
Abstract
Spinal cord injury (SCI) is a devastating event that results in significant physical disabilities for affected individuals. Apart from local injury within the spinal cord, SCI patients develop a variety of complications characterized by multiple organ dysfunction or failure. These disorders, such as neurogenic pain, depression, lung injury, cardiovascular disease, liver damage, kidney dysfunction, urinary tract infection, and increased susceptibility to pathogen infection, are common in injured patients, hinder functional recovery, and can even be life threatening. Multiple lines of evidence point to pathological connections emanating from the injured spinal cord, post-injury systemic inflammation, and immune suppression as important multifactorial mechanisms underlying post-SCI complications. SCI triggers systemic inflammatory responses marked by increased circulation of immune cells and pro-inflammatory mediators, which result in the infiltration of inflammatory cells into secondary organs and persistence of an inflammatory microenvironment that contributes to organ dysfunction. SCI also induces immune deficiency through immune organ dysfunction, resulting in impaired responsiveness to pathogen infection. In this review, we summarize current evidence demonstrating the relevance of inflammatory conditions and immune suppression in several complications frequently seen following SCI. In addition, we highlight the potential pathways by which inflammatory and immune cues contribute to multiple organ failure and dysfunction and discuss current anti-inflammatory approaches used to alleviate post-SCI complications. A comprehensive review of this literature may provide new insights into therapeutic strategies against complications after SCI by targeting systemic inflammation.
Similar content being viewed by others

Background
Spinal cord injury (SCI) causes disastrous damage to patients. While intraspinal infections, ischemia, and tumors can give rise to non-traumatic SCI, the majority of SCI is caused by physical trauma to the spine from sports injuries, car accidents, falls, and gunshots. Traumatic SCI induced by contusion of the spinal cord has a two-phase pathology characterized by primary and secondary injuries [1]. Primary injury can result from physical compression of the spinal cord, stretching of the nervous tissue, or disruption of local blood supply. This trauma causes the spine to deform and narrows the spinal canal, leading to dramatic changes in spinal cord volume. Mechanical damage may also impact blood vessels, immediately inducing intraspinal hemorrhage or reducing blood supply. Pathologically, primary injury occurs in a short window of time and within a limited area; represents direct damage of neurons, glial, or endothelial cells due to mechanical insults; and is characterized by hemorrhage, edema, and ischemia.
Secondary injury is a compilation of complex events subsequent to the initial trauma that develops minutes to weeks after SCI. Several mechanisms underlie the pathogenesis of secondary injury, including neurodegeneration, gliosis, and inflammation. Progressive enlargement of the affected regions exacerbates dysfunction by inducing apoptosis in nearby intact neural tissues. Proliferation or hypertrophy of activated glial cells, such as microglia and astrocytes, leads to the formation of glial scars. The inflammatory microenvironment following SCI is mediated by activated microglia and astrocytes, and infiltrating macrophages greatly contribute to the progression of secondary injury [2–6]. Angiogenic responses and remodeling of vascular structure also contribute to the development of secondary injury. It is well-acknowledged that effective restraint of secondary injury plays a fundamental role in minimizing neurodegeneration and significantly improves functional recovery after SCI. As such, much effort has been devoted to developing strategies that ameliorate secondary injury and facilitate neuroregeneration, such as inhibiting inflammation, blocking endogenous axon growth inhibitors (Nogo and CSPG), and reducing glial scars.
The functional consequences of SCI are largely determined by the level and completeness of the injury. First, the effect of SCI on loss of motor and non-motor function depends on the site of the injury. Nerves controlled by spinal cord segments below the injury site often lose their connections, and thus, the body-brain communication through descending motor pathways and ascending sensory pathways is disrupted in SCI. Due to the anatomical organization of the spinal cord, more rostral injuries are associated with greater levels of functional impairment. Injuries to cervical segments of the spinal cord result in the loss of motor and/or sensory function in the upper and lower limbs and often the trunk (tetraplegia or quadriplegia), whereas injuries to thoracic, lumbar, or sacral segments generally spare upper limb function and involve the lower limbs and trunk to varying degrees (paraplegia) [7]. Second, the extent or completeness of the injury is another determinant of SCI-elicited dysfunction. At the most basic level, SCIs can be classified as either complete or incomplete. Complete SCI represents an absence of motor and sensory function in S4–S5 segments (i.e., no sacral sparing), whereas in incomplete SCI there is preservation of some motor and/or sensory function below the level of injury. The incompleteness of SCI can be further divided according to the American Spinal Injury Association Impairment Scale, which grades injuries based on the amount of function preserved in patients [7].
Multiple organ dysfunction after SCI
Beyond impairments to sensation and voluntary movement, SCI disturbs the autonomic nervous system and induces dysfunction or failure in multiple organs because of the critical role of the spinal cord in coordinating bodily functions [8]. Short- and long-term complications following SCI can occur in the nervous system (such as neurogenic pain and depression), lungs (such as pulmonary edema and respiratory failure), cardiovascular system (such as orthostatic hypotension and autonomic dysreflexia), spleen (such as splenic atrophy and leukopenia), urinary system (such as neurogenic bladder, kidney damage, and urinary tract infection), skeletal muscle (such as muscle spasticity and atrophy), bone and soft tissue (such as osteoporosis and heterotopic ossification), and skin (pressure sores) and include sexual dysfunction, hepatic pathology, neurogenic bowel dysfunction, syringomyelia, and increased susceptibility to infection. Some complications are high risk factors of mortality in SCI patients, e.g., liver, lung, and kidney damage, and therefore, therapeutic interventions that ameliorate post-SCI complications may be as important for prolonging life expectancy and improving life quality as those interventions that promote neuroregeneration and motor function recovery.
Multiple organ dysfunction after SCI is under complex regulation by multiple components. Cranial nerves emanating from brainstem areas (such as the pons and medulla) control the functions of multiple organs, and brainstem reflexes were reported to be changed in human patients with SCI [9]. This suggests a complex relationship between multiple organ dysfunction, the injured spinal cord, and altered brainstem activity. While much consideration should be given to the brainstem’s role in multiple organ dysfunction following SCI, this review focuses on the contributive roles of inflammation and immunity in these systemic complications.
Systemic inflammation following SCI
The local inflammatory microenvironment within the injured spinal cord is a collection of degenerating neurons, degraded myelin sheath, damaged endothelial cells, and activated glial and infiltrating cells, and this microenvironment produces various kinds of pro-inflammatory mediators [10]. In addition to this intraspinal inflammation, SCI can trigger systemic inflammatory response syndrome (SIRS), a life-threatening condition which can affect distal organs [11–15]. Epidemiological analyses have revealed a functional link between systemic inflammation and pathogeneses of post-injury complications: SIRS-positive SCI patients have higher injury severity and a higher incidence of complications than do SIRS-negative patients [16]. Many other factors, such as dysregulation of the neuroendocrine system and altered neuroimmune regulation, are important determinants of the onset and progression of post-SCI systemic inflammation. For instance, SCI activates the hypothalamic-pituitary-adrenal axis, leading to increased macrophage migration inhibitory factor production by the pituitary gland [17]. Macrophage migration inhibitory factor is extensively involved in systemic inflammation, suggesting that SCI-elicited neuroendocrine changes contribute to the progression of systemic inflammation. Chronic activation of microglia, the neuroimmune cells of the central nervous system, occurs in the hippocampus and cerebral cortex after SCI, indicating neuroimmune dysregulation is involved in systemic inflammation following SCI [18].
Immune suppression induced by SCI
Another consequence of post-injury spinal cord-immune system interplay is SCI-induced immune depression syndrome (SCI-IDS) which is due, at least in part, to dysregulation of the sympathetic nervous system and immune organ dysfunction [19–21]. SCI can lead to sympathetic nervous system dysfunction directly by severing thoracolumbar spinal cord projections to sympathetic ganglion or indirectly by disrupting supraspinal control through the hypothalamic-pituitary-adrenal axis. SCI-IDS, indicated by the loss of splenocytes and leukopenia, is a putative self-defense mechanism that lowers potential autoimmunity to self-antigens produced by damage in the central nervous system [22, 23]. In contrast to this protective effect, accumulating evidence has shown that SCI-IDS worsens neurological conditions and impairs the functional recovery of SCI patients. Riegger et al. reported significant decreases in the number of circulating cells involved in both innate and adaptive immunity in the acute phase following rat SCI [24]. Similar observations were made in a pilot study involving 16 SCI patients and ten healthy controls: decreased monocytes, T lymphocytes, and B lymphocytes, but not granulocytes, were observed in peripheral blood within 24 h after SCI [25]. SCI-IDS has important clinical relevance, as SCI patients display increased susceptibility to various infections (e.g., pneumonia and wound infections) [26] and poor functional recovery [27]. Autonomic dysreflexia and the expansion of myeloid-derived suppressor cells following SCI may play causal roles in SCI-IDS [28, 29], though the underlying mechanisms of SCI-IDS are still largely unknown.
Role of inflammation and immunity in post-SCI complications
Nervous system
Hyperesthesia, i.e., increased sensitivity to somatosensory stimuli, frequently occurs after SCI. Many brain regions that participate in nociception, e.g., the thalamus and nucleus accumbens, undergo neuronal changes following SCI [30]. For example, spinal cord contusion results in increased responsiveness of thalamic neurons to somatosensory stimuli [31], and this effect is partially mediated by upregulation of sodium channel Nav1.3 [32]. In addition, the inflammatory response plays an important role in chronic pain after SCI [33]. Progressively activated microglia have been observed in the thalamus not only in the acute phase (several days after SCI) but also in the chronic phase (several weeks after SCI) [34]. Chemokines CCL2 and CCL3, key players in neuropathic pain, were detected in the thalamus and hippocampus in the chronic phase after severe SCI [35, 36]. Chemokine CCL21, induced in lumbar dorsal horn neurons after SCI, may mediate remote activation of cerebral microglia. Neutralization of CCL21 suppressed microglia activation and subsequent hyperexcitability of thalamic neurons [37, 38]. Meanwhile, upregulation of proteins involved in the cell cycle (e.g., cyclin D1) has been associated with microglia activation, and thus, ablation of cell cycle signaling could significantly reduce neuroinflammation and ameliorate motor dysfunction and post-traumatic hyperesthesia [34, 39]. Decreased expression of pro-inflammatory cytokines by progesterone, a neuroactive steroid, also alleviated chronic pain after SCI [40].
Cognitive impairment is associated with extensive cerebral inflammation after SCI [41]. Mice undergoing traumatic SCI exhibited impaired learning and memory associated with elevated neuroinflammation in the hippocampus and cortex, whereas interventions that attenuated inflammatory responses facilitated cognitive function recovery [18, 42, 43]. Nonetheless, a better understanding of the methods and consequences of efficient inhibition of inflammation is necessary to yield significant improvements in human cognitive function following SCI.
Depression is another common complication in SCI patients, especially those with an early onset [44, 45]. To date, the relationship between depression and neuroinflammation after SCI remains elusive. It has been noted that the serum concentration of corticosterone, an inducer of depressive-like phenotypes in animal models, was elevated shortly after SCI and remained so for at least 1 month after experimental SCI in rats [11]. Wu et al. also reported depressive-like behaviors in mice with spinal cord contusion [18]. In a rat model of SCI, high levels of pro-inflammatory cytokines were associated with comorbidity of depression and anxiety, and there was no correlation between these comorbidities and trauma severity [46]. These results help to explain how activation of microglia and astrocytes may contribute to psychiatric complications; furthermore, they underscore the therapeutic potential of targeting these cells. Recently, a randomized clinical trial revealed the effectiveness of targeting inflammation to improve mood in SCI patients by reducing IL-1β and increasing the levels of a neuroactive compound involved in the kynurenine pathway [47]. Taken together, brain dysfunction and neurodegeneration, common complications of SCI, may be closely related to cerebral inflammation, characterized by elevated pro-inflammatory cytokines and activation of microglia and astrocytes. Although a better understanding of this relationship is needed, targeting inflammation in the brain may serve as an important therapeutic approach to improve the overall quality of life for SCI patients.
Lung
Pulmonary complications, such as respiratory failure and pulmonary infection, are quite common in SCI patients and largely contribute to morbidity and mortality in these affected individuals [48, 49]. SCI patients have an increased likelihood of developing pulmonary dysfunction, including acute respiratory distress syndrome and acute lung injury [50]. While impaired vascular and muscular functions could explain, at least in part, the development of pulmonary complications, the fact that patients with lower thoracic SCI still develop pneumonia and respiratory dysfunction suggests that the mechanisms involved in respiratory complications induced by SCI are complex [51]. Insights into lung inflammation after SCI may inspire the development and adoption of novel strategies to treat pulmonary complications.
The lungs are a major target of SCI-induced acute inflammation. Using a rat model of spinal cord compression, Gris et al. reported significant neutrophil activation and lung tissue invasion several hours after SCI, and substantial macrophage infiltration was also found in the lungs 3 days post injury [52]. Notably, the early onset of pulmonary inflammation is consistent with the development of lung dysfunction in the early stage of SCI [51]. Two cross-sectional studies involving chronic SCI patients unveiled a correlation between pulmonary inflammation and dysfunction: SCI patients with higher levels of inflammation markers IL-6 and C-reactive protein had reduced lung volume measurements [53, 54]. Consequently, pharmaceutical interventions that decrease systemic inflammation in the lungs may alleviate pulmonary dysfunction [55–57]. Resveratrol, an anti-inflammatory agent [58] which exhibits neuroprotective effects in spinal cord injuries [59–61], successfully reduced pulmonary infiltration of neutrophils and the production of pro-inflammatory mediators, suppressed NF-κB activation, and ameliorated pulmonary edema [62]. This suggests that antagonizing inflammation in the lungs promotes recovery of impaired pulmonary functions after SCI. Indeed, a small-molecule agonist of dopamine D1 receptor [63] effectively attenuated pulmonary edema and lung damage following SCI [64] via inhibition of NLRP3 inflammasome activation.
Cardiovascular system
Patients with spinal cord damage often suffer from cardiovascular disease, a leading cause of death for these individuals [65]. Following SCI, autonomic nervous system impairment results in blood pressure and heart rate dysregulation [66]. Autonomic dysreflexia, characterized by acute hypertension after afferent stimulation, is frequently seen in SCI patients, especially those with high-level injuries [67]. The development of autonomic dysreflexia in SCI mice has also been demonstrated [68]. Blockade of inflammation-related receptors, e.g., CD11d, CXCR1, and CXCR2, attenuated the development of autonomic dysreflexia after SCI [69–71], highlighting anti-inflammatory treatments as potential therapeutics against cardiovascular dysfunction post injury.
Liver
While few investigations have examined the effect of SCI on liver function, Sipski and colleagues have reported hepatic abnormalities in chronic SCI patients [72]. It is reasonable to suspect that liver dysfunction can be associated with spinal cord trauma, as the liver plays an essential role in the metabolic dysfunction commonly observed after SCI. Indeed, animal studies have revealed that traumatic injury to the spinal cord triggers neutrophil infiltration, macrophage activation, and the expression of pro-inflammatory cytokines and chemokines in the liver [73, 74]. This inflammation appears as early as 30 min after injury [75], and its severity is correlated with lesion level [76]. Of note, considerable lipid accumulation has been detected in rodent livers after SCI [77]. Given the pro-inflammatory and cytotoxic effects of myelin-laden macrophages associated with lipid accumulation in the injured spinal cord [2–4, 78], macrophage-mediated inflammation may substantially contribute to hepatic dysfunction after SCI.
Spleen
The spleen—innervated by the autonomic nervous system and controlled by the high thoracic spinal cord—is an important lymphoid organ and source of infiltrating monocytes in the injured spinal cord. In a mouse SCI model, significant dysfunction of the spleen was observed following high thoracic SCI (T3 section), whereas lower thoracic SCI (T9 section) preserved the majority of splenic function. Spleen dysfunction after T3 SCI was indicated by splenic atrophy with reduced spleen size, decreased splenic leukocyte numbers, and increased splenic norepinephrine [21, 28]. SCI mice challenged with viral infection show impaired protective immune responses and decreased survival, and these outcomes were associated with deficient CD4+ and CD8+ T cell functions, suppressed activation of macrophages, and deficient primary antibody response [79–81]. This suggests that splenic dysfunction may largely contribute to immune suppression in SCI patients. Notably, post-SCI mRNA levels of pro-inflammatory cytokines IL-17 and IL-23 were upregulated in rat splenic tissue through STAT3 signaling [82], and thus, crosstalk between the peripheral spleen and injured spinal cord may be mediated by neuroinflammation.
Gastrointestinal tract
Gastrointestinal dysfunction, e.g., severe constipation, difficulty with evacuation, painful defecation or incontinence, is a common complication following SCI and is quite restrictive for patients, limiting their diet and outdoor activity [83]. Although the function of the gastrointestinal tract is primarily determined by its own intrinsic nervous system and autonomic control from the brainstem, spinal cord trauma may damage the neuronal control of gastrointestinal sensory and motor functions, resulting in neurogenic bowel dysfunction (NBD). As approximately half of SCI patients suffer from moderate to severe NBD [84], it seems that abnormal bowel function exerts a highly negative impact on life quality for SCI patients [85, 86]. The clinical manifestations of NBD with SCI include decreased colonic motility, prolonged bowel transit time, and anorectal dysfunction [87]. Although inflammation has been observed in colonic lesions of SCI patients [88], the relationship between systemic inflammation and NBD largely remains unclear. Utilizing a rat model of NBD after SCI, Guo et al. reported that upregulation of neuronal nitric oxide synthase contributed to colonic dysfunction [89], thus establishing inflammation as a potential target for alleviating post-SCI NBD.
Urinary system
SCI may impair supraspinal control of the bladder and result in neurogenic bladder, characterized by dysfunction in bladder storage and emptying [90–92]. Accordingly, SCI patients have an increased risk of developing urinary tract infections and renal damage, both of which can be life-threatening [93–96]. Besides the direct loss of neuronal input after injury, inflammation has been implicated in the pathogenesis of urinary system dysfunction in SCI patients. Such inflammation may include infiltration of immune cells, production of pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α), upregulation of myeloperoxidase, inducible nitric oxide synthase and cyclooxygenase-2, and activation of NF-κB [52, 94, 97–100]. Many potential therapeutic approaches have been investigated to alleviate bladder dysfunction and kidney damage. Blockade of pro-inflammatory integrin signaling by antibodies targeting CD11d or CD49d has been shown to reduce kidney inflammation induced by SCI [55, 57]. Application of antioxidant vitamin C and activation of adrenoreceptors protected rat kidneys from SCI-induced damage by suppressing NF-κB signaling and pro-inflammatory cytokine expression [94, 97]. Oral administration of anti-inflammatory small-molecule S-nitrosoglutathione can promote recovery of neurogenic bladder via inhibition of inflammatory responses [98]. Oxidants such as dantrolene and quercetin are able to ameliorate urinary bladder lesions after SCI by decreasing bladder hemorrhage and immune cell infiltration [101, 102]. The aforementioned results are a promising sign for the treatment of SCI-induced urinary system dysfunction.
Skeletal muscle
Skeletal muscles controlled by the spinal cord below the injured area become paralyzed and develop atrophy after SCI [103, 104]. Physiological tests have revealed numerous changes in the properties of disabled muscles from SCI patients, including decreased muscle cross-sectional area, reduced muscle mass, increased susceptibility to fatigue, and an increased proportion of fast glycolytic muscular fibers [105–107]. While it is well-known that paralysis-induced disuse is the primary cause of muscular atrophy, emerging evidence suggests that the involvement of inflammation in post-SCI muscle dysfunction may not be trivial. Muscular inflammation can be observed in the acute phase of SCI even before obvious muscle atrophy is seen [108]. In long-term SCI, muscle atrophy is associated with a significant elevation of inflammatory mediators (e.g., IL-1β, IL-6, and TNF-α) and activation of NF-κB signaling [109, 110], a key regulator of the inflammatory state in muscle atrophy [111]. In a mouse transection SCI model, administration of glutamine—which is extensively used to improve clinical outcomes—has been shown to decrease pro-inflammatory cytokine expressions (IL-6 and TNF-α), attenuate loss of myofibrillar proteins in muscle, and mitigate muscular fatigability [112]. Therefore, administration of anti-inflammatory agents may serve as a promising therapeutic approach to accelerate muscular function recovery after SCI.
Bone
Osteoporosis, characterized by the loss of bone mineral density (BMD), is a well-defined consequence of SCI [113, 114]. The distal femur, proximal tibia, and distal boney sites at sublesional levels are the most susceptible to BMD loss [115, 116]. The decrease in BMD is progressive after SCI and increases patients’ risk of fracture [117], and many factors contribute to the pathogenesis of osteoporosis following SCI. In addition to deficiencies in neuronal control, hormonal regulation, and vascular function [118–120], the inflammatory microenvironment in bone also mediates osteoclast differentiation and bone loss [121–123]. Elevated levels of IL-6 have been detected in conditioned medium from bone marrow cultures of SCI patients [124]. Similarly, significant increases in serum IL-6 (approximately 2.5-fold compared to sham) and IL-6 mRNA (approximately sixfold compared to sham) were observed in the femurs of rats post-SCI [125]. IL-6 signaling stimulates osteoclast progenitors to differentiate into osteoclasts [126–129] and suppresses osteoblast differentiation [130, 131], resulting in bone resorption. Blockade of IL-6 by its neutralizing antibody inhibited osteoclast-like cell formation in SCI patient-derived bone marrow cultures [124]. In addition, resveratrol significantly suppressed IL-6 expression in femurs and reduced osteoclastogenesis in SCI rats [125]. Hence, inflammatory mediators are active players in osteoporosis after SCI and may be important anti-osteoporotic targets.
Soft tissue
Neurogenic heterotopic ossification (HO), a process by which new bone formation occurs outside of the skeleton and preferentially around soft tissues, is an irreversible complication of SCI more frequently seen in young patients [132–135]. Cervical and thoracic SCIs induce more HO than do lumbar injuries, and the hip joint is the major ossified area. Regulation of HO development is multifactorial [136–138], and the inflammatory response is an important contributing factor in the early stage of HO. Among the multiple therapeutic choices, non-steroidal anti-inflammatory drugs are effective prophylactic treatments against HO when administered soon after SCI [134, 139–143]. Estrores and colleagues reported that increased levels of C-reactive protein, a commonly used marker of acute inflammation in SCI [144–146], were associated with early occurrence of HO and the concentration of C-reactive protein declined when HO symptoms were alleviated in later stages [147]. The lack of animal models that reproduce the clinical features of HO observed in SCI patients has long hindered the study of underlying HO mechanisms; however, in 2015 a well-characterized mouse HO model showed that phagocytic macrophages play a critical role in driving the development of HO [148]. This study highlighted the impact of the inflammatory microenvironment on HO and may benefit the discovery of novel anti-inflammatory treatments against HO.
Syringomyelia
Syringomyelia is a relatively rare sequela defined by cavity formation in the injured spinal cord via enlargement of the central canal [149]. Syringomyelia induces devastating symptoms including muscle weakness, loss of sensitivity, stiffness, and pain. In SCI rats, induced inflammatory conditions exacerbated syrinx formation, indicating a potential role of inflammation in the pathogenesis of syringomyelia [150].
Conclusions
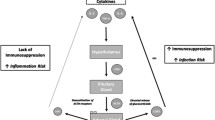
Post-SCI multiple organ dysfunction is influenced by multifactorial mechanisms, and the extent to which systemic inflammation and immune depression contribute to SCI-associated complications is still an open question. Nevertheless, a growing body of evidence demonstrates the involvement of inflammatory conditions in the damage or dysfunction of multiple organ systems secondary to SCI. Systemic inflammatory responses following SCI induce infiltration of inflammatory cells into secondary tissues, activation of resident immune cells, and stimulation of pro-inflammatory cytokine production, all of which contribute to the pathogenesis of multiple organ dysfunction after SCI. Meanwhile, immune suppression subsequent to SCI significantly increases susceptibility to post-injury infection due to impaired innate and adaptive immunity in SCI patients, leading to worsened multiple organ damage and mortality. Therefore, inflammation and immunity not only contribute to the progression of intraspinal injury but also are important determinants of multiple organ dysfunction after SCI (Fig. 1).

Schematic diagram of systemic inflammation- and immune depression-associated multiple organ dysfunction following SCI. SCI triggers an acute increase of inflammatory cells (such as neutrophils and macrophages) in the circulation and elevates serum concentrations of pro-inflammatory mediators. Subsequent infiltration of inflammatory cells from the blood into secondary organs initiates a series of events that mediate inflammatory responses in these organs. Activation of resident immune cells (microglia) in the brain is also found after SCI. SCI itself interrupts innervation of immune organs by the sympathetic nervous system, causing immune depression syndrome. Suppressed immunity leads to an increased susceptibility of the whole body to post-injury pathogen infections through decreased immune cell quantities (such as monocytes, T cells, and B cells)
Many anti-inflammatory strategies that attempt to ameliorate local intraspinal inflammation and promote neural tissue repair may find their true value in alleviating dysfunction in multiple organs secondary to the injured spinal cord. Such therapeutics may include immunomodulators of inflammation-associated pathways, e.g., estrogen, IL-33, IL-37, and adiponectin signaling pathways [151–154]. Small-molecule agonists or antagonists and blocking antibodies that specifically recognize and deactivate a variety of receptors involved in transduction of inflammatory signals—e.g., interleukin receptors, toll-like receptors, integrins, and estrogen receptors—are promising tools to mitigate complications after SCI [55–57, 92, 155–158]. Intracellular components of inflammatory machinery, including enzymes and transcription factors, may also serve as therapeutic targets to resolve inflammation in multiple organs following SCI [159–164].
Co-application of anti-inflammatory strategies with other treatment approaches after SCI may provide a therapeutic benefit for patients, though there is a lack of human clinical trials employing such strategies. In the future, well-designed experimental studies utilizing reliable animal models are needed to better understand the detailed mechanisms of how post-SCI complications develop with systemic inflammation and suppressed immunity and to suggest effective immunoregulatory approaches to mitigate SCI-induced multiple organ dysfunction. Such studies should be taken into consideration with the ultimate goal of developing therapies to improve the total body health of SCI patients.
Abbreviations
- BMD:
-
Bone mineral density
- HO:
-
Heterotopic ossification
- NBD:
-
Neurogenic bowel dysfunction
- SCI:
-
Spinal cord injury
- SCI-IDS:
-
SCI-induced immune depression syndrome
- SIRS:
-
Systemic inflammatory response syndrome
References
Oyinbo CA. Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol Exp (Wars). 2011;71:281–99.
Zhou X, He X, Ren Y. Function of microglia and macrophages in secondary damage after spinal cord injury. Neural Regen Res. 2014;9:1787–95.
Wang X, Cao K, Sun X, Chen Y, Duan Z, Sun L, Guo L, Bai P, Sun D, Fan J, et al. Macrophages in spinal cord injury: phenotypic and functional change from exposure to myelin debris. Glia. 2015;63:635–51.
Guo L, Rolfe AJ, Wang X, Tai W, Cheng Z, Cao K, Chen X, Xu Y, Sun D, Li J, et al. Rescuing macrophage normal function in spinal cord injury with embryonic stem cell conditioned media. Mol Brain. 2016;9:48.
Ren Y, Young W. Managing inflammation after spinal cord injury through manipulation of macrophage function. Neural Plast. 2013;2013:945034.
Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41:369–78.
Kirshblum SC, Burns SP, Biering-Sorensen F, Donovan W, Graves DE, Jha A, Johansen M, Jones L, Krassioukov A, Mulcahey MJ, et al. International standards for neurological classification of spinal cord injury (revised 2011). J Spinal Cord Med. 2011;34:535–46.
Stein DM, Menaker J, McQuillan K, Handley C, Aarabi B, Scalea TM. Risk factors for organ dysfunction and failure in patients with acute traumatic cervical spinal cord injury. Neurocrit Care. 2010;13:29–39.
Kumru H, Kofler M. Effect of spinal cord injury and of intrathecal baclofen on brainstem reflexes. Clin Neurophysiol. 2012;123:45–53.
Hasturk A, Atalay B, Calisaneller T, Ozdemir O, Oruckaptan H, Altinors N. Analysis of serum pro-inflammatory cytokine levels after rat spinal cord ischemia/reperfusion injury and correlation with tissue damage. Turk Neurosurg. 2009;19:353–9.
Popovich PG, Stuckman S, Gienapp IE, Whitacre CC. Alterations in immune cell phenotype and function after experimental spinal cord injury. J Neurotrauma. 2001;18:957–66.
Anthony DC, Couch Y. The systemic response to CNS injury. Exp Neurol. 2014;258:105–11.
Bigford GE, Bracchi-Ricard VC, Keane RW, Nash MS, Bethea JR. Neuroendocrine and cardiac metabolic dysfunction and NLRP3 inflammasome activation in adipose tissue and pancreas following chronic spinal cord injury in the mouse. ASN Neuro. 2013;5:243–55.
Bigford GE, Bracchi-Ricard VC, Nash MS, Bethea JR. Alterations in mouse hypothalamic adipokine gene expression and leptin signaling following chronic spinal cord injury and with advanced age. PLoS One. 2012;7:e41073.
Bao F, Bailey CS, Gurr KR, Bailey SI, Rosas-Arellano MP, Dekaban GA, Weaver LC. Increased oxidative activity in human blood neutrophils and monocytes after spinal cord injury. Exp Neurol. 2009;215:308–16.
Kesani AK, Urquhart JC, Bedard N, Leelapattana P, Siddiqi F, Gurr KR, Bailey CS. Systemic inflammatory response syndrome in patients with spinal cord injury: does its presence at admission affect patient outcomes? Clinical article. J Neurosurg Spine. 2014;21:296–302.
Lerch JK, Puga DA, Bloom O, Popovich PG. Glucocorticoids and macrophage migration inhibitory factor (MIF) are neuroendocrine modulators of inflammation and neuropathic pain after spinal cord injury. Semin Immunol. 2014;26:409–14.
Wu J, Zhao Z, Sabirzhanov B, Stoica BA, Kumar A, Luo T, Skovira J, Faden AI. Spinal cord injury causes brain inflammation associated with cognitive and affective changes: role of cell cycle pathways. J Neurosci. 2014;34:10989–1006.
Kopp MA, Druschel C, Meisel C, Liebscher T, Prilipp E, Watzlawick R, Cinelli P, Niedeggen A, Schaser KD, Wanner GA, et al. The SCIentinel study—prospective multicenter study to define the spinal cord injury-induced immune depression syndrome (SCI-IDS)—study protocol and interim feasibility data. BMC Neurol. 2013;13:168.
Furlan JC, Krassioukov AV, Fehlings MG. Hematologic abnormalities within the first week after acute isolated traumatic cervical spinal cord injury: a case-control cohort study. Spine (Phila Pa 1976). 2006;31:2674–83.
Lucin KM, Sanders VM, Jones TB, Malarkey WB, Popovich PG. Impaired antibody synthesis after spinal cord injury is level dependent and is due to sympathetic nervous system dysregulation. Exp Neurol. 2007;207:75–84.
Schwab JM, Zhang Y, Kopp MA, Brommer B, Popovich PG. The paradox of chronic neuroinflammation, systemic immune suppression, autoimmunity after traumatic chronic spinal cord injury. Exp Neurol. 2014;258:121–9.
Popovich P, McTigue D. Damage control in the nervous system: beware the immune system in spinal cord injury. Nat Med. 2009;15:736–7.
Riegger T, Conrad S, Liu K, Schluesener HJ, Adibzahdeh M, Schwab JM. Spinal cord injury-induced immune depression syndrome (SCI-IDS). Eur J Neurosci. 2007;25:1743–7.
Riegger T, Conrad S, Schluesener HJ, Kaps HP, Badke A, Baron C, Gerstein J, Dietz K, Abdizahdeh M, Schwab JM. Immune depression syndrome following human spinal cord injury (SCI): a pilot study. Neuroscience. 2009;158:1194–9.
Brommer B, Engel O, Kopp MA, Watzlawick R, Muller S, Pruss H, Chen Y, DeVivo MJ, Finkenstaedt FW, Dirnagl U, et al. Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level. Brain. 2016;139:692–707.
Failli V, Kopp MA, Gericke C, Martus P, Klingbeil S, Brommer B, Laginha I, Chen Y, DeVivo MJ, Dirnagl U, Schwab JM. Functional neurological recovery after spinal cord injury is impaired in patients with infections. Brain. 2012;135:3238–50.
Zhang Y, Guan Z, Reader B, Shawler T, Mandrekar-Colucci S, Huang K, Weil Z, Bratasz A, Wells J, Powell ND, et al. Autonomic dysreflexia causes chronic immune suppression after spinal cord injury. J Neurosci. 2013;33:12970–81.
Wang L, Yu WB, Tao LY, Xu Q. Myeloid-derived suppressor cells mediate immune suppression in spinal cord injury. J Neuroimmunol. 2016;290:96–102.
Gustin SM, Wrigley PJ, Siddall PJ, Henderson LA. Brain anatomy changes associated with persistent neuropathic pain following spinal cord injury. Cereb Cortex. 2010;20:1409–19.
Hubscher CH, Johnson RD. Chronic spinal cord injury induced changes in the responses of thalamic neurons. Exp Neurol. 2006;197:177–88.
Hains BC, Saab CY, Waxman SG. Changes in electrophysiological properties and sodium channel Nav1.3 expression in thalamic neurons after spinal cord injury. Brain. 2005;128:2359–71.
Sandhir R, Gregory E, He YY, Berman NE. Upregulation of inflammatory mediators in a model of chronic pain after spinal cord injury. Neurochem Res. 2011;36:856–62.
Wu J, Raver C, Piao C, Keller A, Faden AI. Cell cycle activation contributes to increased neuronal activity in the posterior thalamic nucleus and associated chronic hyperesthesia after rat spinal cord contusion. Neurotherapeutics. 2013;10:520–38.
Knerlich-Lukoschus F, Noack M, von der Ropp-Brenner B, Lucius R, Mehdorn HM, Held-Feindt J. Spinal cord injuries induce changes in CB1 cannabinoid receptor and C-C chemokine expression in brain areas underlying circuitry of chronic pain conditions. J Neurotrauma. 2011;28:619–34.
Knerlich-Lukoschus F, Juraschek M, Blomer U, Lucius R, Mehdorn HM, Held-Feindt J. Force-dependent development of neuropathic central pain and time-related CCL2/CCR2 expression after graded spinal cord contusion injuries of the rat. J Neurotrauma. 2008;25:427–48.
Zhao P, Waxman SG, Hains BC. Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci. 2007;27:8893–902.
Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol. 2008;212:337–47.
Wu J, Sabirzhanov B, Stoica BA, Lipinski MM, Zhao Z, Zhao S, Ward N, Yang D, Faden AI. Ablation of the transcription factors E2F1-2 limits neuroinflammation and associated neurological deficits after contusive spinal cord injury. Cell Cycle. 2015;14:3698–712.
Coronel MF, Raggio MC, Adler NS, De Nicola AF, Labombarda F, Gonzalez SL. Progesterone modulates pro-inflammatory cytokine expression profile after spinal cord injury: implications for neuropathic pain. J Neuroimmunol. 2016;292:85–92.
Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol. 2016;173:681–91.
Wu J, Stoica BA, Luo T, Sabirzhanov B, Zhao Z, Guanciale K, Nayar SK, Foss CA, Pomper MG, Faden AI. Isolated spinal cord contusion in rats induces chronic brain neuroinflammation, neurodegeneration, and cognitive impairment. Involvement of cell cycle activation. Cell Cycle. 2014;13:2446–58.
Allison DJ, Josse AR, Gabriel DA, Klentrou P, Ditor DS. Targeting inflammation to influence cognitive function following spinal cord injury: a randomized clinical trial. Spinal Cord. 2016. http://www.nature.com/sc/journal/vaop/ncurrent/full/sc201696a.html.
Dryden DM, Saunders LD, Rowe BH, May LA, Yiannakoulias N, Svenson LW, Schopflocher DP, Voaklander DC. Depression following traumatic spinal cord injury. Neuroepidemiology. 2005;25:55–61.
Tate DG, Forchheimer MB, Karana-Zebari D, Chiodo AE, Kendall Thomas JY. Depression and pain among inpatients with spinal cord injury and spinal cord disease: differences in symptoms and neurological function. Disabil Rehabil. 2013;35:1204–12.
Maldonado-Bouchard S, Peters K, Woller SA, Madahian B, Faghihi U, Patel S, Bake S, Hook MA. Inflammation is increased with anxiety- and depression-like signs in a rat model of spinal cord injury. Brain Behav Immun. 2016;51:176–95.
Allison DJ, Ditor DS. Targeting inflammation to influence mood following spinal cord injury: a randomized clinical trial. J Neuroinflammation. 2015;12:204.
Cotton BA, Pryor JP, Chinwalla I, Wiebe DJ, Reilly PM, Schwab CW. Respiratory complications and mortality risk associated with thoracic spine injury. J Trauma. 2005;59:1400–7. discussion 1407–1409.
Tollefsen E, Fondenes O. Respiratory complications associated with spinal cord injury. Tidsskr Nor Laegeforen. 2012;132:1111–4.
Veeravagu A, Jiang B, Rincon F, Maltenfort M, Jallo J, Ratliff JK. Acute respiratory distress syndrome and acute lung injury in patients with vertebral column fracture(s) and spinal cord injury: a nationwide inpatient sample study. Spinal Cord. 2013;51:461–5.
Yong T, Lili Y, Wen Y, Xinwei W, Xuhui Z. Pulmonary edema and hemorrhage, possible causes of pulmonary infection and respiratory failure in the early stage of lower spinal cord injury. Med Hypotheses. 2012;79:299–301.
Gris D, Hamilton EF, Weaver LC. The systemic inflammatory response after spinal cord injury damages lungs and kidneys. Exp Neurol. 2008;211:259–70.
Garshick E, Stolzmann KL, Gagnon DR, Morse LR, Brown R. Systemic inflammation and reduced pulmonary function in chronic spinal cord injury. PM R. 2011;3:433–9.
Hart JE, Morse L, Tun CG, Brown R, Garshick E. Cross-sectional associations of pulmonary function with systemic inflammation and oxidative stress in individuals with chronic spinal cord injury. J Spinal Cord Med. 2016;39:344–52.
Bao F, Omana V, Brown A, Weaver LC. The systemic inflammatory response after spinal cord injury in the rat is decreased by alpha4beta1 integrin blockade. J Neurotrauma. 2012;29:1626–37.
Weaver LC, Bao F, Dekaban GA, Hryciw T, Shultz SR, Cain DP, Brown A. CD11d integrin blockade reduces the systemic inflammatory response syndrome after traumatic brain injury in rats. Exp Neurol. 2015;271:409–22.
Bao F, Brown A, Dekaban GA, Omana V, Weaver LC. CD11d integrin blockade reduces the systemic inflammatory response syndrome after spinal cord injury. Exp Neurol. 2011;231:272–83.
Das S, Das DK. Anti-inflammatory responses of resveratrol. Inflamm Allergy Drug Targets. 2007;6:168–73.
Kaplan S, Bisleri G, Morgan JA, Cheema FH, Oz MC. Resveratrol, a natural red wine polyphenol, reduces ischemia-reperfusion-induced spinal cord injury. Ann Thorac Surg. 2005;80:2242–9.
Kiziltepe U, Turan NN, Han U, Ulus AT, Akar F. Resveratrol, a red wine polyphenol, protects spinal cord from ischemia-reperfusion injury. J Vasc Surg. 2004;40:138–45.
Yang YB, Piao YJ. Effects of resveratrol on secondary damages after acute spinal cord injury in rats. Acta Pharmacol Sin. 2003;24:703–10.
Liu J, Yi L, Xiang Z, Zhong J, Zhang H, Sun T. Resveratrol attenuates spinal cord injury-induced inflammatory damage in rat lungs. Int J Clin Exp Pathol. 2015;8:1237–46.
DeNinno MP, Schoenleber R, MacKenzie R, Britton DR, Asin KE, Briggs C, Trugman JM, Ackerman M, Artman L, Bednarz L, et al. A68930: a potent agonist selective for the dopamine D1 receptor. Eur J Pharmacol. 1991;199:209–19.
Jiang W, Li M, He F, Bian Z, Liu J, He Q, Wang X, Sun T, Zhu L. Dopamine D1 receptor agonist A-68930 inhibits NLRP3 inflammasome activation and protects rats from spinal cord injury-induced acute lung injury. Spinal Cord. 2016. http://www.nature.com/sc/journal/vaop/ncurrent/full/sc201652a.html.
Squair JW, West CR, Krassioukov AV. Neuroprotection, plasticity manipulation, and regenerative strategies to improve cardiovascular function following spinal cord injury. J Neurotrauma. 2015;32:609–21.
Phillips AA, Krassioukov AV. Contemporary cardiovascular concerns after spinal cord injury: mechanisms, maladaptations, and management. J Neurotrauma. 2015;32:1927–42.
Weaver LC, Marsh DR, Gris D, Brown A, Dekaban GA. Autonomic dysreflexia after spinal cord injury: central mechanisms and strategies for prevention. Prog Brain Res. 2006;152:245–63.
Jacob JE, Pniak A, Weaver LC, Brown A. Autonomic dysreflexia in a mouse model of spinal cord injury. Neuroscience. 2001;108:687–93.
Marsh DR, Flemming JM. Inhibition of CXCR1 and CXCR2 chemokine receptors attenuates acute inflammation, preserves gray matter and diminishes autonomic dysreflexia after spinal cord injury. Spinal Cord. 2011;49:337–44.
Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, Weaver LC. Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neurosci. 2004;24:4043–51.
Gris D, Marsh DR, Dekaban GA, Weaver LC. Comparison of effects of methylprednisolone and anti-CD11d antibody treatments on autonomic dysreflexia after spinal cord injury. Exp Neurol. 2005;194:541–9.
Sipski ML, Estores IM, Alexander CJ, Guo X, Chandralapaty SK. Lack of justification for routine abdominal ultrasonography in patients with chronic spinal cord injury. J Rehabil Res Dev. 2004;41:101–8.
Campbell SJ, Zahid I, Losey P, Law S, Jiang Y, Bilgen M, van Rooijen N, Morsali D, Davis AE, Anthony DC. Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology. 2008;55:780–7.
Campbell SJ, Perry VH, Pitossi FJ, Butchart AG, Chertoff M, Waters S, Dempster R, Anthony DC. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am J Pathol. 2005;166:1487–97.
Hundt H, Fleming JC, Phillips JT, Lawendy A, Gurr KR, Bailey SI, Sanders D, Bihari R, Gray D, Parry N, et al. Assessment of hepatic inflammation after spinal cord injury using intravital microscopy. Injury. 2011;42:691–6.
Fleming JC, Bailey CS, Hundt H, Gurr KR, Bailey SI, Cepinskas G, Lawendy AR, Badhwar A. Remote inflammatory response in liver is dependent on the segmental level of spinal cord injury. J Trauma Acute Care Surg. 2012;72:1194–201. discussion 1202.
Sauerbeck AD, Laws JL, Bandaru VV, Popovich PG, Haughey NJ, McTigue DM. Spinal cord injury causes chronic liver pathology in rats. J Neurotrauma. 2015;32:159–69.
Sun X, Wang X, Chen T, Li T, Cao K, Lu A, Chen Y, Sun D, Luo J, Fan J, et al. Myelin activates FAK/Akt/NF-kappaB pathways and provokes CR3-dependent inflammatory response in murine system. PLoS One. 2010;5:e9380.
Oropallo MA, Held KS, Goenka R, Ahmad SA, O'Neill PJ, Steward O, Lane TE, Cancro MP. Chronic spinal cord injury impairs primary antibody responses but spares existing humoral immunity in mice. J Immunol. 2012;188:5257–66.
Held KS, Steward O, Blanc C, Lane TE. Impaired immune responses following spinal cord injury lead to reduced ability to control viral infection. Exp Neurol. 2010;226:242–53.
Zha J, Smith A, Andreansky S, Bracchi-Ricard V, Bethea JR. Chronic thoracic spinal cord injury impairs CD8+ T-cell function by up-regulating programmed cell death-1 expression. J Neuroinflammation. 2014;11:65.
Zong S, Zeng G, Fang Y, Peng J, Tao Y, Li K, Zhao J. The role of IL-17 promotes spinal cord neuroinflammation via activation of the transcription factor STAT3 after spinal cord injury in the rat. Mediators Inflamm. 2014;2014:786947.
Han TR, Kim JH, Kwon BS. Chronic gastrointestinal problems and bowel dysfunction in patients with spinal cord injury. Spinal Cord. 1998;36:485–90.
Liu CW, Huang CC, Yang YH, Chen SC, Weng MC, Huang MH. Relationship between neurogenic bowel dysfunction and health-related quality of life in persons with spinal cord injury. J Rehabil Med. 2009;41:35–40.
Correa GI, Rotter KP. Clinical evaluation and management of neurogenic bowel after spinal cord injury. Spinal Cord. 2000;38:301–8.
Stiens SA, Bergman SB, Goetz LL. Neurogenic bowel dysfunction after spinal cord injury: clinical evaluation and rehabilitative management. Arch Phys Med Rehabil. 1997;78:S86–102.
Fajardo NR, Pasiliao RV, Modeste-Duncan R, Creasey G, Bauman WA, Korsten MA. Decreased colonic motility in persons with chronic spinal cord injury. Am J Gastroenterol. 2003;98:128–34.
Han SJ, Kim CM, Lee JE, Lee TH. Colonoscopic lesions in patients with spinal cord injury. J Spinal Cord Med. 2009;32:404–7.
Guo J, Zhu Y, Yang Y, Wang X, Chen B, Zhang W, Xie B, Zhu Z, Yue Y, Cheng J: Electroacupuncture at Zusanli (ST36) ameliorates colonic neuronal nitric oxide synthase upregulation in rats with neurogenic bowel dysfunction following spinal cord injury. Spinal Cord. 2016. http://www.nature.com/sc/journal/vaop/ncurrent/full/sc201676a.html.
Horst M, Heutschi J, van den Brand R, Andersson KE, Gobet R, Sulser T, Courtine G, Eberli D. Multisystem neuroprosthetic training improves bladder function after severe spinal cord injury. J Urol. 2013;189:747–53.
Cameron AP, Rodriguez GM, Schomer KG. Systematic review of urological followup after spinal cord injury. J Urol. 2012;187:391–7.
David BT, Sampath S, Dong W, Heiman A, Rella CE, Elkabes S, Heary RF. A toll-like receptor 9 antagonist improves bladder function and white matter sparing in spinal cord injury. J Neurotrauma. 2014;31:1800–6.
Balsara ZR, Ross SS, Dolber PC, Wiener JS, Tang Y, Seed PC. Enhanced susceptibility to urinary tract infection in the spinal cord-injured host with neurogenic bladder. Infect Immun. 2013;81:3018–26.
Wang WG, Xiu RJ, Xu ZW, Yin YX, Feng Y, Cao XC, Wang PS. Protective effects of vitamin C against spinal cord injury-induced renal damage through suppression of NF-kappaB and proinflammatory cytokines. Neurol Sci. 2015;36:521–6.
Shunmugavel A, Khan M, Te Chou PC, Dhindsa RK, Martin MM, Copay AG, Subach BR, Schuler TC, Bilgen M, Orak JK, Singh I. Simvastatin protects bladder and renal functions following spinal cord injury in rats. J Inflamm (Lond). 2010;7:17.
Chaudhry R, Madden-Fuentes RJ, Ortiz TK, Balsara Z, Tang Y, Nseyo U, Wiener JS, Ross SS, Seed PC. Inflammatory response to Escherichia coli urinary tract infection in the neurogenic bladder of the spinal cord injured host. J Urol. 2014;191:1454–61.
Shou-Shi W, Ting-Ting S, Ji-Shun N, Hai-Chen C. Preclinical efficacy of dexmedetomidine on spinal cord injury provoked oxidative renal damage. Ren Fail. 2015;37:1190–7.
Shunmugavel A, Khan M, Hughes Jr FM, Purves JT, Singh A, Singh I. S-Nitrosoglutathione protects the spinal bladder: novel therapeutic approach to post-spinal cord injury bladder remodeling. Neurourol Urodyn. 2015;34:519–26.
Wognum S, Lagoa CE, Nagatomi J, Sacks MS, Vodovotz Y. An exploratory pathways analysis of temporal changes induced by spinal cord injury in the rat bladder wall: insights on remodeling and inflammation. PLoS One. 2009;4:e5852.
Ersahin M, Cevik O, Akakin D, Sener A, Ozbay L, Yegen BC, Sener G. Montelukast inhibits caspase-3 activity and ameliorates oxidative damage in the spinal cord and urinary bladder of rats with spinal cord injury. Prostaglandins Other Lipid Mediat. 2012;99:131–9.
Torres B, Serakides R, Caldeira F, Gomes M, Melo E. The ameliorating effect of dantrolene on the morphology of urinary bladder in spinal cord injured rats. Pathol Res Pract. 2011;207:775–9.
Cevik O, Ersahin M, Sener TE, Tinay I, Tarcan T, Cetinel S, Sener A, Toklu HZ, Sener G. Beneficial effects of quercetin on rat urinary bladder after spinal cord injury. J Surg Res. 2013;183:695–703.
Biering-Sorensen B, Kristensen IB, Kjaer M, Biering-Sorensen F. Muscle after spinal cord injury. Muscle Nerve. 2009;40:499–519.
Qin W, Bauman WA, Cardozo C. Bone and muscle loss after spinal cord injury: organ interactions. Ann N Y Acad Sci. 2010;1211:66–84.
Castro MJ, Apple Jr DF, Hillegass EA, Dudley GA. Influence of complete spinal cord injury on skeletal muscle cross-sectional area within the first 6 months of injury. Eur J Appl Physiol Occup Physiol. 1999;80:373–8.
Castro MJ, Apple Jr DF, Rogers S, Dudley GA. Influence of complete spinal cord injury on skeletal muscle mechanics within the first 6 months of injury. Eur J Appl Physiol. 2000;81:128–31.
Wu Y, Zhao J, Zhao W, Pan J, Bauman WA, Cardozo CP. Nandrolone normalizes determinants of muscle mass and fiber type after spinal cord injury. J Neurotrauma. 2012;29:1663–75.
Thakore NP, Samantaray S, Park S, Nozaki K, Smith JA, Cox A, Krause J, Banik NL. Molecular changes in sub-lesional muscle following acute phase of spinal cord injury. Neurochem Res. 2016;41:44–52.
Yarar-Fisher C, Bickel CS, Kelly NA, Stec MJ, Windham ST, McLain AB, Oster RA, Bamman MM. Heightened TWEAK-NF-kappaB signaling and inflammation-associated fibrosis in paralyzed muscles of men with chronic spinal cord injury. Am J Physiol Endocrinol Metab. 2016;310:E754–61.
Graham ZA, Collier L, Peng Y, Saez JC, Bauman WA, Qin W, Cardozo CP. A soluble activin receptor IIB fails to prevent muscle atrophy in a mouse model of spinal cord injury. J Neurotrauma. 2016;33:1128–35.
Jackman RW, Cornwell EW, Wu CL, Kandarian SC. Nuclear factor-kappaB signalling and transcriptional regulation in skeletal muscle atrophy. Exp Physiol. 2013;98:19–24.
Chamney C, Godar M, Garrigan E, Huey KA. Effects of glutamine supplementation on muscle function and stress responses in a mouse model of spinal cord injury. Exp Physiol. 2013;98:796–806.
Qin W, Sun L, Cao J, Peng Y, Collier L, Wu Y, Creasey G, Li J, Qin Y, Jarvis J, et al. The central nervous system (CNS)-independent anti-bone-resorptive activity of muscle contraction and the underlying molecular and cellular signatures. J Biol Chem. 2013;288:13511–21.
Dudley-Javoroski S, Shields RK. Muscle and bone plasticity after spinal cord injury: review of adaptations to disuse and to electrical muscle stimulation. J Rehabil Res Dev. 2008;45:283–96.
Maimoun L, Couret I, Mariano-Goulart D, Dupuy AM, Micallef JP, Peruchon E, Ohanna F, Cristol JP, Rossi M, Leroux JL. Changes in osteoprotegerin/RANKL system, bone mineral density, and bone biochemicals markers in patients with recent spinal cord injury. Calcif Tissue Int. 2005;76:404–11.
Coupaud S, McLean AN, Purcell M, Fraser MH, Allan DB. Decreases in bone mineral density at cortical and trabecular sites in the tibia and femur during the first year of spinal cord injury. Bone. 2015;74:69–75.
Bauman WA, Cardozo CP. Osteoporosis in individuals with spinal cord injury. PM R. 2015;7:188–201. quiz 201.
Jiang SD, Jiang LS, Dai LY. Mechanisms of osteoporosis in spinal cord injury. Clin Endocrinol (Oxf). 2006;65:555–65.
Tan CO, Battaglino RA, Morse LR. Spinal cord injury and osteoporosis: causes, mechanisms, and rehabilitation strategies. Int J Phys Med Rehabil. 2013;1:127.
Alexandre C, Vico L. Pathophysiology of bone loss in disuse osteoporosis. Joint Bone Spine. 2011;78:572–6.
Matzelle MM, Gallant MA, Condon KW, Walsh NC, Manning CA, Stein GS, Lian JB, Burr DB, Gravallese EM. Resolution of inflammation induces osteoblast function and regulates the Wnt signaling pathway. Arthritis Rheum. 2012;64:1540–50.
Baum R, Gravallese EM. Impact of inflammation on the osteoblast in rheumatic diseases. Curr Osteoporos Rep. 2014;12:9–16.
Romas E, Gillespie MT. Inflammation-induced bone loss: can it be prevented? Rheum Dis Clin North Am. 2006;32:759–73.
Demulder A, Guns M, Ismail A, Wilmet E, Fondu P, Bergmann P. Increased osteoclast-like cells formation in long-term bone marrow cultures from patients with a spinal cord injury. Calcif Tissue Int. 1998;63:396–400.
Wang HD, Shi YM, Li L, Guo JD, Zhang YP, Hou SX. Treatment with resveratrol attenuates sublesional bone loss in spinal cord-injured rats. Br J Pharmacol. 2013;170:796–806.
Kurihara N, Bertolini D, Suda T, Akiyama Y, Roodman GD. IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J Immunol. 1990;144:4226–30.
Axmann R, Bohm C, Kronke G, Zwerina J, Smolen J, Schett G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009;60:2747–56.
Tamura T, Udagawa N, Takahashi N, Miyaura C, Tanaka S, Yamada Y, Koishihara Y, Ohsugi Y, Kumaki K, Taga T, et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc Natl Acad Sci U S A. 1993;90:11924–8.
Udagawa N, Takahashi N, Katagiri T, Tamura T, Wada S, Findlay DM, Martin TJ, Hirota H, Taga T, Kishimoto T, Suda T. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J Exp Med. 1995;182:1461–8.
Peruzzi B, Cappariello A, Del Fattore A, Rucci N, De Benedetti F, Teti A. c-Src and IL-6 inhibit osteoblast differentiation and integrate IGFBP5 signalling. Nat Commun. 2012;3:630.
Kaneshiro S, Ebina K, Shi K, Higuchi C, Hirao M, Okamoto M, Koizumi K, Morimoto T, Yoshikawa H, Hashimoto J. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J Bone Miner Metab. 2014;32:378–92.
Sautter-Bihl ML, Hultenschmidt B, Liebermeister E, Nanassy A. Fractionated and single-dose radiotherapy for heterotopic bone formation in patients with spinal cord injury. A phase-I/II study. Strahlenther Onkol. 2001;177:200–5.
Riklin C, Baumberger M, Wick L, Michel D, Sauter B, Knecht H. Deep vein thrombosis and heterotopic ossification in spinal cord injury: a 3 year experience at the Swiss Paraplegic Centre Nottwil. Spinal Cord. 2003;41:192–8.
Sullivan MP, Torres SJ, Mehta S, Ahn J. Heterotopic ossification after central nervous system trauma: a current review. Bone Joint Res. 2013;2:51–7.
Edwards DS, Clasper JC. Heterotopic ossification: a systematic review. J R Army Med Corps. 2015;161:315–21.
Zychowicz ME. Pathophysiology of heterotopic ossification. Orthop Nurs. 2013;32:173–7. quiz 178–179.
Balboni TA, Gobezie R, Mamon HJ. Heterotopic ossification: pathophysiology, clinical features, and the role of radiotherapy for prophylaxis. Int J Radiat Oncol Biol Phys. 2006;65:1289–99.
Sakellariou VI, Grigoriou E, Mavrogenis AF, Soucacos PN, Papagelopoulos PJ. Heterotopic ossification following traumatic brain injury and spinal cord injury: insight into the etiology and pathophysiology. J Musculoskelet Neuronal Interact. 2012;12:230–40.
Ranganathan K, Loder S, Agarwal S, Wong VW, Forsberg J, Davis TA, Wang S, James AW, Levi B. Heterotopic ossification: basic-science principles and clinical correlates. J Bone Joint Surg Am. 2015;97:1101–11.
Banovac K, Sherman AL, Estores IM, Banovac F. Prevention and treatment of heterotopic ossification after spinal cord injury. J Spinal Cord Med. 2004;27:376–82.
Teasell RW, Mehta S, Aubut JL, Ashe MC, Sequeira K, Macaluso S, Tu L. A systematic review of the therapeutic interventions for heterotopic ossification after spinal cord injury. Spinal Cord. 2010;48:512–21.
Aubut JA, Mehta S, Cullen N, Teasell RW. A comparison of heterotopic ossification treatment within the traumatic brain and spinal cord injured population: an evidence based systematic review. NeuroRehabilitation. 2011;28:151–60.
Schuetz P, Mueller B, Christ-Crain M, Dick W, Haas H. Amino-bisphosphonates in heterotopic ossification: first experience in five consecutive cases. Spinal Cord. 2005;43:604–10.
Liang H, Mojtahedi MC, Chen D, Braunschweig CL. Elevated C-reactive protein associated with decreased high-density lipoprotein cholesterol in men with spinal cord injury. Arch Phys Med Rehabil. 2008;89:36–41.
Morse LR, Stolzmann K, Nguyen HP, Jain NB, Zayac C, Gagnon DR, Tun CG, Garshick E. Association between mobility mode and C-reactive protein levels in men with chronic spinal cord injury. Arch Phys Med Rehabil. 2008;89:726–31.
Gibson AE, Buchholz AC, Martin Ginis KA. C-Reactive protein in adults with chronic spinal cord injury: increased chronic inflammation in tetraplegia vs paraplegia. Spinal Cord. 2008;46:616–21.
Estrores IM, Harrington A, Banovac K. C-reactive protein and erythrocyte sedimentation rate in patients with heterotopic ossification after spinal cord injury. J Spinal Cord Med. 2004;27:434–7.
Genet F, Kulina I, Vaquette C, Torossian F, Millard S, Pettit AR, Sims NA, Anginot A, Guerton B, Winkler IG, et al. Neurological heterotopic ossification following spinal cord injury is triggered by macrophage-mediated inflammation in muscle. J Pathol. 2015;236:229–40.
Jaksche H, Schaan M, Schulz J, Bosczcyk B. Posttraumatic syringomyelia—a serious complication in tetra- and paraplegic patients. Acta Neurochir Suppl. 2005;93:165–7.
Seki T, Fehlings MG. Mechanistic insights into posttraumatic syringomyelia based on a novel in vivo animal model. Laboratory investigation J Neurosurg Spine. 2008;8:365–75.
Pomeshchik Y, Kidin I, Korhonen P, Savchenko E, Jaronen M, Lehtonen S, Wojciechowski S, Kanninen K, Koistinaho J, Malm T. Interleukin-33 treatment reduces secondary injury and improves functional recovery after contusion spinal cord injury. Brain Behav Immun. 2015;44:68–81.
Dinarello CA, Nold-Petry C, Nold M, Fujita M, Li S, Kim S, Bufler P. Suppression of innate inflammation and immunity by interleukin-37. Eur J Immunol. 2016;46:1067–81.
Yau SY, Li A, Hoo RL, Ching YP, Christie BR, Lee TM, Xu A, So KF. Physical exercise-induced hippocampal neurogenesis and antidepressant effects are mediated by the adipocyte hormone adiponectin. Proc Natl Acad Sci U S A. 2014;111:15810–5.
Amini Pishva A, Akbari M, Farahabadi A, Arabkheradmand A, Beyer C, Dashti N, Moradi F, Hassanzadeh G. Effect of estrogen therapy on TNF-alpha and iNOS gene expression in spinal cord injury model. Acta Med Iran. 2016;54:296–301.
David BT, Ratnayake A, Amarante MA, Reddy NP, Dong W, Sampath S, Heary RF, Elkabes S. A toll-like receptor 9 antagonist reduces pain hypersensitivity and the inflammatory response in spinal cord injury. Neurobiol Dis. 2013;54:194–205.
Dicpinigaitis PV, Spungen AM, Bauman WA, Absgarten A, Almenoff PL. Inhibition of bronchial hyperresponsiveness by the GABA-agonist baclofen. Chest. 1994;106:758–61.
Chakrabarti M, Haque A, Banik NL, Nagarkatti P, Nagarkatti M, Ray SK. Estrogen receptor agonists for attenuation of neuroinflammation and neurodegeneration. Brain Res Bull. 2014;109:22–31.
Murakami T, Kanchiku T, Suzuki H, Imajo Y, Yoshida Y, Nomura H, Cui D, Ishikawa T, Ikeda E, Taguchi T. Anti-interleukin-6 receptor antibody reduces neuropathic pain following spinal cord injury in mice. Exp Ther Med. 2013;6:1194–8.
Dulin JN, Karoly ED, Wang Y, Strobel HW, Grill RJ. Licofelone modulates neuroinflammation and attenuates mechanical hypersensitivity in the chronic phase of spinal cord injury. J Neurosci. 2013;33:652–64.
Park SW, Yi JH, Miranpuri G, Satriotomo I, Bowen K, Resnick DK, Vemuganti R. Thiazolidinedione class of peroxisome proliferator-activated receptor gamma agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J Pharmacol Exp Ther. 2007;320:1002–12.
Pearse D, Jarnagin K. Abating progressive tissue injury and preserving function after CNS trauma: the role of inflammation modulatory therapies. Curr Opin Investig Drugs. 2010;11:1207–10.
Qu WS, Tian DS, Guo ZB, Fang J, Zhang Q, Yu ZY, Xie MJ, Zhang HQ, Lu JG, Wang W. Inhibition of EGFR/MAPK signaling reduces microglial inflammatory response and the associated secondary damage in rats after spinal cord injury. J Neuroinflammation. 2012;9:178.
Rafati DS, Geissler K, Johnson K, Unabia G, Hulsebosch C, Nesic-Taylor O, Perez-Polo JR. Nuclear factor-kappaB decoy amelioration of spinal cord injury-induced inflammation and behavior outcomes. J Neurosci Res. 2008;86:566–80.
Esposito E, Rinaldi B, Mazzon E, Donniacuo M, Impellizzeri D, Paterniti I, Capuano A, Bramanti P, Cuzzocrea S. Anti-inflammatory effect of simvastatin in an experimental model of spinal cord trauma: involvement of PPAR-alpha. J Neuroinflammation. 2012;9:81.
Acknowledgements
We thank Yijie Chi, Hongkai Xiang, and Qishuang Zhou for literature search.
Funding
This work was supported by the Guangdong Natural Science Foundation (2016A030313105), the Fundamental Research Funds for the Central Universities to Jinan University (21616339), the National Natural Science Foundation of China (31571408), the Program of Introducing Talents of Discipline to Universities (B14036), and the National Institutes of Health (R01GM100474-4).
Availability of data and materials
The data supporting the conclusions of this article are included within the “References” section.
Authors’ contributions
XS, ZBJ, and YR wrote the paper. XMC, LZ, and KFS participated in the discussion of the manuscript. All the authors reviewed and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sun, X., Jones, Z.B., Chen, Xm. et al. Multiple organ dysfunction and systemic inflammation after spinal cord injury: a complex relationship. J Neuroinflammation 13, 260 (2016). https://doi.org/10.1186/s12974-016-0736-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-016-0736-y