IL-6, IL-11, leukaemia inhibitory factor (LIF) and ciliary neurotrophic factor (CNTF) [52]. In CD8+ T lymphocytes, IL-6 binding to gp80 leads to the formation of a gp130 homodimer complex that results in activation of STAT3 and consequent effector functions mediated by IFNγ and granzyme B production [53,54,55]. CRPS patients have elevated IL-6 in blister fluid and CSF, as well as reduced plasma levels of soluble gp130, that counteracts IL-6 signalling [14, 17, 56, 57]. Therefore, it is possible increased gp130 expression by CD8+ lymphocytes may be related to increased IL-6 signalling resulting in effector functions that enhance cellular damage in CRPS. These cell signalling patterns indicate activation of the expanded central memory CD8+ T lymphocyte population and are consistent with their pathogenic role in CRPS.
CD4+ Th1 lymphocytes
Central memory Th1 lymphocytes, but not other naïve or effector CD4+ T lymphocytes, show a selective expansion in CRPS. Relative to effector T lymphocytes, central memory CD4+ T cells are activated by lower levels of antigen presentation through MHC class II, have strong IL-2 mediated proliferative capacity but reduced production of effector cytokines, IL-4 or IFN-γ [58]. Under certain stimulation parameters, Th1 cells are much more likely to enter the memory pool than other helper T subtypes, such as Th17 [59]. In CRPS, the exact antigen that drives central memory Th1 lymphocyte expansion and consequently whether these cells are auto-reactive remains to be determined. Th1 responses are highly dependent on NF κ B signalling, being impaired by overexpression of the negative regulator, IκB, as well as in the absence of p65 where IFNγ production was diminished [50, 60]. Thus, it is unsurprising to see that these cells have increased phosphorylation of p65 in CRPS. Increased tumour necrosis factor (TNF), IL-2 and soluble IL-2 receptor (sIL-2R) in the blood of CRPS patients provide supporting evidence of a Th1 response [19, 27]. An increase in central memory CD4+ T lymphocytes is also found in carpal tunnel syndrome [61], whilst in vivo adoptive transfer of Th1 cells in rats lacking mature T lymphocytes potentiated neuropathic pain symptoms [62]. Our findings, together with previous literature, support a role for central memory Th1 lymphocytes in the exaggerated nociceptive signalling associated with CRPS.
CD4+ regulatory T lymphocytes
Central memory CD4+ Tregs are expanded and have increased phosphorylation of p65 and PLCγ2 in CRPS. Although NFκB signalling has been shown to regulate development of Tregs [63], deletion of Ubc13, a positive regulator of IκB, impairs their suppressive function and renders them sensitive to acquisition by Th1 cells [64]. Phosphorylated PLCγ2 is able to cleave phosphatidylinositol 4,5-bisphosphate (PIP2) into the second messengers phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and diacylglycerol (DAG). DAG activates protein kinase C (PKC), which activates NFκB signalling. Several PLCγ2 mutations have been linked to inflammatory and auto-immune diseases [65, 66].
A recent study that supports our findings reported an increase in CD39+ Tregs in CRPS, with concomitant decrease in Th17 cells, which they supress [29]. A similar imbalance of Th17 and Tregs was observed in chronic lower back pain and neuropathic pain [45, 67], although whether elevated Tregs contribute to pain or represent a compensatory response to persistent pain, stress and inflammation is unclear. Interestingly, in vivo expansion of Tregs has been shown to reduce symptoms of neuropathic pain, whilst Treg depletion increases pain hypersensitivity [68, 69]. Clearly, increased numbers of Tregs in CRPS patients is insufficient to suppress pain, which may be due to increased PLCγ2 and NFκB signalling, which renders them sensitive to Th1 conversion.
Dendritic cells
It is believed that in CRPS, on the production of damaged tissue, there is a release of substances such as damage-associated molecular patterns (DAMPS), which activate immature DCs in the skin and tissue [48, 70, 71]. This is supported by evidence of accumulation of dermal resident dendritic cells (i.e. Langerhans cells) in the affected limbs of CRPS patients with severe pain compared to the limbs of non-CRPS affected patients who had recovered from nerve injury, and in the tibial fracture mouse model of CRPS [21, 72]. These cells then become phenotypically mature and translocate from their resident position to the draining lymph node (axilla and groin) where they can activate CD8+ and CD4+ T lymphocytes [73,74,75].
We found a reduced number of circulating CD1+ mDCs, but with increased p38 phosphorylation. CD1c+ mDCs are strong stimulators of naive CD4+ T lymphocytes, and phosphorylation of p38 critically regulates the production of IL-12, a Th1 priming cytokine, in DCs [76]. Therefore, since increased CD1c+ mDCs p38 signalling correlated with increased NFκB and STAT1 signalling in central memory CD4+ Th1 and CD8+ T lymphocytes respectively, we hypothesise that increased phospho-p38 expression in mDCs is related to T lymphocyte activation. Moreover, reduced numbers of circulating CD1+ mDCs could be indicative of trafficking to tissues or lymph nodes, given that reduced immature DCs have been reported in neurodegenerative diseases following brain trafficking [77]. Our findings together with those of others provide evidence of expansion of activated CD8+ and CD4+ T lymphocytes; activation of DCs, but with fewer found in the circulation, and larger numbers sequestrated in the affected peripheral tissue and draining lymph nodes [21, 72].
Correlation of immune changes with pain and psychological variables
Activated CD4+ and CD8+ T lymphocytes contribute to nociceptive signalling and cellular damage through the release of cytokines or direct infiltration into peripheral nerves, dorsal root ganglia and the spinal cord [78, 79]. It is well known that cytokines and immune cells can signal to the brain through neural, humoral and cellular pathways and modulate mood [80, 81]. Although, it is yet to be confirmed whether T lymphocytes migrate to supraspinal sites in CRPS. Despite this, T lymphocytes can indirectly modulate pain processing in critical brain regions through peripheral inflammatory cascades [82,83,84].
The number of central memory CD8+ T lymphocytes positively correlated with stress scores, whilst the number of Th1 cells and their NFκB signalling negatively correlated with anxiety scores and pain catastrophising, respectively. These results suggest that central memory CD8+ T lymphocyte expansion, rather than Th1 lymphocyte expansion and activation, may drive mood changes in CRPS. Elevated CD8+ T lymphocytes and decreased CD4+ T lymphocytes have been previously reported in human immunodeficiency virus (HIV) and liver cirrhosis patients with co-morbid depression [85,86,87,88]. Further, plasma levels of the archetypal pro-inflammatory cytokine TNF correlated with depression scores in painful neuropathy [89]. Therefore, our findings provide further evidence that peripheral and central inflammatory cascades may mediate the co-morbidity of pain and depression [44, 82, 84, 90].
Limitations
We wish to emphasise that immune changes are only one aspect of the complex pathophysiology of CRPS and we have not addressed these multiple other areas of investigation such as neural, glial and cortical changes. We refer the reader to several current models that describe these changes in much detail [7, 9, 91]. The major limitations of this study are the small CRPS cohort, and despite being non-significant, the larger proportion of females in the CRPS group relative to the healthy control group. In particular, sex differences in immune function and pain processing could represent a confounding factor in our findings, given that in female mice T lymphocyte function appears to be more critical to the development of neuropathic pain than in male mice [92, 93]. Therefore, whilst expanded T lymphocyte populations in the CRPS group could be related to pain, one could also argue that such differences are exaggerated due to the female dominant make-up of the CRPS group, relative to the male dominant healthy control group.
It is also worth considering that our data demonstrate immune associations with CRPS, rather than causation. Immune changes could represent a pre-existing baseline due to infection or an auto-immune condition that is unmasked by trauma, or trauma-related markers rather than CRPS markers. Therefore, an injury control group where participants experienced a similar trauma but did not go onto develop CRPS would be important to demonstrate that immune changes are CRPS specific. Another factor worth considering is that the immune changes could be related to the stress which occurs due to living with a painful condition such as CRPS, as cortisol is known to modulate T lymphocyte polarisation [94]. This fact is further highlighted given the similar Treg profiles reported in other pain conditions [45, 67].
In light of these limitations, our findings should be considered preliminary and we recommend a large confirmatory study with balanced sexes and an injury control group be performed in the future. Such a study could also seek to immunophenotype patients across different phases of CRPS, and investigate the effects of medication to confirm if a 7-day wash-out period is significant.
Conclusions
We have demonstrated a significant disruption to lymphocyte homeostasis in 14 participants in the chronic phase of CRPS. Expansion of central memory CD8+ and CD4+ T lymphocytes with distinct signalling activation patterns provides preliminary evidence of a chronic antigen-mediated T lymphocyte response in CRPS. Moreover, reduced numbers, but with increased p38 signalling in CD1+ mDCs, could implicate this dendritic cell type in T lymphocyte activation.
Change history
03 April 2019
Following publication of the original article [1], the authors reported an error in Figure 4 as the wrong figure was used.
Abbreviations
- AKT:
-
Protein kinase B
- CD:
-
Cluster of differentiation
- CNTF:
-
Ciliary neurotrophic factor
- CRPS:
-
Complex regional pain syndrome
- DAG:
-
Diacylglycerol
- DAMPS:
-
Damage-associated molecular patterns
- DASS21:
-
Short form depression, anxiety and stress scale
- EDTA:
-
Ethylenediaminetetraacetic acid
- ERK:
-
Extracellular signal-regulated kinase
- FACS buffer:
-
Fluorescence-activated cell sorting buffer
- FCS:
-
Fetal calf serum
- .fcs:
-
Flow cytometry standard
- FoxP3:
-
Forkhead box P3
- gp130/CD130:
-
Glycoprotein130
- HLA-DR:
-
Human leukocyte antigen-DR isotype
- IFNγ :
-
Interferon-gamma
- IL:
-
Interleukin
- IκB:
-
Inhibitor of NFkB
- LIF:
-
Leukaemia inhibitory factor
- M2:
-
Muscarinic-2 acetylcholine receptor
- MAPKAPK2:
-
Mitogen-activated protein kinase-activated protein kinase 2
- mDCs:
-
Myeloid dendritic cells
- MHC:
-
Major histocompatibility complex
- NFkB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- p38:
-
p38 mitogen-activated protein kinase
- p65:
-
Subunit of NFκB
- PBMCs:
-
Peripheral blood mononuclear cells
- PBS:
-
Phosphate-buffered saline
- PCS:
-
Pain catastrophizing scale
- PE:
-
Phycoerythrin
- PFA:
-
Paraformaldehyde
- PIP2:
-
Phosphatidylinositol 4,5-bisphosphate
- PIP3:
-
Phosphatidylinositol (3,4,5)-trisphosphate
- PKC:
-
Protein kinase C
- PLCγ2:
-
1-Phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2
- PSEQ:
-
Pain self-efficacy questionnaire
- S.E.M.:
-
Standard error of the mean
- SF-MPQ-2:
-
Short form McGill pain questionnaire
- sIL-2R:
-
Soluble IL-2 receptor
- SPADE:
-
Spanning tree progression of density-normalised events
- STAT:
-
Signal transducer and activator of transcription
- T-bet:
-
T-box protein expressed in T lymphocytes
- TCRγδ + :
-
T lymphocyte receptor γδ + sub-unit
- Th:
-
T helper lymphocytes
- TNF:
-
Tumour necrosis factor
- TREM:
-
Triggering receptor expressed on myeloid cells
- TSK:
-
Tampa scale for kinesiophobia
- VAS:
-
Visual analogue scale
References
Bass C. Complex regional pain syndrome medicalises limb pain. BMJ. 2014;348:g2631.
Del Pinal F. Editorial. I have a dream ... reflex sympathetic dystrophy (RSD or complex regional pain syndrome—CRPS I) does not exist. J Hand Surg Eur. 2013;38:595–7.
Geertzen JH, Bodde MI, van den Dungen JJ, Dijkstra PU, den Dunnen WF. Peripheral nerve pathology in patients with severely affected complex regional pain syndrome type I. Int J Rehabil Res. 2015;38:121–30.
Oaklander AL, Rissmiller JG, Gelman LB, Zheng L, Chang Y, Gott R. Evidence of focal small-fiber axonal degeneration in complex regional pain syndrome-I (reflex sympathetic dystrophy). Pain. 2006;120:235–43.
Albrecht PJ, Hines S, Eisenberg E, Pud D, Finlay DR, Connolly MK, Pare M, Davar G, Rice FL. Pathologic alterations of cutaneous innervation and vasculature in affected limbs from patients with complex regional pain syndrome. Pain. 2006;120:244–66.
Goebel A, Barker C, Birklein F, et al. Standards for the diagnosis and management of complex regional pain syndrome: Results of a European Pain Federation task force. Eur J Pain. 2019;00:1–11. https://doi.org/10.1002/ejp.1362.
Birklein F, Ibrahim A, Schlereth T, Kingery WS. The rodent tibia fracture model: a critical review and comparison with the complex regional pain syndrome literature. J Pain. 2018;19:1102 e1101–19.
Goebel A, Blaes F. Complex regional pain syndrome, prototype of a novel kind of autoimmune disease. Autoimmun Rev. 2013;12:682–6.
Russo M, Georgius P, Santarelli DM. A new hypothesis for the pathophysiology of complex regional pain syndrome. Med Hypotheses. 2018;119:41–53.
Sigler JW, Ensign DC. ACTH and cortisone in the treatment of the shoulder-hand syndrome. J Mich State Med Soc. 1951;50:1038–44.
Birklein F, Schlereth T. Complex regional pain syndrome-significant progress in understanding. Pain. 2015;156(Suppl 1):S94–103.
Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. 2006;51:240–64.
Pinho-Ribeiro FA, Verri WA Jr, Chiu IM. Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol. 2017;38:5–19.
Alexander GM, van Rijn MA, van Hilten JJ, Perreault MJ, Schwartzman RJ. Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. Pain. 2005;116:213–9.
Konig S, Schlereth T, Birklein F. Molecular signature of complex regional pain syndrome (CRPS) and its analysis. Expert Rev Proteomics. 2017;14:857–67.
Kramer HH, Eberle T, Uceyler N, Wagner I, Klonschinsky T, Muller LP, Sommer C, Birklein F. TNF-alpha in CRPS and ‘normal’ trauma—significant differences between tissue and serum. Pain. 2011;152:285–90.
Parkitny L, McAuley JH, Di Pietro F, Stanton TR, O'Connell NE, Marinus J, van Hilten JJ, Moseley GL. Inflammation in complex regional pain syndrome: a systematic review and meta-analysis. Neurology. 2013;80:106–17.
Schinkel C, Gaertner A, Zaspel J, Zedler S, Faist E, Schuermann M. Inflammatory mediators are altered in the acute phase of posttraumatic complex regional pain syndrome. Clin J Pain. 2006;22:235–9.
Uceyler N, Eberle T, Rolke R, Birklein F, Sommer C. Differential expression patterns of cytokines in complex regional pain syndrome. Pain. 2007;132:195–205.
Kingery WS. Role of neuropeptide, cytokine, and growth factor signaling in complex regional pain syndrome. Pain Med. 2010;11:1239–50.
Li WW, Guo TZ, Shi X, Birklein F, Schlereth T, Kingery WS, Clark JD. Neuropeptide regulation of adaptive immunity in the tibia fracture model of complex regional pain syndrome. J Neuroinflammation. 2018;15:105.
Huygen FJ, De Bruijn AG, De Bruin MT, Groeneweg JG, Klein J, Zijlstra FJ. Evidence for local inflammation in complex regional pain syndrome type 1. Mediat Inflamm. 2002;11:47–51.
Dirckx M, Schreurs MW, de Mos M, Stronks DL, Huygen FJ. The prevalence of autoantibodies in complex regional pain syndrome type I. Mediat Inflamm. 2015;2015:718201.
Dubuis E, Thompson V, Leite MI, Blaes F, Maihofner C, Greensmith D, Vincent A, Shenker N, Kuttikat A, Leuwer M, Goebel A. Longstanding complex regional pain syndrome is associated with activating autoantibodies against alpha-1a adrenoceptors. Pain. 2014;155:2408–17.
Kohr D, Singh P, Tschernatsch M, Kaps M, Pouokam E, Diener M, Kummer W, Birklein F, Vincent A, Goebel A, et al. Autoimmunity against the beta2 adrenergic receptor and muscarinic-2 receptor in complex regional pain syndrome. Pain. 2011;152:2690–700.
Shubayev V, Strongin A, Yaksh T Structural homology of myelin basic protein and muscarinic acetylcholine receptor: Significance in the pathogenesis of complex regional pain syndrome. Molecular Pain. 14:1–5. https://doi.org/10.1177/1744806918815005.
Bharwani KD, Dirckx M, Stronks DL, Dik WA, Schreurs MWJ, Huygen F. Elevated plasma levels of sIL-2R in complex regional pain syndrome: a pathogenic role for T-lymphocytes? Mediat Inflamm. 2017;2017:2764261.
Clark JD, Tawfik VL, Tajerian M, Kingery WS. Autoinflammatory and autoimmune contributions to complex regional pain syndrome. Mol Pain. 2018;14:1744806918799127.
Heyn J, Azad SC, Luchting B. Altered regulation of the T-cell system in patients with CRPS. Inflamm Res. 2019;68:1-6. https://doi.org/10.1007/s00011-018-1182-3.
Kaufmann I, Eisner C, Richter P, Huge V, Beyer A, Chouker A, Schelling G, Thiel M. Lymphocyte subsets and the role of TH1/TH2 balance in stressed chronic pain patients. Neuroimmunomodulation. 2007;14:272–80.
Ribbers GM, Oosterhuis WP, van Limbeek J, de Metz M. Reflex sympathetic dystrophy: is the immune system involved? Arch Phys Med Rehabil. 1998;79:1549–52.
Ritz BW, Alexander GM, Nogusa S, Perreault MJ, Peterlin BL, Grothusen JR, Schwartzman RJ. Elevated blood levels of inflammatory monocytes (CD14+ CD16+ ) in patients with complex regional pain syndrome. Clin Exp Immunol. 2011;164:108–17.
Cheng Y, Newell EW. Deep profiling human T cell heterogeneity by mass cytometry. Adv Immunol. 2016;131:101–34.
Gaudilliere B, Fragiadakis GK, Bruggner RV, Nicolau M, Finck R, Tingle M, Silva J, Ganio EA, Yeh CG, Maloney WJ, et al. Clinical recovery from surgery correlates with single-cell immune signatures. Sci Transl Med. 2014;6:255ra131.
Leipold MD, Newell EW, Maecker HT. Multiparameter phenotyping of human PBMCs using mass cytometry. Methods Mol Biol. 2015;1343:81–95.
Newell EW, Cheng Y. Mass cytometry: blessed with the curse of dimensionality. Nat Immunol. 2016;17:890–5.
Simoni Y, Chng MHY, Li S, Fehlings M, Newell EW. Mass cytometry: a powerful tool for dissecting the immune landscape. Curr Opin Immunol. 2018;51:187–96.
Spitzer MH, Nolan GP. Mass cytometry: single cells, many features. Cell. 2016;165:780–91.
Harden R, Bruehl S. Diagnostic criteria: the statistical derivation of the four criterion factors. In: Wilson PR, Stanton-Hicks M, Harden RN, editors. CRPS: current diagnosis and therapy. Seattle: IASP Press; 2005. p. 45–58.
Rahman AH, Tordesillas L, Berin MC. Heparin reduces nonspecific eosinophil staining artifacts in mass cytometry experiments. Cytometry A. 2016;89:601–7.
Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, Pe'er D, Nolan GP, Bendall SC. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83:483–94.
Qiu P, Simonds EF, Bendall SC, Gibbs KD Jr, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29:886–91.
Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87:636–45.
Austin PJ, Berglund AM, Siu S, Fiore NT, Gerke-Duncan MB, Ollerenshaw SL, Leigh SJ, Kunjan PA, Kang JW, Keay KA. Evidence for a distinct neuro-immune signature in rats that develop behavioural disability after nerve injury. J Neuroinflammation. 2015;12:96.
Luchting B, Rachinger-Adam B, Heyn J, Hinske LC, Kreth S, Azad SC. Anti-inflammatory T-cell shift in neuropathic pain. J Neuroinflammation. 2015;12:12.
Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305.
Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211.
Tajerian M, Hung V, Khan H, Lahey LJ, Sun Y, Birklein F, Krämer HH, Robinson WH, Kingery WS, Clark JD. Identification of KRT16 as a target of an autoantibody response in complex regional pain syndrome. Exp Neurol. 2017;287:14–20.
Goebel A, Vogel H, Caneris O, Bajwa Z, Clover L, Roewer N, Schedel R, Karch H, Sprotte G, Vincent A. Immune responses to campylobacter and serum autoantibodies in patients with complex regional pain syndrome. J Neuroimmunol. 2005;162:184–9.
Balasubramani A, Shibata Y, Crawford GE, Baldwin AS, Hatton RD, Weaver CT. Modular utilization of distal cis-regulatory elements controls Ifng gene expression in T cells activated by distinct stimuli. Immunity. 2010;33:35–47.
Quigley M, Huang X, Yang Y. STAT1 signaling in CD8 T cells is required for their clonal expansion and memory formation following viral infection in vivo. J Immunol. 2008;180:2158–64.
Murakami M, Hibi M, Nakagawa N, Nakagawa T, Yasukawa K, Yamanishi K, Taga T, Kishimoto T. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science. 1993;260:1808–10.
Atsumi T, Sato M, Kamimura D, Moroi A, Iwakura Y, Betz UAK, Yoshimura A, Nishihara M, Hirano T, Murakami M. IFN-γ expression in CD8+ T cells regulated by IL-6 signal is involved in superantigen-mediated CD4+ T cell death. Int Immunol. 2009;21:73–80.
Bottcher JP, Schanz O, Garbers C, Zaremba A, Hegenbarth S, Kurts C, Beyer M, Schultze JL, Kastenmuller W, Rose-John S, Knolle PA. IL-6 trans-signaling-dependent rapid development of cytotoxic CD8+ T cell function. Cell Rep. 2014;8:1318–27.
Yang R, Masters AR, Fortner KA, Champagne DP, Yanguas-Casas N, Silberger DJ, Weaver CT, Haynes L, Rincon M. IL-6 promotes the differentiation of a subset of naive CD8+ T cells into IL-21-producing B helper CD8+ T cells. J Exp Med. 2016;213:2281–91.
Alexander GM, Perreault MJ, Reichenberger ER, Schwartzman RJ. Changes in immune and glial markers in the CSF of patients with complex regional pain syndrome. Brain Behav Immun. 2007;21:668–76.
Alexander GM, Peterlin BL, Perreault MJ, Grothusen JR, Schwartzman RJ. Changes in plasma cytokines and their soluble receptors in complex regional pain syndrome. J Pain. 2012;13:10–20.
Catron DM, Rusch LK, Hataye J, Itano AA, Jenkins MK. CD4+ T cells that enter the draining lymph nodes after antigen injection participate in the primary response and become central-memory cells. J Exp Med. 2006;203:1045–54.
Pepper M, Linehan JL, Pagán AJ, Zell T, Dileepan T, Cleary PP, Jenkins MK. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat Immunol. 2009;11:83.
Aronica MA, Mora AL, Mitchell DB, Finn PW, Johnson JE, Sheller JR, Boothby MR. Preferential role for NF-κB/Rel signaling in the type 1 but not type 2 T cell-dependent immune response in vivo. J Immunol. 1999;163:5116–24.
Moalem-Taylor G, Baharuddin B, Bennett B, Krishnan AV, Huynh W, Kiernan MC, Shin-Yi Lin C, Shulruf B, Keoshkerian E, Cameron B, Lloyd A. Immune dysregulation in patients with carpal tunnel syndrome. Sci Rep. 2017;7:8218.
Moalem G, Xu K, Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience. 2004;129:767–77.
Ruan Q, Chen YH. Nuclear factor-κB in immunity and inflammation: the Treg and Th17 connection. In: Lambris JD, Hajishengallis G, editors. Current topics in innate immunity II. New York: Springer New York; 2012. p. 207–21.
Chang JH, Xiao Y, Hu H, Jin J, Yu J, Zhou X, Wu X, Johnson HM, Akira S, Pasparakis M, et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat Immunol. 2012;13:481–90.
Yu P, Constien R, Dear N, Katan M, Hanke P, Bunney TD, Kunder S, Quintanilla-Martinez L, Huffstadt U, Schroder A, et al. Autoimmunity and inflammation due to a gain-of-function mutation in phospholipase C gamma 2 that specifically increases external Ca2+ entry. Immunity. 2005;22:451–65.
Zhou Q, Lee GS, Brady J, Datta S, Katan M, Sheikh A, Martins MS, Bunney TD, Santich BH, Moir S, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. 2012;91:713–20.
Luchting B, Rachinger-Adam B, Zeitler J, Egenberger L, Mohnle P, Kreth S, Azad SC. Disrupted TH17/Treg balance in patients with chronic low back pain. PLoS One. 2014;9:e104883.
Austin PJ, Kim CF, Perera CJ, Moalem-Taylor G. Regulatory T cells attenuate neuropathic pain following peripheral nerve injury and experimental autoimmune neuritis. Pain. 2012;153:1916–31.
Lees JG, Duffy SS, Perera CJ, Moalem-Taylor G. Depletion of Foxp3+ regulatory T cells increases severity of mechanical allodynia and significantly alters systemic cytokine levels following peripheral nerve injury. Cytokine. 2015;71:207–14.
Heil M, Land WG. Danger signals - damaged-self recognition across the tree of life. Front Plant Sci. 2014;5:578.
Nace G, Evankovich J, Eid R, Tsung A. Dendritic cells and damage-associated molecular patterns: endogenous danger signals linking innate and adaptive immunity. J Innate Immun. 2012;4:6–15.
Calder JS, Holten I, McAllister RM. Evidence for immune system involvement in reflex sympathetic dystrophy. J Hand Surg Br. 1998;23:147–50.
Bousso P, Robey E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol. 2003;4:579–85.
Linderman JJ, Riggs T, Pande M, Miller M, Marino S, Kirschner DE. Characterizing the dynamics of CD4+ T cell priming within a lymph node. J Immunol. 2010;184:2873.
Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science. 2002;296:1873–6.
Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T, Pulendran B. Cutting edge: different toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol. 2003;171:4984–9.
Bossu P, Spalletta G, Caltagirone C, Ciaramella A. Myeloid dendritic cells are potential players in human neurodegenerative diseases. Front Immunol. 2015;6:632.
Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. 2010;229:26–50.
Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 2012;11:629–42.
Capuron L, Miller AH. Immune system to brain signalling: Neuropsychopharmacological implications. Pharmacol Ther. 2011;130:226–38.
Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest. 2017;127:3210–9.
Fiore NT, Austin PJ. Are the emergence of affective disturbances in neuropathic pain states contingent on supraspinal neuroinflammation. Brain Behav Immun. 2016;56:397–411.
Linnman C, Becerra L, Borsook D. Inflaming the brain: CRPS a model disease to understand neuroimmune interactions in chronic pain. J NeuroImmune Pharmacol. 2013;8:547–63.
Walker AK, Kavelaars A, Heijnen CJ, Dantzer R. Neuroinflammation and comorbidity of pain and depression. Pharmacol Rev. 2014;66:80–101.
Cruess DG, Douglas SD, Petitto JM, Leserman J, Ten Have T, Gettes D, Dube B, Evans DL. Association of depression, CD8+ T lymphocytes, and natural killer cell activity: implications for morbidity and mortality in human immunodeficiency virus disease. Curr Psychiatry Rep. 2003;5:445–50.
Evans DL, Ten Have TR, Douglas SD, Gettes DR, Morrison M, Chiappini MS, Brinker-Spence P, Job C, Mercer DE, Wang YL, et al. Association of depression with viral load, CD8 T lymphocytes, and natural killer cells in women with HIV infection. Am J Psychiatry. 2002;159:1752–9.
Ickovics JR, Hamburger ME, Vlahov D, Schoenbaum EE, Schuman P, Boland RJ, Moore J, Group ftHERS. Mortality, CD4 cell count decline, and depressive symptoms among HIV-seropositive WomenLongitudinal analysis from the HIV epidemiology research study. JAMA. 2001;285:1466–74.
Ko FY, Tsai SJ, Yang AC, Zhou Y, Xu LM. Association of CD8 T cells with depression and anxiety in patients with liver cirrhosis. Int J Psychiatry Med. 2013;45:15–29.
Uceyler N, Rogausch JP, Toyka KV, Sommer C. Differential expression of cytokines in painful and painless neuropathies. Neurology. 2007;69:42–9.
Fiore NT, Austin PJ. Glial-cytokine-neuronal adaptations in the ventral hippocampus of rats with affective behavioral changes following peripheral nerve injury. Neuroscience. 2018;390:119–40.
Wei T, Guo TZ, Li WW, Kingery WS, Clark JD. Acute versus chronic phase mechanisms in a rat model of CRPS. J Neuroinflammation. 2016;13:14.
Lopes DM, Malek N, Edye M, Jager SB, McMurray S, McMahon SB, Denk F. Sex differences in peripheral not central immune responses to pain-inducing injury. Sci Rep. 2017;7:16460.
Mapplebeck JC, Beggs S, Salter MW. Sex differences in pain: a tale of two immune cells. Pain. 2016;157(Suppl 1):S2–6.
Calcagni E, Elenkov I. Stress system activity, innate and T helper cytokines, and susceptibility to immune-related diseases. Ann N Y Acad Sci. 2006;1069:62–76.
Acknowledgements
The authors would like to thank Mr. Thomas Ashhurst from Sydney Cytometry at the University of Sydney for his guidance on analysis of high-dimensional mass cytometry data.
Funding
This study has been supported by a Commercial Development and Industry Partnership between Genesis Research Services and the University of Sydney, as well as the NWG Macintosh Memorial Fund from the Discipline of Anatomy & Histology, the University of Sydney.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
MAR contributed to study and panel design, participant recruitment, data interpretation and writing of the manuscript. NTF contributed to blood processing, running the mass cytometry samples and performed FlowSOM and SPADE analyses. CV conjugated antibodies to metal isotypes, optimised all antibody concentrations and contributed to panel design, blood processing, running of the samples, data analysis and interpretation and critically evaluated the manuscript. DB recruited participants, processed questionnaire data and took blood samples. DMS processed questionnaire data and contributed to writing of the manuscript. HMM contributed to panel design, running of the samples, data analysis and interpretation and critically evaluated the manuscript. BF contributed to panel design, data analysis and presentation, and critically evaluated the manuscript. PJA performed blood processing and mass cytometry staining, running of the samples and FlowJo analysis and contributed to study and panel design, data interpretation and writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the University of Sydney Human Ethics committee (Approval #2017/019). Study participation was on a completely voluntary basis with all participants signed informed consent. All demographic information and blood samples were de-identified from the study team.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
The original version of this article was revised: In the incorrect Figure 4, panels b-i were missing the numerical scale values.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Russo, M.A., Fiore, N.T., van Vreden, C. et al. Expansion and activation of distinct central memory T lymphocyte subsets in complex regional pain syndrome. J Neuroinflammation 16, 63 (2019). https://doi.org/10.1186/s12974-019-1449-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-019-1449-9