
Abstract
Chimeric antigen receptors (CARs) are engineered to target T cells specifically to tumor cells, resulting in the engineered T cell killing the tumor cell. This technology has been developed to target a range of cancers, with the most notable successes in the treatment of B-cell malignancies where four approved therapies, all targeting CD19, are on the market. These four products differ in the costimulation domains, with axicabtagene ciloleucel (Yescarta) and brexucabtagene autoleucel (Tecartus) both utilizing the CD28 costimulation domain whilst tisagenlecleucel (Kymriah) and lisocabtagene maraleucel (Breyanzi) both utilizing the 4-1BB costimulation domain. There are clearly defined differences in how the CD28 and 4-1BB domains signal, yet it is difficult to ascertain which domain affords a superior mechanism of action given many other differences between these products, including overall CAR architecture and manufacturing methods. Additionally, while in vitro and preclinical in vivo studies have compared CARs with different costimulation domains, it remains a challenge to extrapolate differences observed in this biology across different experimental systems to the overall product performance. While there has been extensive preclinical and clinical work looking at CARs with a variety of targeting domains and architectures, this review will focus on the differences between the four marketed anti-CD19 CAR-Ts, with an additional focus on the impact of hinge and transmembrane domain on CAR activity and interaction with the target cell as well as other proteins on the surface of the T-cell.
Similar content being viewed by others

Background
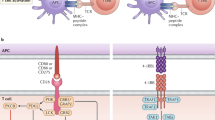
Chimeric antigen receptors (CARs) have shown exceptional promise in the clinic in the treatment of hematological malignancies, introducing engineered receptors into a patient’s own T cells to target cancer cells. CARs are designed with an extracellular domain that binds to a tumor cell-surface antigen and cytosolic domains that drive T-cell activation, with the goal of trying to recapitulate the process of T cell activation in response to a foreign antigen, while redirecting the T cell to killing a tumor cell. An effective T-cell response to a target antigen requires two signals [1]. The first comes from engagement of the T-cell receptor (TCR) complex with the MHC-peptide complex on the antigen presenting cell. This signal provides recognition to a specific antigen. However, this signal alone will induce T-cell anergy, a critical mechanism for establishing tolerance to self-antigens [2]. Professional antigen presenting cells are able to provide a second signal via engagement with costimulatory receptors such as CD28 [3] and 4-1BB [4] on the surface of the T-cell, thus initiating a response to the antigen being presented by the MHC (Fig. 1a). CD28 and 4-1BB do initiate different signaling pathways (Fig. 1b), though with significant overlap with the downstream signals from the TCR complex. The overall outcomes (IL-2 production, T cell proliferation and anti-apoptotic signals) are broadly similar [5].

a The involvement of native CD28 and 4-1BB costimulation in T cell activation. T cell activation requires two signals. The first is provided by recognition of a specific peptide antigen held by MHC class II on the surface of an antigen presenting cell (APC) by the T cell receptor (TCR) complex. Professional APCs also provide a second signal through engagement of costimulatory molecules on the surface of the T cell; this second signal is essential for survival and expansion of the T cell. This figure highlights two costimulators; CD28, which is recognized by APC surface proteins CD80 and CD86 and 4-1BB, which is recognized by APC surface protein 4-1BBL. Created with BioRender.com. b A simplified representation of the integration of TCR signaling with costimulatory signals. The primary contact between the T cell and the APC is via the TCR/CD3 complex on the T cell and the MHC/peptide complex on the APC, initiating ‘signal 1’. CD28 and 4-1BB signaling (‘signal 2’) is then triggered by engagement with their ligands on the surface of the APC. This triggers dimerization of CD28, or trimerization of 4-1BB, resulting in recruitment of a variety of signaling molecules on the cytosolic side of the T cell membrane. It is important to note that the downstream signals from the TCR/CD3 complex overlap with those from CD28 and 4-1BB, notably via the PKCθ and AKT pathways. This in turn ultimately leads to transcription and translation of genes required to drive cytokine production (particularly IL2 and IFNγ), T cell proliferation and anti-apoptotic signals. Created with BioRender.com
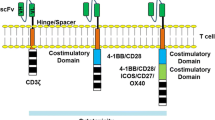
First generation CARs consisted of extracellular domains including a tumor-antigen specific single chain antibody fragment (scFv) linked to the CD3ζ signaling domain [6, 7]. While first-generation CARs induced an antigen-specific signal, anti-tumor activity in preclinical models was limited. Inclusion of a costimulation domain in second generation CARs provided the extra signal required to fully activate the T cell and drive an anti-tumor response [8], and clearly demonstrated superiority over first generation CARs in rodent models [9,10,11,12] (Fig. 2). Third generation CARs attempted to further enhance the costimulatory signal by stacking two or more costimulatory domains into a single CAR [13]. There was nonetheless mixed success with third generation CARs (reviewed in [14]), possibly underscored by the complex spatial requirements for effective engagement of the tandem cytosolic costimulatory domains with multiple effectors [15]. Several different costimulatory domains, including OX40, CD2 and CD27 have been studied [8], however the most common domains used in the field are CD28 and 4-1BB. The four marketed anti-CD19 CARs use either CD28 or 4-1BB costimulatory domains in a second-generation format.

First, second and third generation CARs. First generation CARs consist of an antigen-binding scFv fused via a hinge to the transmembrane and activation domain from CD3ζ. Second generation CARs use a variety of hinges and transmembrane domains, plus a single costimulatory domain in addition to the antigen-binding scFv and CD3ζ activation domain. This enables them to recapitulate the two signals required for full T-cell activation. Third generation CARs build on the basic structure of the second generation CAR by adding a second costimulation domain. Created with BioRender.com
Comparing the four approved anti-CD19 CAR-T products
The architecture and manufacturing processes used for the four approved anti-CD19 CAR-T therapies are summarized in Table 1. All four CARs utilize the same binder, an scFv derived from a murine anti-human CD19 antibody known as FMC63 [16], and the same activation domain, CD3ζ [13]. The rest of the architecture is significantly different. Axi-cel [17,18,19,20] and brexu-cel [21,22,23] use the same CAR, which has hinge, transmembrane and costimulation domains derived from CD28. Tisa-cel [24,25,26] uses hinge and transmembrane domains derived from CD8α, and the 4-1BB costimulation domain. Liso-cel [27, 28] uses a hinge derived from IgG4, the CD28 transmembrane domain and the 4-1BB costimulation domain.
The native CD28 and 4-1BB receptors differ in the downstream signaling cascades, predominantly at the level of distinct protein binding partners recruited to the cytosolic domains following receptor-ligand engagement (Fig. 1b). However, analyses of activation and phosphorylation events following stimulation reveal few, if any, converged signaling differences [29]. Even an unbiased mass spectroscopic phosphoproteomic analysis looking at over 26,000 phosphorylation events across 4849 proteins revealed that a majority of phosphorylation events were shared between both costimulation domains (when the rest of the CAR architecture was held constant) [5]. The key difference was instead in the magnitude and kinetics of the signals, with the CD28-based anti-CD19 CAR inducing a significantly faster signal with greater amplitude. The CD28 CAR also exhibited a low level of basal CD28 phosphorylation that correlated with increased levels of Lck associated with the CAR, which was not seen in the 4-1BB-based anti-CD19 CAR. While this did correlate with potent cytotoxicity in vitro, the CD28-based CARs use in this study were not as effective at controlling a tumor in a rodent model as their 4-1BB-based counterpart, with the former cells becoming more rapidly exhausted and failing to persist. The authors do note the limitations of their analyses, with the in vitro activation being performed with antigen coated beads, thus excluding the contribution of other costimulators that would be found on tumor cells. Also, they only looked at one architecture (FMC63 anti-CD19 scFv plus IgG4 hinge plus CD28 transmembrane domain). A recent study indicated differences in T-cell dysfunction arising from chronic in vitro stimulation via a CAR with a CD28 costimulation domain, which exhibited a classical T-cell exhaustion profile, versus a CAR with a 4-1BB costimulation domain which exhibited a novel FOXO3-mediated dysfunction profile [30]. The authors showed a similar transcriptional profile in Tisa-cel CAR-T cells taken from a single patient who failed to respond. Further studies are needed to validate this observation in additional patients, and to understand if this novel profile is seen in other CAR-T cells that utilize a 4-1BB costimulation domain.
Functional impact of the hinge and transmembrane domains within CD19-directed CARs
While there has been much focus on the impact of CD28 versus 4-1BB costimulation domains on CAR activity, the choice of hinge and transmembrane domain can have a profound impact [31]. This is evident from the range of results seen in preclinical rodent studies that have directly compared different costimulatory domains in anti-CD19 CARs, with some studies indicating CD28 costimulation yields a better response [32,33,34,35,36], while others indicate a better response when a 4-1BB costimulation domain is used [12, 37, 38]. It is important to note that it is impossible to draw clear conclusions from these studies as to which costimulatory domain in general is better (CD28 or 4-1BB) given the additional differences (hinge and transmembrane domains) in architectures used, as well as the different model systems in which they were tested. If we start to look at the various components of CAR architecture at a molecular level, we can begin to determine their relative contributions, and how they interact with each other, in defining the properties of a given CAR.
The approved CD19 CARs cover the range of widely used hinges, utilizing immunoglobin (Ig) family derived domains. Ig domains are found in over 750 extracellular proteins, not just immunoglobulins (or antibodies), and consist of around 125 amino acids forming an approximately globular structure. They are frequently found in membrane proteins where they form stable modular components of the extracellular domain. These favorable properties have supported their use in CAR hinge domains. These domains provide both structure and function. Ig domains from IgG1 antibodies mediate engagement with Fc receptors on myeloid cells, a property that has negated their use as CAR hinges [39, 40]. Other classes of antibody, such as IgG4, do not bind to cell surface receptors and have been used successfully as CAR hinges as in the case of liso-cel [40]. Another alternative is to use immunoglobulin-like domains from non-antibody proteins such as CD28 (axi-cel and brexu-cel) or CD8α (tisa-cel). Initially these sequences were considered to be largely inert, beyond providing appropriate presentation of the scFv to its cognate antigen on the target cell. However, there is an increasing body of evidence pointing towards the choice of hinge and transmembrane domain greatly impacting CAR activity [41,42,43,44].
A comparison of CD28 and CD8α-derived hinge and transmembrane domains with an anti-VEGFR2 scFv was performed by Fujiwara et al., showing that first-generation CARs with CD28-derived sequences had greater cytotoxic and cytokine-producing activity than equivalent CARs with CD8α derived hinge and transmembrane sequences in mouse CAR-T cells [41]. They then tested second-generation CARs using CD28 costimulation domains, and either CD28- or CD8α-derived hinge and transmembrane domains in human CAR-T cells. In this case the CARs with CD28-derived hinge and transmembrane domain had higher cytotoxicity activity, when controlling for CAR expression [41]. These data suggest that axi-cel may mediate pronounced cytotoxicity relative to liso-cel and tisa-cel, which do not harbor a CD28 hinge.
What could be the mechanism driving this difference? Further work from the same group has indicated that differences in glycosylation and the propensity for intermolecular disulfide bridging can influence antigen-dependent and independent activity [45]. This should not be surprising as native CD28 and 4-1BB receptors require multimerization (induced by engagement with their ligands) to be activated. In fact, first generation CARs require inclusion of CD3ζ transmembrane elements for activity, presumably enabling CAR dimerization and incorporation of the CAR into the immune synapse [46]. Dimerization of endogenous CD28 following engagement with its ligands, CD80 and CD86, results in recruitment of signaling molecules such as PI3 kinase, Lck and GRB2/GRAP2 to the cytosolic costimulatory domain, driving the second signal required for T cell activation [3] (Fig. 1b). The costimulatory domain in a CAR generates an equivalent signal when the CAR engages with an antigen on a tumor cell. It has been demonstrated that CD28 hinge/ transmembrane domain CARs can heterodimerize with native CD28 on the T cell surface [47]. Mueller et al. compared CARs consisting of FMC63 scFv and 4-1BB costimulatory domain (chosen to avoid any potential CD28 CAR cytoplasmic domain-driven association with native CD28) with combinations of IgG4, CD28 and CD8α hinges and CD28 and CD8α transmembrane domains. These CARs were introduced into T cells by CRISPR gene editing to better control equivalent levels of expression. Formation of CAR-CD28 heterodimers reduced the amount of free CD28 monomers on the T cell surface. Additionally, the heterodimers were unresponsive to the native CD28 ligands CD80 and CD86 (which is reassuring as this was viewed as a potential route to CD19-independent CAR activation). The recruitment of native CD28 to the CAR, and thus the immune synapse, may explain the stronger signal and increased sensitivity to lower antigen levels when using the CD28 hinge and transmembrane domains. A similar pairing, but to a lesser degree was seen with the CD8α hinge and transmembrane domain, while it was completely absent with the IgG4 hinge. Three-dimensional modeling of the structures of the various ECDs does suggest a structural basis for preference of CD28 hinge and transmembrane domain > CD8α hinge and transmembrane domain > IgG4 hinge, with potential stabilization of the native CD28 - CAR hinge-TM domain heterodimer mediated through access to an unpaired cysteine in the extracellular domain of native CD28 by an equivalent residue in either the CD28 hinge and transmembrane domain or the CD8α hinge and transmembrane domain. The IgG4 hinge lacks a suitable free cysteine in the interface region. In contrast the modeling shows how the three different CAR hinges can homodimerize following engagement of their associated binding domains with their cognate targets.
Majzner et al. have investigated how these differences manifest at a mechanistic level [48]. Utilizing confocal microscopy, they showed that FMC63-derived CARs had similar distribution on the T cell surface, irrespective of the hinge used, but that CD28 hinge/ transmembrane domains induced faster synapse formation than CARs incorporating a CD8α hinge/ transmembrane domain. This was assessed by measuring recruitment of a ZAP70-red fluorescent protein fusion to the intracellular side of the synapse. This is not to say that choice of costimulation domain does not impact activity; in this study it was shown that T cell activation kinetics (with FMC63 as a binder with CD28 hinge and transmembrane domains) were faster with the CD28 costimulatory domain than 4-1BB. Additionally, CD28 hinge and transmembrane domains conferred greater sensitivity to lower antigen levels than the CD8α hinge and transmembrane domains, suggesting higher potency in conditions where target expression may be lower. This has clinical impact that when targeting a population of tumor cells with a range of target antigen expression even cells expressing low levels of antigen will be eliminated [49]. It is notable that using either the 4-1BB or CD28 costimulatory domain in the context of the CD28 hinge and transmembrane domain resulted in CAR-Ts that had equivalent anti-tumor activity in an in vivo model of a CD19-low expressing leukemia. For CD19, where on target/ off tumor cytotoxicity results in the clinically manageable outcome of B cell aplasia, this could provide a significant advantage in driving a complete response. However, many solid tumor targeting concepts rely on differential expression of antigen between healthy tissue and tumor for establishing a therapeutic window [50,51,52].
Expression of the CAR on the cell surface is dependent on the CAR protein folding into its correct three-dimensional shape as it is produced inside the cell and then being transported to the cell surface. While the choice of hinge and transmembrane domain can influence how efficiently the CAR folds, the choice of scFv can have a more profound impact on CAR folding. Not all antibodies can be successfully converted into an scFv, resulting in mis-alignment between the heavy and light chain variable regions that can reveal hydrophobic ‘sticky patches’ that can cause the CAR to aggregate within the cell and not reach the surface [53,54,55,56]. In instances when a sub-optimally folded CAR gets to the cell surface this can result in ‘tonic signaling’ or signaling independent of antigen engagement as a result of CAR aggregation [57,58,59]. As all approved anti-CD19 CAR-Ts use the same scFv this can be excluded as a variable in impacting CAR expression or tonic signaling.
Clinical data comparison of 41BB vs. CD28 CARs
As already noted with preclinical studies, it is difficult to infer patterns driven solely by choice of costimulatory domain across clinical studies for a number of reasons [60]. We have described how the rest of the CAR architecture has a strong influence on overall activity, even if the same scFv (as is the case with the four marketed anti-CD19 CAR-T therapies) is used. The method of cell manufacturing (summarized in Table 1) [61], including choice of vector, pre-transduction processing of apheresed material, activation method and length of culture can have a profound impact on the final product in terms of phenotype and overall potency. Patient baseline characteristics are critical, from the type and burden of disease, prior treatments, use of bridging chemotherapy, the lymphodepletion regimen used [17, 24, 62,63,64] as well as CAR T dose. Finally, there is a lack of consistency in how durable responses are assessed, including follow-up which may be complicated by the patient receiving consolidative allogenic hematopoietic stem cell transplantation. In addition, a range of different scales for grading adverse events are used [65,66,67]. A detailed summary reviewing clinical data across multiple trials for the four marketed anti-CD19 CAR-T therapies has recently been published by Cappell and Kochenderfer [60]. In general overall complete response (CR) rates are comparable; indeed a real-world retrospective analysis of B cell non-Hodgkins lymphoma patients who received either axi-cel or tisa-cel reported similar CR rates with both products [68].
Despite the challenges in comparing both responses and adverse events, Davey et al. [69] performed an analysis of a number of trials looking at different anti-CD19 CARs [17, 24, 62, 63, 70,71,72,73,74,75,76,77,78,79,80,81,82,83,84]. In this analysis of published trial data, they calculated CR rate as the number of patients who achieved CR at any point post-infusion expressed as a percentage of the total patients in the trial. Overall, FMC63-CD28-CD28-CD28-3ζ CARs have a slightly increased CR rate relative to FMC63-CD8α-CD8α-41BB-3ζ CARs in the studies included in this analysis (60% versus 50%). Interestingly a single trial with a FMC63-CD28-CD28-41BB-3ζ CAR had a very high CR rate (80%), but this only covered 10 patients [73]. In terms of CRS, there is a higher incidence at grades 1 and 2 with CARs using the CD28 hinge and transmembrane domains (73% versus 39% for CARs using other architectures), though there is insufficient data to determine if this differs based on the costimulatory domain used, given that only 10 patients in a single trial received a FMC63-CD28-CD28-41BB-3ζ CAR, versus 501 patients with published data who received FMC63-CD28-CD28-CD28-3ζ CAR. Interestingly the rate of CRS at grades 3 and higher is similar across all architectures (approximately 13%). A similar pattern can be seen for neurotoxicity, though as the authors note there are significant differences in the methods used for defining neurotoxicity events, particularly at low grade [85]. That being said, there is a clear trend in this analysis towards severe neurotoxicity events in patients treated with CARs utilizing the CD28 hinge and transmembrane domains. In addition, onset of CRS is more rapid with CARs containing a CD28 costimulatory domain (1–3 days) [17, 63] versus CARs containing 4-1BB (3–5 days) [62, 64]. It should be noted however that incidence and severity of both CRS and neurotoxicity can also be influenced by disease burden, strength and method of lymphodepletion and CAR-T dose (reviewed in [86]).
Cellular kinetics of axi-cel and tisa-cel are well characterized in B-ALL and LBCL and generally exhibit rapid expansion within 2 weeks post-infusion followed by multiexponential decline. For tisa-cel and liso-cel, peak expansion occurs at 12 days, and persists in the peripheral blood for up to 2 years [25, 64]. In comparison, axi-cel expands more robustly upfront to a higher peak at 7 days in patients with DLBCL [17]. While peak levels of tisa-cel are considerably lower than axi-cel, dosing is up to 3-fold higher [62, 87]. Across products, cell expansion strongly correlates with objective response that is further impacted by tumor burden [64, 87, 88]. Persistence is considered a contributor to overall efficacy of CAR-T products. Multiple reports of persisting CAR-Ts with 4-1BB costimulatory domains in patients with ongoing CRs up to 2 years post-infusion are reported [24, 62, 64, 72, 76, 89], while many early trials reporting limited persistence of CAR-Ts with CD28 costimulatory domains [18, 19, 71, 83]. Consistently, axi-cel does not persist as readily as liso-cel and tisacel, and while some axi-cel patients maintain detectable low levels of CAR, persistence in the periphery is not required for durable response for axi-cel. Of course, these differences can also be influenced by the same range of variables beyond the costimulation domain previously discussed. In particular, axi-cel and brexu-cel are delivered by a gamma retrovirus that can only infect proliferating cells, while tisa-cel and liso-cel are delivered by lentivirus, which can infect non-proliferating cells that may be in a less differentiated state and have a greater potential to survive. That being said, in one trial 51% of patients treated with axi-cel showed complete remissions lasting over three years, with remissions of over 9 years still ongoing [90]. This is also borne out by data from the ZUMA-1 trial [91] and is comparable with the outcome of the ELIANA trial of tisa-cel with an overall survival rate of 55% (2022 EHA Congress, Abstracts #S112, #3782).
While the majority of trials test a single CAR there have been efforts to make direct clinical comparisons, albeit on a small scale, between CARs utilizing either CD28 or 4-1BB costimulatory domains. Cheng et al. infused seven patients with a 1:1 mixture of CAR-Ts produced with either FMC63-CD8α-CD28-CD28-3ζ or FMC63-CD8α-CD8α-41BB-3ζ CARs [33]. It is important to note that not only do the costimulatory domains differ, but so do the transmembrane regions, though the CD28 heterodimerization motif is absent as both CARs utilize a CD8α hinge. The authors transduced PBMCs with gamma retroviral vectors encoding either CAR separately, then combined the resulting products in a 1:1 ratio. They showed that the FMC63-CD8α-CD28-CD28-3ζ and mixed products produced more interferon gamma from the CD8 population and were more potent than the FMC63-CD8α-CD8α-41BB-3ζ product in a Raji mouse model. Following infusion with manufactured, mixed cells, five of seven patients achieved CR within one month, with one patient remaining in CR for 18 months, and a second patient in ongoing CR at 15 months at time of publication. In vivo expansion kinetics were monitored by qPCR and indicated that the FMC63-CD8α-CD28-CD28-3ζ cells peaked first around day 9, with the FMC63-CD8α-CD8α-41BB-3ζ cells peaking around day 13. These differences in kinetics are comparable to those seen with other 28-3ζ and 4-1BB-3ζ CARs in the clinic [17, 62,63,64]. There was no overall difference in expansion and persistence profiles for one CAR-T over the other, though this was a small sample size.
A more direct comparison was attempted by Li et al. [35]. In this study two CARs with IgG4 hinge, CD28 transmembrane and CD3ζ activation domains and either a CD28 costimulatory domain or 4-1BB costimulatory domain were compared in five patients each. While there was a higher peak of CAR-T cells in the 4-1BB group this was not statistically significant. In 9/10 patients CAR-T cells persisted for less than a month, probably contributing to all the patients, including the 7 complete responders, relapsing. The authors consider that their manufacturing process, based on OKT3 and IL2 activation, created a highly differentiated product that limited the potential for long term persistence. As such it is not possible to draw any meaningful comparison between the relative contributions of each costimulatory domain from this study.
Zhao et al. professed to comparing axi-cel and tisa-cel in both a preclinical and a clinical study [92] but this needs to be looked at with a degree of skepticism as the manufacturing method for the “axi-cel” material was based on lentiviral transduction of enriched T-cells, rather than gamma retroviral transduction of PBMCs. However, removing the variability in manufacturing method does allow a more direct comparison of the constructs on a similar background; it is important in a case like this to fully understand the question being asked, as in this case this is not a true comparison of the commercial products, rather it is focused on the CARs themselves. In this instance it was found that the tisa-cel-like CAR, containing the 4-1BB costimulatory domain was more potent and persistent in a mouse Nalm6 B-ALL model and in a small clinical trial with 36 r/r B-ALL patients (18 patients for each treatment). The authors note that their observations differ from earlier studies, notably that by Li et al. [35], but acknowledge that differences in architecture and manufacturing continue to make it difficult to compare across studies.
Conclusions
CAR-T cell activity is dependent on a wide range of factors. While there are clear biological differences between CD28 and 4-1BB costimulation, these quickly become difficult to parse out from other variables such as the rest of the CAR architecture (including binder, hinge, and transmembrane domain) which can influence how the CAR interacts with not only the target antigen, but also other proteins on the T cell surface. Manufacturing methods, including starting material, vector, activation, and culture conditions can greatly influence the phenotype of the final product, which can impact potency and persistence. Finally, the differences in clinical trial design, including patient population, prior treatment, lymphodepletion methods, dose, and criteria for assessing response and adverse events, make it impossible to draw all but the broadest comparisons across different CARs to determine which costimulatory domain is more effective. In the future, the development of high throughput screening platforms coupled with predictive, biologically relevant assays will facilitate identifying optimal CAR architectures through a holistic consideration of the entire molecule.
Availability of data and materials
Not applicable.
Abbreviations
- APC:
-
Antigen presenting cell
- CAR:
-
Chimeric antigen receptor
- TCR:
-
T-cell receptor
- MHC:
-
Major histocompatibility complex
- ScFv:
-
Single chain antibody variable region fragment
- Ig:
-
Immunoglobulin
- CRS:
-
Cytokine release syndrome
- CR:
-
Complete response
- PBMC:
-
Peripheral blood mononuclear cell
- Axi-cel:
-
Axicabtagene ciloleucel
- Brexu-cel:
-
Brexucabtagene autoleucel
- Tisa-cel:
-
Tisagenlecleucel
- Liso-cel:
-
Lisocabtagene maraleucel
References
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–42.
Schwartz RH, Mueller DL, Jenkins MK, Quill H. T-cell clonal anergy. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 2):605–10.
Boomer JS, Green JM. An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol. 2010;2(8):a002436.
Pollok KE, Kim YJ, Zhou Z, Hurtado J, Kim KK, Pickard RT, et al. Inducible T cell antigen 4-1BB. Analysis of expression and function. J Immunol. 1993;150(3):771–81.
Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. 2018;11:544.
Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–4.
Hwu P, Shafer GE, Treisman J, Schindler DG, Gross G, Cowherd R, et al. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J Exp Med. 1993;178(1):361–6.
van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509.
Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13(18 Pt 1):5426–35.
Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8 + T cell-mediated tumor eradication. Mol Ther. 2010;18(2):413–20.
Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106(9):3360–5.
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–64.
Guedan S, Calderon H, Posey AD Jr, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev. 2019;12:145–56.
Stoiber S, Cadilha BL, Benmebarek MR, Lesch S, Endres S, Kobold S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. 2019. https://doi.org/10.3390/cells8050472.
Zuccolotto G, Penna A, Fracasso G, Carpanese D, Montagner IM, Dalla Santa S, et al. PSMA-Specific CAR-Engineered T cells for prostate Cancer: CD28 outperforms combined CD28-4-1BB “Super-Stimulation. Front Oncol. 2021;11:708073.
Nicholson IC, Lenton KA, Little DJ, Decorso T, Lee FT, Scott AM, et al. Construction and characterisation of a functional CD19 specific single chain fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol Immunol. 1997;34(16–17):1157–65.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44.
Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–102.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20.
Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32(7):689–702.
Anderson MK, Torosyan A, Halford Z. Brexucabtagene Autoleucel: a novel chimeric Antigen receptor T-cell therapy for the treatment of Mantle cell lymphoma. Ann Pharmacother. 2022;56(5):609–19.
Frey NV. Approval of brexucabtagene autoleucel for adults with relapsed and refractory acute lymphocytic leukemia. Blood. 2022;140(1):11–5.
Mian A, Hill BT. Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma. Expert Opin Biol Ther. 2021;21(4):435–41.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48.
Mueller KT, Waldron E, Grupp SA, Levine JE, Laetsch TW, Pulsipher MA, et al. Clinical pharmacology of Tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24(24):6175–84.
Stein AM, Grupp SA, Levine JE, Laetsch TW, Pulsipher MA, Boyer MW, et al. Tisagenlecleucel model-based cellular kinetic analysis of chimeric antigen receptor-T cells. CPT Pharmacometr Syst Pharmacol. 2019;8(5):285–95.
Iragavarapu C, Hildebrandt G. Lisocabtagene Maraleucel for the treatment of B-cell lymphoma. Expert Opin Biol Ther. 2021;21(9):1151–6.
Kharfan-Dabaja MA, Yassine F, Alhaj Moustafa M, Iqbal M, Murthy H. Lisocabtagene maraleucel in relapsed or refractory diffuse large B cell lymphoma: what is the evidence? Hematol Oncol Stem Cell Ther. 2021. https://doi.org/10.1016/j.hemonc.2021.09.004.
Karlsson H, Svensson E, Gigg C, Jarvius M, Olsson-Strömberg U, Savoldo B, et al. Evaluation of Intracellular Signaling downstream chimeric Antigen receptors. PLoS ONE. 2015;10(12):e0144787.
Selli ME, Landmann JH, Terekhova M, Lattin J, Heard A, Hsu YS, et al. Costimulatory domains direct distinct fates of CAR-driven T-cell dysfunction. Blood. 2023;141(26):3153–65.
Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25(11):2452–65.
Amatya C, Pegues MA, Lam N, Vanasse D, Geldres C, Choi S, et al. Development of CAR T cells expressing a suicide gene plus a chimeric antigen receptor targeting signaling lymphocytic-activation molecule F7. Mol Ther. 2021;29(2):702–17.
Cheng Z, Wei R, Ma Q, Shi L, He F, Shi Z, et al. In vivo expansion and antitumor activity of coinfused CD28- and 4-1BB-engineered CAR-T cells in patients with B cell leukemia. Mol Ther. 2018;26(4):976–85.
Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112–6.
Li S, Zhang J, Wang M, Fu G, Li Y, Pei L, et al. Treatment of acute lymphoblastic leukaemia with the second generation of CD19 CAR-T containing either CD28 or 4-1BB. Br J Haematol. 2018;181(3):360–71.
Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–28.
Li G, Boucher JC, Kotani H, Park K, Zhang Y, Shrestha B, et al. 4–1BB enhancement of CAR T function requires NF-κB and TRAFs. JCI Insight. 2018. https://doi.org/10.1172/jci.insight.12132.
Salter AI, Pont MJ, Riddell SR. Chimeric antigen receptor-modified T cells: CD19 and the road beyond. Blood. 2018;131(24):2621–9.
Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17(10):1206–13.
Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid Fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23(4):757–68.
Fujiwara K, Tsunei A, Kusabuka H, Ogaki E, Tachibana M, Okada N. Hinge and Transmembrane domains of chimeric Antigen receptor regulate receptor expression and signaling threshold. Cells. 2020. https://doi.org/10.3390/cells9051182.
Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–67.
Xu H, Hamburger AE, Mock JY, Wang X, Martin AD, Tokatlian T, et al. Structure-function relationships of chimeric antigen receptors in acute T cell responses to antigen. Mol Immunol. 2020;126:56–64.
Calderon H, Mamonkin M, Guedan S. Analysis of CAR-mediated tonic signaling. Methods Mol Biol. 2020;2086:223–36.
Hirobe S, Imaeda K, Tachibana M, Okada N. The effects of chimeric antigen receptor (CAR) hinge domain post-translational modifications on CAR-T cell activity. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms23074056.
Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 2010;184(12):6938–49.
Muller YD, Nguyen DP, Ferreira LMR, Ho P, Raffin C, Valencia RVB, et al. The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. Front Immunol. 2021;12: 639818.
Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 2020;10(5):702–23.
Song MK, Park BB, Uhm JE. Resistance mechanisms to CAR T-cell therapy and overcoming strategy in B-cell hematologic malignancies. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20205010.
Mao R, Hussein MS, He Y. Chimeric antigen receptor engineered T cells and their application in the immunotherapy of solid tumours. Expert Rev Mol Med. 2022;24:e7.
Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12(1):81.
Miao L, Zhang Z, Ren Z, Tang F, Li Y. Obstacles and coping strategies of CAR-T cell immunotherapy in solid tumors. Front Immunol. 2021;12: 687822.
Burns WR, Zhao Y, Frankel TL, Hinrichs CS, Zheng Z, Xu H, et al. A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res. 2010;70(8):3027–33.
Whitlow M, Bell BA, Feng SL, Filpula D, Hardman KD, Hubert SL, et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. 1993;6(8):989–95.
Landoni E, Fuca G, Wang J, Chirasani VR, Yao Z, Dukhovlinova E, et al. Modifications to the framework regions eliminate chimeric antigen receptor tonic signaling. Cancer Immunol Res. 2021;9(4):441–53.
Fujiwara K, Masutani M, Tachibana M, Okada N. Impact of scFv structure in chimeric antigen receptor on receptor expression efficiency and antigen recognition properties. Biochem Biophys Res Commun. 2020;527(2):350–7.
Desplancq D, King DJ, Lawson AD, Mountain A. Multimerization behaviour of single chain fv variants for the tumour-binding antibody B72.3. Protein Eng. 1994;7(8):1027–33.
Hege K. Context matters in CAR T cell tonic signaling. Nat Med. 2021;27(5):763–4.
Lamarthée B, Marchal A, Charbonnier S, Blein T, Leon J, Martin E, et al. Transient mTOR inhibition rescues 4-1BB CAR-Tregs from tonic signal-induced dysfunction. Nat Commun. 2021;12(1):6446.
Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. 2021;18(11):715–27.
Watanabe N, Mo F, McKenna MK. Impact of manufacturing procedures on CAR T cell functionality. Front Immunol. 2022;13: 876339.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-Cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–42.
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–52.
Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–38.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95.
Porter D, Frey N, Wood PA, Weng Y, Grupp SA. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol. 2018;11(1):35.
Sesques P, Ferrant E, Safar V, Wallet F, Tordo J, Dhomps A, et al. Commercial anti-CD19 CAR T cell therapy for patients with relapsed/refractory aggressive B cell lymphoma in a european center. Am J Hematol. 2020;95(11):1324–33.
Davey AS, Call ME, Call MJ. The influence of chimeric antigen receptor structural domains on clinical outcomes and associated toxicities. Cancers (Basel). 2020. https://doi.org/10.3390/cancers13010038.
Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. 2019;25(6):947–53.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17.
Ma F, Ho JY, Du H, Xuan F, Wu X, Wang Q, et al. Evidence of long-lasting anti-CD19 activity of engrafted CD19 chimeric antigen receptor-modified T cells in a phase I study targeting pediatrics with acute lymphoblastic leukemia. Hematol Oncol. 2019;37(5):601–8.
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8(355):355ra116.
Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8 + composition in adult B cell all patients. J Clin Invest. 2016;126(6):2123–38.
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric Antigen receptor T cells in Refractory B-Cell Lymphomas. N Engl J Med. 2017;377(26):2545–54.
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139.
Westin JR, Kersten MJ, Salles G, Abramson JS, Schuster SJ, Locke FL, et al. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am J Hematol. 2021;96(10):1295–312.
Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019;134(26):2361–8.
Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–59.
Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32(2):169–80.
Sauter CS, Senechal B, Rivière I, Ni A, Bernal Y, Wang X, et al. CD19 CAR T cells following autologous transplantation in poor-risk relapsed and refractory B-cell non-hodgkin lymphoma. Blood. 2019;134(7):626–35.
Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum Interleukin-15 levels. J Clin Oncol. 2017;35(16):1803–13.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28.
Maziarz RT, Schuster SJ, Romanov VV, Rusch ES, Li J, Signorovitch JE, et al. Grading of neurological toxicity in patients treated with tisagenlecleucel in the JULIET trial. Blood Adv. 2020;4(7):1440–7.
Hay KA. Cytokine release syndrome and neurotoxicity after CD19 chimeric antigen receptor-modified (CAR-) T cell therapy. Br J Haematol. 2018;183(3):364–74.
Awasthi R, Pacaud L, Waldron E, Tam CS, Jager U, Borchmann P, et al. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv. 2020;4(3):560–72.
Locke FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020;4(19):4898–911.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73.
Cappell KM, Sherry RM, Yang JC, Goff SL, Vanasse DA, McIntyre L, et al. Long-term follow-up of anti-CD19 chimeric antigen receptor T-cell therapy. J Clin Oncol. 2020;38(32):3805–15.
Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42.
Zhao X, Yang J, Zhang X, Lu XA, Xiong M, Zhang J, et al. Efficacy and safety of CD28- or 4-1BB-based CD19 CAR-T cells in B cell acute lymphoblastic leukemia. Mol Ther Oncolytics. 2020;18:272–81.
Acknowledgements
We thank Francesco Marincola, Frank Neumann, John Langowski and David Barrett for their comments and feedback.
Funding
Both authors are employees and shareholders of Kite, a Gilead Company.
Author information
Authors and Affiliations
Contributions
Both authors contributed to the writing of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Both authors are employees and shareholders of Kite, a Gilead Company.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Smith, R., Shen, R. Complexities in comparing the impact of costimulatory domains on approved CD19 CAR functionality. J Transl Med 21, 515 (2023). https://doi.org/10.1186/s12967-023-04372-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04372-4