
Abstract
Background
Comitant strabismus (CS) is a heterogeneous disorder that is a major contributing factor to unilateral childhood-onset visual impairment. Studies have confirmed that genetic factors play an important role in the development of CS. The aim of this study was to identify the genetic cause of non-syndromic familial CS.
Methods
Fourteen unrelated CS families were recruited for the study. Twelve affected and 2 unaffected individuals from a large four-generation family (CS08) were selected to perform whole genome-wide linkage analysis. Parallel whole-exome sequencing (WES) was conducted in the same family (9 patients and 1 unaffected member) and 31 additional CS cases from 13 other unrelated families. Sanger sequencing was used to determine whether any of the remaining variants co-segregated with the disease phenotype in the corresponding family.
Results
Based on linkage analysis, CS in family CS08 mapped to a novel region of 34.17 centimorgan (cM) on chromosome 2q22.3-2q32.1 between markers D2S151 and D2S364, with a maximum log odds (LOD) score of 3.54 (theta = 0) at D2S142. Parallel WES identified a heterozygous variant, LRP2 c.335 A > G (p.Q112R), located in such a linkage interval that completely co-segregated with the disease in the family. Furthermore, another novel heterozygous variant (c.7274A > G, p.D2425G) in LRP2 that co-segregated was detected in 2 additional affected individuals from another unrelated family by WES. Both variants are predicted to be damaging by PolyPhen-2, SIFT and MutationTaster, and were absent in 100 ethnically matched normal controls.
Conclusion
LRP2 is a novel candidate genetic cause of non-syndromic familial CS.
Similar content being viewed by others

Introduction
Strabismus is clinically defined as a condition in any misalignment of the eyes in coordination, which is a major ocular abnormality in children and often accompanied by adverse effects on binocularity, stereopsis, and depth of perception. Epidemiological data indicate a prevalence of approximately 1–4% approximately in some populations [1, 2]. According to the change in the magnitude of misalignment in different gaze directions, strabismus can be subclassified as comitant strabismus (CS; constant in all directions) and incomitant strabismus (various). CS is the most common form of strabismus and a major contributing factor to unilateral childhood-onset visual impairment, especially amblyopia [2, 3]. In addition, strabismus affects normal binocular vision function even in the absence of amblyopia, subsequently affecting daily physiological and psychosocial performance unless successfully treated [4, 5].
CS is highly heterogeneous and influenced by genetic and environmental factors, though the pathogenesis remains unclear [6, 7]. Based on the inheritance pattern, numerous studies have been performed to investigate the genetic causation of CS using different methods. Considering the complex influencing background and high prevalence of this disease, two genome-wide association studies were recently carried out for strabismus, two variants in TSPAN10 (rs6420484 and rs397693108) and a variant in WRB (rs2244352) were found to increase the susceptibility to strabismus [8, 9]. However, previous family, twin, and pedigree studies have confirmed that some CS families show an autosomal dominant (AD) or autosomal recessive (AR) pattern of inheritance [10,11,12]. Linkage analysis has also implicated several associated loci, with the most significant being chromosome 7p22.1 (STBMS1 locus, OMIM: 185100), transmitting in both AR and AD models [13, 14]. Moreover, two other susceptibility loci, 4q28.3 and 7q31.2, were recently identified in the Japanese population in association with the candidate genes MGST2 and WNT2 [15, 16]. In addition, variants of AHI1 and PAX3 have been detected by whole-exome sequencing (WES) in Chinese families, contributing to strabismus [17, 18]. Anyway, these studies indicate that CS may manifest as a rare and monogenic subtype.
In the current study, we recruited 14 unrelated non-syndromic CS-affected Chinese families, including a large four-generation family, CS08. WES and genome-wide linkage analysis were performed synchronously to identify a rare heterozygous variant, c.335 A > G (p.Q112R) in the LRP2 gene, located in the corresponding linkage interval (2q22.3-2q32.1) and co-segregating with the disease in the family CS08. WES also detected another heterozygous variant (c.7274A > G, p.D2425G) in LRP2 in two additional affected individuals from another unrelated CS family (CS06).
Materials and methods
Families and clinical examinations
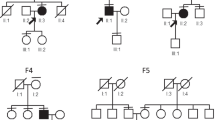
Forty-seven non-syndromic CS-affected and 18 CS-unaffected siblings from 14 unrelated families (1 with esotropia and 13 with exotropia; Fig. 1a and 3a, and Additional file 2a-l), including a large four-generation family (CS08), were recruited and clinically followed-up at the First Affiliated Hospital with Nanjing Medical University. Written informed consent was obtained from the participants and the parents of each child for sample collection and genetic analysis, and this study was approved by the ethics committee of the First Affiliated Hospital with Nanjing Medical University (2019-SR-134) in accordance with the Declaration of Helsinki principles.

Pedigree of family CS08 and haplotype reconstruction for the mapped region on chromosome 2 and clinical evaluations of the proband in family CS08. a Affected and unaffected members are indicated by filled and open symbols respectively. The black arrow indicates the proband. Haplotypes for tested short tandem repeat (STR) markers and genotypes for LRP2 c.335, rs2683454 and rs2683454, are given for all participants. Black bars represent the ancestral haplotype associated with the disease. *Individuals on whom WES was performed, §Individuals on whom sanger sequencing was performed. Abbreviation: M1, mutation c.335A > G b Ocular positions and movements. White arrow, exotropia phenotype of right eye. c Fundus photograph of right eye. d Ocular MRI. e–f Brain MRI for patient III:19
Before they received any treatment, routine ocular examinations were performed on available participants, including visual acuity, slit-lamp biomicroscopy, and funduscopic evaluations. Angles of deviation at a distance (5 m) and at near (0.3 m) with the cover/uncover test, alternate prism and cover test or Krimsky test (in young or uncooperative patients) were also carried out. Refractive errors were measured using an autorefractometer. Ocular and brain magnetic resonance imaging (MRI) was performed on the proband of family CS08. Renal function was investigated using urine and blood samples from the two probands of families CS08 and CS06. Data for the history of strabismus treatment, including prior surgery or patching, were obtained before the examination from available participants themselves or confirmed by telephone conversations.
Strabismus was defined if any tropia was present at a distance or near, with or without wearing spectacles, and classified according to the primary direction (esotropia, exotropia, vertical) of the tropia. Strabismus can be subclassified according to the change in magnitude of misalignment in different directions of gaze as CS (comitant) and incomitant strabismus (various). Strabismus is considered constant tropia if constant at both near and distance fixation; otherwise, it is considered intermittent tropia. To minimize the effect of environmental and syndromic factors, cases according to the following criteria were excluded [13]: (i) any secondary strabismus; (ii) any incomitant strabismus; (iii) individual with known CS risk factors such as prematurity (< 35 weeks of age), low birth weight (< 1.8 kg); (iv) strabismus caused by deprivation or myasthenia gravis.
Another 100 unrelated ethnically matched normal controls free of brain and ocular diseases were also recruited. Genomic DNA was isolated from peripheral venous blood (5 ml) using TIANamp Genomic DNA Kit (TIANGEN, Beijing, China).
Whole genome-wide linkage analysis
Whole genome-wide linkage screening was performed on the largest family CS08, including 12 patients (Fig. 1a; II:1, II:8, II:12, III:1, III:6, III:9, III:14, III:17, III:19, IV:1, IV:4 and IV:5) and two unaffected members (Fig. 1a; III:12 and IV:3). Moreover, 366 microsatellite markers and 3 single nucleotide polymorphisms (SNPs) spanning the entire human genome with an interval of approximately 10 cM (Weber set 6.0) were amplified by polymerase chain reaction (PCR) using primers labelled with Fam (Additional file 1). The PCR products were appropriately pooled according to allele size and labelling, mixed with GeneScanTM—500 Liz Size Standard (Applied Biosystems, Foster City, CA), denatured, loaded onto 6% standard denaturing polyacrylamide gels, and processed using an ABI 3730xl Analyzer (Applied Biosystems) for fluorescent detection. The pedigree displayed male-to-male transmission (Fig. 1a; II:1 and III:1) of the disease and almost equal numbers of affected males and females, indicating an AD pattern of inheritance (Fig. 1a). The multipoint LOD score was calculated using an AD inheritance model with 0.0001 and 0.01 disease allele frequencies and a penetrance range from 80 to 100%, respectively. Genotyping data were collected and analysed using the Genemapper 4.1 software package (Applied Biosystems). Multipoint linkage analysis was performed with the MERLIN program (http://www.sph.umich.edu/csg/abecasis/Merlin/index.html). Family and haplotype data were generated using Cyrillic, Version 2.1 program.
Whole exome sequencing and Sanger sequencing
Parallel WES was carried out using genomic DNA from 9 patients (Fig. 1a; excluding III:6, IV:1, and IV:5 compared with linkage screening) and 1 unaffected member (III:12) of family CS08. WES was performed on genomic DNA from 31 additional patients from 13 unrelated families (14 males and 17 females, Additional file 2). WES was performed with a SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, CA) and sequenced on the HiSeq 2000 platform (Illumina, San Diego, CA). CASAVA v1.8.2 was used to convert Illumina BCL files to FASTQ files. Low-quality bases and adapters were filtered with Trimmomatic version 0.32 [19]. Then, sequence reads were mapped to the human reference sequence (GRCh37) with default parameters by Burrows-Wheeler Aligner (BWA-MEM v0.7.15-r1140) [20]. Single nucleotide variants (SNVs) and small insertions and deletions (INDELs) were called using GATK Best Practice pipelines [21]. Base-quality score recalibration and local alignment around INDELs were refined by the GATK suite version 3.5.0. We used snpEff (v4.3–3) to annotate variants with population frequency, phylogenetic conservation scores, gene regions, and exonic functions, after which all annotated variants were loaded into the GEMINI (v0.19.1) [22]. Sanger sequencing and intrafamilial co-segregation analysis on the variants shared among all patients and absent in unaffected members.
In silico analyses
Pathogenicity prediction was performed by using three online mutational pathogenicity evaluation software programs: SIFT (http://sift.jcvi.org/); PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/); and Mutationtaster (http://www.mutationtaster.org/). Evolutionary conservation of the mutated residue was analysed with Ensemble Genome Server database BLAST / BLAT Tools by aligning the protein sequence of human LRP2 with sequences of the following orthologous proteins: Pan troglodytes (XP_515882.2), Macaca mulatta (XP_001104179.2), Bos Taurus (XP_002685354.2), Mus musculus (NP_001074557.1), Rattus norvegicus (NP_110454.1), Gallus gallus (XP_004942820.1), and Danio rerio (NP_001181916.1). Crystal structural models of wild-type and mutant LRP2 were constructed using the SWISS-MODEL online server. Predicted structures were displayed with PyMol software (version 1.5).
Results
Characterizations of the four-generation family CS08
The pedigree of the family CS08 is depicted in Fig. 1a. All of the affected individuals exhibited the strabismus phenotype with comitant exotropia. A total of 12 patients and two unaffected individuals in this family were recruited, comprising six males and eight females. The proband (III:19) was a 22-year-old female with intermittent exotropia, which occurred at approximately 5 years of age. The amount of tropia before surgery displayed—30 PD (near), and—25 PD (far) with best-corrected visual acuity (BCVA) was 1.0 in both eyes (Fig. 1b and Table 1). The refractive error status was slight astigmatism in the left eye (-1.00 D). No abnormality was found by slit-lamp biomicroscopy and funduscopic evaluations of the two eyes (Fig. 1c). Both ocular and brain MRI appeared normal (Fig. 1d–f). The proband denied any renal symptoms, and normal results were confirmed by renal function detection. In addition to the patient, III:6 presented constant exotropia in the right eye; the remaining 11 patients presented different degrees of intermittent exotropia. All patients were born via normal pregnancy and delivery. None of the patients, except for the proband, had a prior history of treatment. Moreover, no other syndromic feature was observed in any of the individuals. The ocular clinical data of this family are summarized in Table 1.
Linkage analysis of initially located pathogenic genes on chromosome 2
To determine the genetic cause of non-syndromic comitant exotropia in family CS08, we performed the whole genome-wide linkage scan using genomic DNA from family CS08 (12 patients and 2 unaffected members). We calculated the multipoint LOD score for family CS08 under 0.0001 and 0.01 disease allele frequencies and penetrance varying from 80 to 100%. Multipoint linkage analysis identified a 34.17 centimorgan (cM) candidate region co-segregating with the disease on chromosome 2q22.3-2q32.1 using the dominant model. The critical interval is flanked by markers D2S151 and D2S364, with a maximum LOD score of 3.54 (theta = 0) at D2S142 under a model in which the disease allele frequency was 0.01 and penetrance is 100% (Fig. 2a, b, Additional file 3). Haplotype construction is illustrated in Fig. 1a.

Multipoint linkage analysis results and analysis of mutation c.335 A > G (p.Q112R) in LPR2. a The locus for chromosomal region 2q22.3-2q32.1. Relative order of genotyped microsatellite markers is shown at the bottom next to an ideogram of chromosome 2. b Results from multipoint linkage analysis and genetic locations for the markers genotyped. The horizontal axis represents position of Chromosome 2. The vertical axis represents LOD score. bt., between. c Sanger sequencing showing heterozygous c.335 A > G mutation in III:19 (patient) and III:12 (unaffected) respectively. Reference sequences are given at the bottom. Abbreviation: Het., heterozygous; Ref., reference. d Conservation analysis of residue p.Q112 (boxed) of LRP2 across eight species
WES and Sanger sequencing identified a candidate gene
Parallel WES was performed on genomic DNA from 9 affected and 1 unaffected member of the CS08 family pedigree (Fig. 1a). Initially, a total of 112,804 variants were detected; each sample had a mean depth of 188 × , with at least 5 × coverage over 98.98% of the reference genome. As shown in Additional file 4, after bioinformatics analysis and filtering, only one heterozygous missense variant LRP2 c.335 A > G, (p.Q112R) remained. This missense mutation is predicted to be disease-causing by prediction tools (SIFT, PolyPhen-2, and MutationTaster) (Table 2). This variant is located inside the linkage interval on 2q22.3-2q32.1, with complete co-segregation with the disease in this family based on Sanger sequencing, which was conducted for all 14 CS08 members (Fig. 1a, 2c). Furthermore, Sanger sequencing of the same site was performed in 100 normal controls, with no positive result. Therefore, we believe that LRP2 is likely to be the causative gene of CS in this family.
Detection of LRP2 mutations in 13 additional families with CS
To assess the possibility of the genetic contribution of the LRP2, we further performed WES on 31 affected members of the other 13 unrelated CS families. An additional heterozygous variant (c.7274A > G, p.D2425G) in LRP2 was detected in the proband of family CS06 (Fig. 3a). This variant was confirmed to co-segregate with the phenotype by Sanger sequencing, which was performed on all family members available (II:4, II:5, II:7, II:8, III:1, III:5, III:8, III:10, III:12, IV:2, IV:3). The proband III:8 was affected by comitant esotropia, which occurred at approximately 2 years of age. The amount of tropia displayed + 25 PD (near) and + 15 PD (distant); BCVA was 1.0 in both eyes (Fig. 3b). Refractive error status was moderate myopia (-2.75 D) in her left eye. Her son (IV:3), a 4-year-old boy with comitant esotropia, showed tropia of + 50 PD (near) and + 45 PD (distant). Sanger sequencing revealed a de novo mutation transmitted from his mother (III:8) that was present in his grandparents (Fig. 3a, c).

Pedigree of family CS06, clinical evaluations of the proband, and analysis of mutation c.7274A > G (p.D2425G) in LPR2. a Pedigree of family CS06. *Individuals on whom WES was performed, §Individuals on whom sanger sequencing was performed. Abbreviation: M2, mutation c.7274A > G. b Ocular positions and movements for patient IV:3. White arrow, esotropia phenotype of right eye. c Sanger sequencing showing heterozygous c.7274A > G mutation in IV:3 (patient) and III:5 (unaffected) respectively. Reference sequences are given at the bottom. Abbreviation: Het., heterozygous; Ref., reference. d Conservation analysis of residue p.D2425 (boxed) of LRP2 across eight species
Pathogenic analysis
Both heterozygous variants are absent or extremely rare in public databases and predicted to be disease-causing by SIFT, PolyPhen-2, and MutationTaster (Table 2). Furthermore, both of the missense variants (Q112R and D2425G) are highly conserved across different species (Fig. 2d, 3d), which supports the pathogenicity of LRP2 variants causing CS.
The structural organization of LRP2 is shown in Fig. 4a; it is composed of complement-type repeats (CRs), epidermal growth factor (EGF)-like repeats, and β-propellers. Mutation Q112R is located in a CR, and D2425G is located between two β-propellers; both mutations are located in the extracellular domain. We performed crystal structural modelling for the mutant LRP2 using SWISS-MODEL to predict the pathogenic effect caused by two mutations (Fig. 4b-g) and found that the hydrogen bonds between residues 2425 and Tyr2434, Tyr2426, Phe2473, as well Asn2641 are eliminated upon the change from a wild-type aspartic to mutant glycine. It is likely that the mutation p.D2425G affects the folding and relevant biological process of LRP2.

Schematic structural of LRP2/megalin and predicted crystal structural models of the wild-type and mutant. a Megalin is composed of a large extracellular domain, a single transmembrane domain, and a short cytoplasmic domain. The extracellular domain harbors four cysteine-rich complement-type ligand binding repeats, which are separated from each other by β-propellers and EGF-like repeats. b-d Crystal structures of wild-type human LRP2 and mutant human LRP2 carrying p.Q112R. c The mutation spot is highlighted red. D, The 112 residue glutamine is replaced by arginine. e–g Crystal structures of wild-type and mutant human LRP2 carrying p.D2425G. e The mutated residue is indicated in green. Amino acids interacted with residue 2425, including Tyr2426, Tyr2434, Phe2473, and Asn2641, are indicated. g The hydrogen bond between residue 2425 and Tyr2434, Tyr2426, Phe2473, as well Asn2641 are eliminated upon the change from wild-type aspartic to mutant glycine
Discussion
In this study, we mapped non-syndromic CS in a four-generation family to a linkage interval on chromosome 2q22.3-2q32.1 (34.17 cM), with a maximum LOD score of 3.54. Furthermore, a rare heterozygous variant in LRP2 (c.335A > G, p.Q112R) located in the corresponding linkage interval that completely co-segregates with the disease in the family was detected by WES. This missense variant LRP2 c.335A > G exhibits a very low allelic frequency of 0.00002389 in gnomAD and was absent in other public databases (1000G and TOPmed, Table 2), indicating it was not a common benign polymorphism represented by these databases. WES also revealed another heterozygous variant of LRP2 (c.7274A > G, p.D2425G) in 2 additional affected individuals from another unrelated family with CS. Both variants are highly conserved, absent or extremely rare in public databases; they were absent in 100 ethnically matched normal controls according to Sanger sequencing. These mutations are predicted to be damaging by PolyPhen-2, SIFT and Mutation Taster. These data indicated that mutations in LRP2 are novel genetic causes of non-syndrome familial CS.
LRP2, located on chromosome 2q31.1, encodes a giant multiligand transmembrane receptor (600 kDa; also named megalin) of the low-density lipoprotein (LDL) receptor gene family [23, 24]. The structure of LRP2/megalin is depicted in Fig. 4a, consisting of a large extracellular domain, a single transmembrane domain, and a short cytoplasmic domain. The extracellular domain harbours four cysteine-rich complement-type ligand binding repeats, which are separated from each other by β-propellers and EGF-like repeats. The single transmembrane domain is connected to the intracellular segment, and the cytoplasmic tail is rich in multiple functional elements. LRP2 is highly expressed in epithelial cells in mammals, including the kidney, brain, eye, lung, and reproductive tissues. It binds many ligands associated with diverse signalling pathways, including Sonic Hedgehog (Shh), bone morphogenic protein (BMP), and retinoid trafficking, etc. [23,24,25]. Most LRP2 mutations to date are associated with Donnai-Barrow syndrome (DBS), also known as facio-oculo-acoustico-renal syndrome, which is a rare autosomal-recessive and multi-system condition involving craniofacial features, ocular abnormalities, developmental delay, agenesis of the corpus callosum (ACC), intellectual disability, sensorineural hearing loss, and proteinuria [23, 26,27,28]. The universal ocular features of DBS are hypertelorism and high myopia; others, such as retinal detachment, iris coloboma, progressive visual loss, and optic nerve hypoplasia, have been mentioned in several cases. In addition, mutations in LRP2 may contribute to Stickler syndrome and autosomal recessive non-syndromic intellectual disability [29, 30]. Interestingly, few cases of DBS, Stickler syndrome, and non-syndromic intellectual disability have been accompanied by strabismus, both exotropia and esotropia [28,29,30]. Nevertheless, it is unknown whether the strabismus exhibited in these patients is a primary phenotype caused by LRP2 deficiency or merely a secondary change from abnormal development and function of the brain and/or ocular organs.
In our study, none of the patients from families CS08 and CS06 showed ocular symptoms or multi-system features other than CS. Despite intrafamilial phenotypic variability, high myopia was often observed in these DBS patients, ranging from -12.5 to -22.0 D, and was accompanied by large eyes. However, the refractive error state of proband III:19 was only slight astigmatism in the left eye (-1.00 D); ocular features of DBS were also absent in all other patients from family CS08. In the other family, CS06, both patient III:8 and her son IV:3 had a normal refractive state, with moderate myopia in the left eye of III:8. None of the members of the two families had anisometropia. In addition, characteristic craniofacial features and bulbophthalmia were absent in the two families. The results of routine renal function detection revealed a lack of proteinuria in both probands. Based on the above, the CS08 and CS06 patients could not be classified as having DBS. Therefore, we assume that LRP2 is a possible genetic contributor to primary strabismus; the two LRP2 mutations in this study are associated with an independent familial CS phenotype.
Multiple LRP2-deficient animal models exhibit frequent ophthalmic eye enlargement and high myopia, comparable to the phenotypes in DBS patients, involving both homozygous and compound heterozygous states [31,32,33,34,35]. Interestingly, adult heterozygous variant fish showed normal eye sizes and slight hyperopia [31,32,33,34,35], similar to the heterozygous carrier cases [28,29,30]. These data may explain the possible reason for the absence of eye enlargement, high myopia, and systemic features in our patients. In additional, neither strabismus was observed in these LRP2-deficient models (mouse and zebrafish). Despite the various phenotypes caused by mutations in LRP2, the choice of model animals (i.e., zebrafish and mice) in the above studies is also another possible explanation for the lack or limited binocular vision. In some cases, fish displayed asymmetry in the level of enlargement of their two eyes. However, anisometropia and bulbophthalmia were absent in the participants with strabismus in our study. Therefore, it is unlikely that ocular misalignment occurred subsequent to those abnormalities.
Overall, the pathogenesis of CS remains unclear, and various hypotheses have been proposed. Among these, ocular misalignment being likely caused by a disruption of binocular vision during the early critical period for development is universal [36, 37]. Normal binocular vision is dependent on interhemispheric connections, which are achieved by the corpus callosum (CC), the major fibre bundle in the mammalian brain [37]. In particular, the visual callosa connect the homologous regions of the visual cortex and combine the two halves of the visual field [37]. A number of observations and experiments in humans and cats have shown alterations in interhemispheric connections via the CC in strabismic eyes [38, 39]. It is worth noting that CC is a major forebrain-derived structure of white matter in the brain; mutations in LRP2 lead to ACC (variable) in humans [23, 26,27,28,29,30]. Reported data confirmed that during forebrain development, LRP2 is the main auxiliary receptor of the SHH signalling pathway, and defect in this protein causes SHH/Patch1/LRP2 complex failure formation, thus affecting downstream signalling pathway activation [24, 25, 40]. Knockout of the LRP2 gene causes non-split deformity of the forebrain in mice [27, 31, 35]. Evidence from the above studies strengthens the idea that LPR2 is likely to be an important participant in regulating the brain and eye movement network.
There are some limitations to our study. First, due to patient rejection of functional MRI and diffusion-based tensor, we could not confirm whether there was an alteration in interhemispheric connection via the CC that existed; nevertheless, the CC structure of the proband appeared normal by brain MRI. Second, besides of the family CS06 and CS08, neither candidate variant was detected in another 12 families, confirming the complex genetic trait of CS.
Conclusions
In summary, based on genome-wide linkage analysis, WES, and pathogenic analysis, we mapped non-syndromic CS to a novel locus on chromosome 2q22.3-2q32.1 and identified the rare heterozygous variant c.335A > G (p.Q112R) and novel heterozygous variants c.7274A > G, (p.D2425G) in LRP2. To the best of our knowledge, our study is the first report that LRP2 is a genetic cause of non-syndromic CS, expanding the phenotypic spectrum of LRP2 mutations. Similar to other known genes associated with CS, LRP2 variants can only explain a small subgroup of CS. Meanwhile, the pathogenicity of LRP2 variants in non-syndromic CS and mechanisms linking LRP2 and CS require further elucidation.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CS:
-
Comitant strabismus
- WES:
-
Whole-exome sequencing
- cM:
-
Centimorgan
- LOD:
-
Log odds
- CS:
-
Comitant strabismus
- AD:
-
Autosomal dominant
- AR:
-
Autosomal recessive
- MRI:
-
Magnetic resonance imaging
- PCR:
-
Polymerase chain reaction
- SNVs:
-
Single nucleotide variants
- INDELs:
-
Insertions and deletions
- BCVA:
-
Best-corrected visual acuity
- CR:
-
Complement-type repeats
- EGF:
-
Epidermal growth factor
- LDL:
-
Low-density lipoprotein
- Shh:
-
Sonic Hedgehog
- DBS:
-
Donnai-Barrow syndrome
- ACC:
-
Agenesis of the corpus callosum
- D:
-
Diopter
References
Group M-ePEDS. Prevalence of amblyopia and strabismus in African American and Hispanic children ages 6 to 72 months: the multi-ethnic pediatric eye disease study. Ophthalmology. 2008;115(7):1229-36 e1. https://doi.org/10.1016/j.ophtha.2007.08.001.
Chen X, Fu Z, Yu J, Ding H, Bai J, Chen J, et al. Prevalence of amblyopia and strabismus in Eastern China: results from screening of preschool children aged 36–72 months. Br J Ophthalmol. 2016;100(4):515–9. https://doi.org/10.1136/bjophthalmol-2015-306999.
Robaei D, Rose KA, Kifley A, Cosstick M, Ip JM, Mitchell P. Factors associated with childhood strabismus: findings from a population-based study. Ophthalmology. 2006;113(7):1146–53. https://doi.org/10.1016/j.ophtha.2006.02.019.
Williams C, Northstone K, Harrad R, Sparrow J, Harvey I. Amblyopia treatment outcomes after preschool screening v school entry screening: observational data from a prospective cohort study. Br J Ophthalmol. 2003;87(8):988–93. https://doi.org/10.1136/bjo.87.8.988.
Stewart CE, Fielder AR, Stephens DA, Moseley MJ. Treatment of unilateral amblyopia: factors influencing visual outcome. Invest Ophthalmol Vis Sci. 2005;46(9):3152–60. https://doi.org/10.1167/iovs.05-0357.
Maconachie GD, Gottlob I, McLean RJ. Risk factors and genetics in common comitant strabismus: a systematic review of the literature. JAMA Ophthalmol. 2013;131(9):1179–86. https://doi.org/10.1001/jamaophthalmol.2013.4001.
Ye X, Pegado V, Patel M, Wasserman W. Strabismus genetics across a spectrum of eye misalignment disorders. Clin Genet. 2014;86(2):103–11. https://doi.org/10.1111/cge.12367.
Shaaban S, MacKinnon S, Andrews C, Staffieri SE, Maconachie GD, Chan W-M, et al. Genome-wide association study identifies a susceptibility locus for comitant Esotropia and suggests a parent-of-origin effect. Invest Ophthalmol Vis Sci. 2018;59(10):4054–64. https://doi.org/10.1167/iovs.18-24082.
Plotnikov D, Shah RL, Rodrigues JN, Cumberland PM, Rahi JS, Hysi PG, et al. A commonly occurring genetic variant within the NPLOC4–TSPAN10–PDE6G gene cluster is associated with the risk of strabismus. Hum Genet. 2019;138(7):723–37. https://doi.org/10.1007/s00439-019-02022-8.
Matsuo T, Yamane T, Ohtsuki H. Heredity versus abnormalities in pregnancy and delivery as risk factors for different types of comitant strabismus. J Pediatr Ophthalmol Strabismus. 2001;38(2):78–82.
Matsuo T, Hayashi M, Fujiwara H, Yamane T, Ohtsuki H. Concordance of strabismic phenotypes in monozygotic versus multizygotic twins and other multiple births. Jpn J Ophthalmol. 2002;46(1):59–64. https://doi.org/10.1016/s0021-5155(01)00465-8.
Kruger JM, Mansouri B, Cestari DM. An update on the genetics of comitant strabismus. Semin Ophthalmol. 2013. https://doi.org/10.3109/08820538.2013.825298.
Parikh V, Shugart YY, Doheny KF, Zhang J, Li L, Williams J, et al. A strabismus susceptibility locus on chromosome 7p. Proc Natl Acad Sci USA. 2003;100(21):12283–8. https://doi.org/10.1073/pnas.2035118100.
Rice A, Nsengimana J, Simmons IG, Toomes C, Hoole J, Willoughby CE, et al. Replication of the recessive STBMS1 locus but with dominant inheritance. Invest Ophthalmol Vis Sci. 2009;50(7):3210–7. https://doi.org/10.1167/iovs.07-1631.
Shaaban S, Matsuo T, Fujiwara H, Itoshima E, Furuse T, Hasebe S, et al. Chromosomes 4q28. 3 and 7q31. 2 as new susceptibility loci for comitant strabismus. Invest Ophthalmol Vis Sci. 2009;50(2):654–61. https://doi.org/10.1167/iovs.08-2437.
Zhang J, Matsuo T. MGST2 and WNT2 are candidate genes for comitant strabismus susceptibility in Japanese patients. PeerJ. 2017;5: e3935. https://doi.org/10.7717/peerj.3935.
Min X, Fan H, Zhao G, Liu G. Identification of 2 potentially relevant gene mutations involved in strabismus using whole-exome sequencing. Med Sci Monit. 2017;23:1719. https://doi.org/10.12659/msm.902823.
Gong H-M, Wang J, Xu J, Zhou Z-Y, Li J-W, Chen S-F. Identification of rare paired box 3 variant in strabismus by whole exome sequencing. Int J Ophthalmol. 2017;10(8):1223. https://doi.org/10.18240/ijo.2017.08.06.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–60. https://doi.org/10.1093/bioinformatics/btp324.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013. https://doi.org/10.1002/0471250953.bi1110s43.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92. https://doi.org/10.4161/fly.19695.
Willnow TE, Christ A. Endocytic receptor LRP2/megalin—of holoprosencephaly and renal Fanconi syndrome. Pflügers Arch. 2017;469(7–8):907–16. https://doi.org/10.1007/s00424-017-1992-0.
Christ A, Herzog K, Willnow TE. LRP2, an auxiliary receptor that controls sonic hedgehog signaling in development and disease. Dev Dyn. 2016;245(5):569–79. https://doi.org/10.1002/dvdy.24394.
Christ A, Christa A, Klippert J, Eule JC, Bachmann S, Wallace VA, et al. LRP2 acts as SHH clearance receptor to protect the retinal margin from mitogenic stimuli. Dev Cell. 2015;35(1):36–48. https://doi.org/10.1016/j.devcel.2015.09.001.
Kantarci S, Ragge NK, Thomas NS, Robinson DO, Noonan KM, Russell MK, et al. Donnai-Barrow syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy. Am J Med Genet A. 2008;146(14):1842–7. https://doi.org/10.1002/ajmg.a.32381.
Pober BR, Longoni M, Noonan KM. A review of Donnai-Barrow and facio-oculo-acoustico-renal (DB/FOAR) syndrome: clinical features and differential diagnosis. Birth Defects Res A. 2009;85(1):76–81. https://doi.org/10.1002/bdra.20534.
Chinta S, Gupta A, Sachdeva V, Kekunnaya R. Persistent pupillary membrane, strabismus, and optic nerve hypoplasia in Donnai-Barrow syndrome. J Am Assoc Pediatr Ophthalmol Strabismus. 2011;15(6):604–5. https://doi.org/10.1016/j.jaapos.2011.07.016.
Schrauwen I, Sommen M, Claes C, Pinner J, Flaherty M, Collins F, et al. Broadening the phenotype of LRP2 mutations: a new mutation in LRP2 causes a predominantly ocular phenotype suggestive of Stickler syndrome. Clin Genet. 2014;86(3):282–6. https://doi.org/10.1111/cge.12265.
Vasli N, Ahmed I, Mittal K, Ohadi M, Mikhailov A, Rafiq MA, et al. Identification of a homozygous missense mutation in LRP2 and a hemizygous missense mutation in TSPYL2 in a family with mild intellectual disability. Psychiatr Genet. 2016;26(2):66–73. https://doi.org/10.1097/YPG.0000000000000114.
Storm T, Heegaard S, Christensen EI, Nielsen R. Megalin–deficiency causes high myopia, retinal pigment epithelium-macromelanosomes and abnormal development of the ciliary body in mice. Cell Tissue Res. 2014;358(1):99–107. https://doi.org/10.1007/s00441-014-1919-4.
Collery RF, Veth KN, Dubis AM, Carroll J, Link BA. Rapid, accurate, and non-invasive measurement of zebrafish axial length and other eye dimensions using SD-OCT allows longitudinal analysis of myopia and emmetropization. PLoS ONE. 2014. https://doi.org/10.1371/journal.pone.0110699.
Storm T, Burgoyne T, Dunaief JL, Christensen EI, Futter C, Nielsen R. Selective ablation of megalin in the retinal pigment epithelium results in megaophthalmos, macromelanosome formation and severe retina degeneration. Invest Ophthalmol Vis Sci. 2019;60(1):322–30. https://doi.org/10.1167/iovs.18-25667.
Cases O, Obry A, Ben-Yacoub S, Augustin S, Joseph A, Toutirais G, et al. (2017) Impaired vitreous composition and retinal pigment epithelium function in the FoxG1: LRP2 myopic mice. Biochimica et Biophysica Acta. 1863;6:1242–54. https://doi.org/10.1016/j.bbadis.2017.03.022.
Cases O, Joseph A, Obry A, Santin MD, Ben-Yacoub S, Pâques M, et al. Foxg1-Cre mediated Lrp2 inactivation in the developing mouse neural retina, ciliary and retinal pigment epithelia models congenital high myopia. PLoS ONE. 2015. https://doi.org/10.1371/journal.pone.0129518.
Das VE. Strabismus and the oculomotor system: insights from macaque models. Annu Rev Vis Sci. 2016;2:37–59. https://doi.org/10.1146/annurev-vision-111815-114335.
Pietrasanta M, Restani L, Caleo M. The corpus callosum and the visual cortex: plasticity is a game for two. Neural plast. 2012. https://doi.org/10.1155/2012/838672.
Berman NE, Payne BR. Alterations in connections of the corpus callosum following convergent and divergent strabismus. Brain Res. 1983;274(2):201–12. https://doi.org/10.1016/0006-8993(83)90697-2.
Bui Quoc E, Ribot J, Quenech’Du N, Doutremer S, Lebas N, Grantyn A, et al. Asymmetrical interhemispheric connections develop in cat visual cortex after early unilateral convergent strabismus: anatomy, physiology, and mechanisms. Front Neuroanat. 2012;5:68. https://doi.org/10.3389/fnana.2011.00068.
Christ A, Christa A, Kur E, Lioubinski O, Bachmann S, Willnow TE, et al. LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev Cell. 2012;22(2):268–78. https://doi.org/10.1016/j.devcel.2011.11.023.
Acknowledgements
We are grateful to all of the patients and their family members who participated in this study.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81673198); the Natural Science Foundation of Jiangsu Province (Grant No. BK20161595); Jiangsu Province’s Key Provincial Talents Program (Grant No. QNRC2016563) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (Grant No. SJCX20_0477). The funding organizations had no role in the design and conduct of this study.
Author information
Authors and Affiliations
Contributions
HL, ZH and XC designed the study. XC, YW, XZ, AZ, RL and ZW collected patients’ data and performed clinical assessments. TJ, YG and WY analysed the data. YW, XZ and AZ conducted the experiments. XC, YW and TJ wrote the manuscript. HL and ZH were responsible for the study supervision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of the First Affiliated Hospital with Nanjing Medical University (2019-SR-134) in accordance with the Declaration of Helsinki principles. Written informed contents were obtained from all participants or their legal guardians before their enrollments.
Consent for publication
Yes.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Information of microsatellite markers used in the study.
Additional file 2:
Figure S1. Pedigree of the other 12 families. A-E. CS01 – CS05. F. CS07. G-L. CS09 – CS14. *Individuals from whom blood samples were collected. §Individuals on whom WES were performed.
Additional file 3:
Table S2. Details of microsatellite markers with LOD score > 0.
Additional file 4:
Figure S2. Flow chart of variant analyses.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Y., Chen, X., Jiang, T. et al. Expanding the phenotypic spectrum of mutations in LRP2: a novel candidate gene of non-syndromic familial comitant strabismus. J Transl Med 19, 495 (2021). https://doi.org/10.1186/s12967-021-03155-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-021-03155-z