
Abstract
Drug resistance is of great concern in cancer treatment because most effective drugs are limited by the development of resistance following some periods of therapeutic administration. The tumor microenvironment (TME), which includes various types of cells and extracellular components, mediates tumor progression and affects treatment efficacy. TME-mediated drug resistance is associated with tumor cells and their pericellular matrix. Noninherent-adaptive drug resistance refers to a non-cell-autonomous mechanism in which the resistance lies in the treatment process rather than genetic or epigenetic changes, and this mechanism is closely related to the TME. A new concept is therefore proposed in which tumor cell resistance to targeted therapy may be due to non-cell-autonomous mechanisms. However, knowledge of non-cell-autonomous mechanisms of resistance to different treatments is not comprehensive. In this review, we outlined TME factors and molecular events involved in the regulation of non-cell-autonomous resistance of cancer, summarized how the TME contributes to non-cell-autonomous drug resistance in different types of antineoplastic treatment, and discussed the novel strategies to investigate and overcome the non-cell-autonomous mechanism of cancer non-cell-autonomous resistance.
Similar content being viewed by others

Introduction
There has been spectacular advances and successes in the development and clinical application of small molecule antineoplastic drugs in the past several decades [1]. While cytotoxic compounds with more potent tumor-killing effects are still being discovered, molecularly targeted drugs are under development following the identification of promising targets in cancers [2]. Both cytotoxic chemotherapeutics and targeted treatments have significantly improved the survival of patients with cancers. As far, the majority of antineoplastic treatments are small-molecules, which have had great success in saving the lives of patients with cancer [3].
However, drug resistance is frequently developed during the clinical application of antineoplastic agents [4]. A substantial percentage of cancer patients exposed to an antineoplastic agent either does not benefit from the treatment (primary resistance) and show reduced responsiveness and undergo tumor relapse progression (secondary resistance) [5]. Although new compounds and combinations of drugs with higher potency in killing cancer cells have been developed, the nearly inevitable development of drug resistance has limited the clinical efficacy and effectiveness of antineoplastic treatment [6].
Both intrinsic and extrinsic biological causes of cancer drug resistance have been postulated. First, the overexpression of several transmembrane transporters in tumor cells, such as p-glycoproteins and multidrug resistance protein family members, reduces the intracellular drug concentration by restricting drug absorption and promoting drug efflux [7,8,9]. Second, changes in drug metabolism and drug targets, such as modifications of drug metabolizing enzymes by mutation and altered expression, lead to the dysregulation of prodrug activation and inactivation of the active form of the drug, thereby subsidizing the drug efficacy and promoting drug resistance [6, 10, 11]. Third, gene amplification in tumor cells increases the number of copies of oncogenes, which then reinforces oncogenic signaling during drug treatment [8]. Mutations in DNA repair systems might also promote resistance to antineoplastic agents by increasing DNA mutations and adapt to the drug [12, 13]. Fourth, pre-existing or acquired tumor cell heterogeneity might lead to variation in the response of cancer cells to antineoplastic agents [11]. For example, cancer stem cells, a subpopulation of cells that possess self-renewal and differentiation abilities, are more resistant to therapy than well-differentiated tumor cells [14].
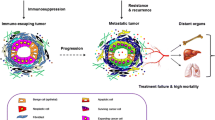
Although most of these mechanisms have been validated in patients, models of tumor cell-derived resistance have apparent limitations. Cancer cells typically interact with stromal cells within solid tumors in vivo, and these interactions extensively contribute to tumor development and therapeutic resistance. Thus, a new concept has been proposed in which tumor cells resistance to antineoplastic agents may be due to both cell-autonomous and non-cell-autonomous mechanisms. While the cell-autonomous mechanisms of cancer resistance have been reviewed elsewhere [6, 11], our knowledge of non-cell-autonomous mechanisms underlying tumor cell resistance to different treatments is incomplete. In particular, previous studies have highlighted the role of the tumor microenvironment (TME) in the development of non-cell-autonomous resistance to antineoplastic agents. Hence, in this review, we outlined the role of the TME in the development of non-cell-autonomous resistance to different antineoplastic agents. Intracellular signaling of tumor cells response to TME was discussed and how TME involved in resistance of each antineoplastic agent was depicted (Fig. 1).

The role of the TME in the development of non–cell-autonomous resistance to antineoplastic agents
Non-cell-autonomous mechanisms of drug resistance in tumors
Cell-autonomous resistance, which is the “intrinsic” mechanism of resistance, involves the activation of alternative signaling pathways, acquisition of secondary mutations in drug targets, amplification of the target genes, and activation of efflux pumps. Extensive strategies to overcome cell-autonomous resistance have been developed, including but not limited to the development of new and more potent compounds, novel combined regimens of treatment and discovery of novel targets. Nonetheless, non-cell-autonomous mechanisms of resistance in tumors have only recently been highlighted, which suggests that drug failures and tumor relapse are closely related to factors in the surrounding TME [5].
Human tumors consist of both epithelial-like tumor cells and their surrounding cells and extracellular components, such as vasculature, fibroblasts, immune cells, endothelial cells and extracellular matrix (ECM). The surrounding components interact with tumor cells to form a microenvironment that favors tumor cell proliferation and survival [15]. The concept of the TME was introduced to illustrate that cancer progression is influenced by factors other than tumor cells. As a result, it was postulated that the TME might mediate the acquisition of resistance when tumors are exposed to antineoplastic agents in vivo [16,17,18]. Indeed, noninherent-adaptive drug resistance refers to non-cell-autonomous resistance, which relies on the treatment process rather than genetic or epigenetic changes and is closely related to the TME [19]. The TME may play a role in the initiation and maintenance of non-cell-autonomous drug resistance through various mechanisms, including hypoxia, extracellular acidity, vascular abnormalities, changes in immune populations, cancer-associated fibroblasts (CAFs) and their secretomes, exosomes, extracellular matrix, and other soluble factors. The overall regulatory mechanisms of non-cell-autonomous cancer resistance involving the TME are shown in Fig. 2 and have been reviewed in detail elsewhere [20, 21]. The mechanism involved in the non-cell-autonomous resistance to specific agents will be discussed later in this manuscript.

The main factors of tumor microenvironment-driven non-cell-autonomous drug resistance
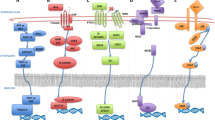
Intracellular signaling in tumor cells in response to the TME
Upon changes in the microenvironment, tumor cells may initiate a series of intracellular signaling cascades to transfer these messages from the aforementioned environmental factors into cytoplasm and nuclei. Signal transduction between the TME and tumor cells may occur through direct cell-cell contact or binding of TME-derived ligands with their specific receptors on tumor cell surface. Intracellular signaling pathways are then altered to regulate the expression and activity of downstream effector molecules that confer drug resistance on tumor cells. These signaling pathways, including the mTOR, NF-κB, AKT, and STAT3 pathways, are generally associated with tumor cell proliferation and survival.
mTOR signaling
As a vital signaling molecule that controls cell proliferation and growth under physiological conditions, mTOR is constitutively active in multiple types of cancer [19]. mTOR signaling is activated by the therapy-induced secretome, a collection of ambiguous components that form in TME after drug administration. Inhibition of mTOR signaling was found to blunt non-cell-autonomous resistance induced by vemurafenib, crizotinib or erlotinib (ERL) [22]. Lactate in the TME is imported by tumor cells and upregulates mTOR signaling via glutamine metabolism during treatment with vascular endothelial growth factor (VEGF) inhibitors. Activation of mTOR signaling initiates metabolic symbiosis in cancer cells, which confers resistance to VEGF inhibitors [23]. The switch towards the senescence-associated secretory phenotype (SASP) of tumor cells is activated by mTOR signaling and promotes non-cell-autonomous resistance. DNA damage-associated signaling through the ATM-TRAF6-TAK1 axis after initial drug treatment is responsible for the activation of mTOR signaling [24].
NF-κB signaling
NF-κB signaling determines the expression of immunomodulatory and secretory factors, which are key factors for cell senescence in the non-cell-autonomous mechanisms of drug resistance [25]. Cell senescence is an antiproliferative program, and antineoplastic therapy often aims to limit cell proliferation [26]. NF-κB-driven IL-6 and IL-8 expression modulate the initiation and persistence of the SASP. In an in vivo model of lymphoma, the SASP is destroyed by NF-κB inhibition, leading to escape from immunosurveillance by natural killer cells and p53 inactivation, thereby producing drug resistance [27].
AKT signaling
Both AKT and its associated signaling pathways are directly or indirectly involved in the oncogenic pathways in most human tumors [28]. Nevertheless, surprisingly results have been observed in which inhibition of AKT signaling in cancer cells confers drug resistance to the tumors. AKT-inhibited cancer cells activate their endomembrane system in a posttranscriptional manner to secrete inflammatory proteins IL-6 and IL-8, and extracellular vesicles (EVs), which enable rapidly growing cells to better withstand the stress conditions induced by drug treatment. These data suggest that AKT inhibition may contribute to the non-cell-autonomous mechanism of drug resistance in vivo [29].
STAT3 signaling
STAT3 signaling is generally considered the pathway in tumor cells that responds to the inflammatory TME [30]. A series of cytokines and chemokines can activate STAT3 in tumor cells, among which IL-6 and IL-1β are the major inducers derived from the TME. Tumor-associated macrophages (TAMs), neutrophils and CAFs in the TME are the major cell types that secrete IL-6 and IL-1β and are responsible for the activation of STAT3 in tumor cells [31]. Several downstream effectors of STAT3 in tumor cells were reported to contribute to non-cell-autonomous resistance to antineoplastic treatment. STAT3 activation may confer drug resistance by initiating epithelial-mesenchymal transition (EMT), suppressing epigenetic tumor suppressor microRNAs (miRNAs) and enhancing the expression of antiapoptotic proteins [32,33,34]. STAT3 activation in the TME may trigger vascular abnormalities, which were shown to abolish the proper delivery of gemcitabine [35]. STAT3 in tumor cells may also enhance the expression of Rab family proteins to facilitate exosome release, which confers cisplatin resistance in ovarian cancer [36].
Antineoplastic drug resistance involving TME-driven non-cell-autonomous mechanisms
TME-driven non-cell-autonomous mechanisms of resistance to various types of antineoplastic agents have been extensively studied. Individual drug may involve unique TME-driven non-cell-autonomous mechanisms, and resistance may come from more than one non-cell-autonomous factor in the TME (Table 1).
DNA-targeted drugs
Platinum-based chemotherapy
Platinum-based chemotherapy, including cisplatin, carboplatin, oxaliplatin, and nedaplatin, is the front-line treatment for several advanced cancers; however, treatment failure due to chemoresistance is common. In addition to the autonomous mechanism of resistance, such as the aberrant expression of antiapoptotic proteins in resistant tumor cells, some non-cell-autonomous resistances mechanisms could be involved. In colorectal cancer treatment, there is the possibility of drug resistance and tumor recurrence in patients treated with oxaliplatin, and the reason underlying this risk may be the changes in CAFs [37]. Oxaliplatin-based chemotherapy can increase hypoxia and the accumulation of CAFs in the TME, and hypoxia-inducible factor (HIF) activation. Moreover, expression of fibroblast growth factor 2 (FGF-2) increases significantly and initiates cancer proliferation and tumor vascular angiogenesis [38]. In esophageal squamous cell carcinoma, cisplatin treatment can promote PAI-1 secretion by CAFs, which acts in a paracrine manner to maintain AKT and ERK1/2 signaling in cancer cells and to promote cell survival [39]. These changes in cytokines are related to the accumulation of CAFs and mediate the mechanism of drug resistance [38, 40].
Conditioned media from ovarian cancer-associated mesenchymal stem cells (MSCs) was found to protect tumor cells by inhibiting endogenous proapoptotic signalings, such as that by XIAP and the caspases cascade [41]. Coculture of breast cancer cells with tumor tissue-derived MSCs (BC-MSCs) led to the development of cisplatin resistance; this process could be associated with the IL-6 secreted by BC-MSCs, which activates STAT3 signaling in breast cancer cells and promotes cell survival [42]. Another study suggested that the upregulation of IL-6 in MSCs could be dependent on cisplatin treatment [43].
EVs may also contribute to cisplatin resistance. Samuel et al. collected EVs from ovarian cancer cells and showed that they could activate the p38 and JNK pathways in bystander tumor cells. EVs uptake promoted in vitro resistance to cisplatin in ovarian cancer cells [44]. Further, it was found that hypoxia triggered ovarian cancer cells to secrete more exosomes, which in turn ameliorated dsDNA damage in cisplatin-treated cells and promote cell survival by activating the STAT3 pathway [36]. In breast cancer cells, exosomal miRNAs, such as miR-222/223, were found to play a role in facilitating the adaptation to a quiescent state during carboplatin-based therapy [45].
Some soluble stromal factors may also contribute to resistance to platinum-based chemotherapy. In epithelial ovarian cancer patients, stromal expression of periostin was associated with cisplatin resistance and clinical treatment outcomes. An in vitro study revealed that periostin caused persistent activation of AKT in A2780 ovarian cancer cells, leading to cell survival under cisplatin treatment [46]. Fibroblast activation protein alpha (FAP) expression in the stroma of epithelial ovarian cancer predicted the poor outcome of patients treated with cisplatin. Ovarian cancer cells treated in vitro with FAP showed significantly improved cell survival when exposed to cisplatin [47]. Platinum compounds may also trigger the ability of tumor cells to shape the immunosuppressive microenvironment, such as inducing M2 polarization of macrophages through the IL-6/STAT3 and NF-κB pathways; these changes indirectly contribute to the chemoresistance of cervical and ovarian cancers [48]. M2 macrophages may also produce nitric oxide to counteract the cisplatin-induced activation of syntaxin 4 and acid sphingomyelinase, thereby conferring tumor cells with chemoresistance [49].
Other alkylating agents
Acquired resistance to temozolomide (TMZ) has been reported in glioblastoma multiforme (GBM). In addition to resistance based on the modulation of DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) [50], the hypoxic TME was recently shown to affect drug sensitivity considerably. Cycling hypoxia was found to induce TMZ resistance in GBM, which was associated with ROS-mediated activation of HIF-1α and NF-κB, resulting in increased expression of the antiapoptotic protein Bcl-xL in GBM cell lines and xenograft tumors [51]. Furthermore, the hypoxic microenvironment could inhibit mitochondrial apoptosis by the HIF-1α-associated induction of miR-26a expression, which directly targets and suppresses proapoptotic Bad and Bax expression to protect mitochondrial function [52]. Besides, non-cell-autonomous resistance to TMZ may stem from factors in the surrounding environment. The perivascular niche that comprises endothelial and stromal cells was shown to support the resistance of GBM cells to TMZ treatment. Coculture of GBM cells with perivascular niche cells led to the activation of MGMT, epidermal growth factor receptor (EGFR), PI3K/AKT and Ras/Raf signaling that promoted TMZ resistance [53]. Direct contact between astrocytes and GBM cells through connexin43-dependent gap junctional communication might protect tumor cells from TMZ-induced apoptosis [54].
Nucleotide analogs and precursor analogs
Gemcitabine resistance is commonly reported in pancreatic cancer. Previous studies have shown that the resistance mechanism may involve non-cell-autonomous changes in signaling pathways within tumor cells due to contact with CAFs [55, 56]. In pancreatic ductal adenocarcinoma, CAFs are activated in response to tissue transglutaminase in the TME and initiate signaling pathways in tumor cells related to gemcitabine resistance. This process could be attributed to the overexpression of SATB-1 in tumor cells near stimulated CAFs, which then upregulate the secretion of SDF-1, a cytokine that plays a crucial role in many types of solid tumors by initiating signaling through its receptor CXCR4 [57, 58]. Another study suggested that gemcitabine resistance may be associated with TAM-derived exosome and exosomal miRNAs. Exosomal miR-365 was identified as the critical mediator of gemcitabine resistance in pancreatic ductal adenocarcinoma; it modulates pyrimidine metabolism and upregulates CDA expression, which inactivates gemcitabine by conversion to dFdUridine [59]. Furthermore, gemcitabine was found to induce inflammasome activation and IL-1β production in myeloid-derived suppressor cells (MDSCs), which in turn induced IL-17 secretion by CD4+ T cells to blunt gemcitabine toxicity [60].
Cytarabine is used for the treatment of acute myelogenous leukemia (AML). A previous study showed that cytarabine resistance might arise from intercellular communication between AML and bone marrow-derived MSCs [61]. AML cells cocultured with MSCs had elevated levels of Mcl-1, which is associated with multidrug resistance [62], and AML cell autophagy induced by MSC conferred cytarabine resistance [63]. In addition, the hypoxic microenvironment downregulated FLT3 expression in AML cells, which was associated with suppression of the PI3K pathway. Reduced FLT3 expression led to a lack of response to cytarabine treatment [64].
Fludarabine is used for the treatment of chronic lymphocytic leukemia (CLL). Bone marrow stromal cells (BMSCs) may create a CXCL12 gradient to promote the migration of CLL B cells, which promotes fludarabine resistance in CLL [65]. This response could be attributed to the binding of environmental CXCL12 to CXCR4 on CLL B cells and the subsequent activation of the AKT/FoxO3a/Bim axis within tumor cells [66]. Trimaco et al. also proved that MSCs isolated from the bone marrow of CLL patients rendered CLL B cells resistant to fludarabine in coculture conditions, which could be related to the presence of cytoprotective cytokines such as IL-8, CCL4, CCL11, and CXCL10 [67]. Furthermore, BMSCs could induce an increased import of cystine and its conversion into cysteine in the TME, and the resulting cysteine could be taken up by CLL B-cells for GSH synthesis. The intracellular redox balance maintained by GSH protects CLL cells from fludarabine toxicity [68].
Cytotoxic drugs
Anthracyclines
Doxorubicin/Adriamycin is an anthracycline widely used to treat various types of cancer, and doxorubicin resistance is frequently observed and involves a non-cell-autonomous mechanism. A study by Tu and colleagues showed that in vitro and in vivo interactions between MSCs and the osteosarcoma cell lines Saos-2 and U2-OS led to doxorubicin resistance through intercellular signal transduction involving the IL-6/STAT3 axis. MSC-derived IL-6 protects tumor cells from doxorubicin-induced apoptosis by activating STAT3 signaling [69]. The intercellular activation of survival signals by MSCs was also observed in multiple myeloma (MM), in which MSCs initiated NF-κB signaling through autophagy-dependent IκB degradation in MM cells [70].
MDSCs of a particular phenotype, with a polymorphonuclear structure and neutrophils in bone marrow were reported to mediate doxorubicin resistance through the secretion of soluble factors [71] including IL-1β, which was shown to activate PI3K/Rac and IL-1RI/β-catenin-dependent BIRC3 transcription in breast cancer cells, and CXCL1/GROα which increased angiogenesis in a mouse model of breast cancer [72, 73].
Zhang and colleagues suggested that the endothelial cell population may play a role in doxorubicin resistance in soft tissue sarcoma by facilitating vascular abnormalities. The overexpression of VEGF induces doxorubicin resistance without overtly impacting tumor cells but promotes endothelial cell proliferation, migration ,and sensitivity to doxorubicin. The addition of an anti-VEGF monoclonal antibody significantly improved doxorubicin sensitivity in soft tissue sarcoma [74].
Coculture of prostate cancer cells with CAFs attenuated doxorubicin-induced DNA damage and cytotoxicity. This effect of CAFs was attributed to the blockade of doxorubicin accumulation in prostate cancer cells due to increased cancer cell glutathione levels, which inhibited doxorubicin-induced ROS production [75].
Besides, non-cell factors in the TME also contribute to non-cell-autonomous doxorubicin resistance. The hypoxia-mediated induction of miR-424 in tumor cells promotes doxorubicin resistance. The HIF-1α-binding sequence in A375 melanoma cells, U251 glioblastoma cells, HCT116 colon cancer cells, A375 cell xenografts, and clinical breast cancer samples directly increased the transcription of miR-424, which suppressed the levels of the apoptosis-associated factor PDCD4 and protected cells from apoptosis [76]. In addition, the accumulation of hyaluronic acid (HA) in the TME plays an essential role in maintaining hypoxia which was shown by TME remodeling in many types of cancer cell lines [77]. In aggressive N-Myc-amplified neuroblastoma cells, the cathepsin family in the ECM contributes to doxorubicin-resistance [78]. Joyce et al. suggested that ECM changes in the breast cancer microenvironment, such as increased stiffness, led to the nuclear translocation of YAP in MDA-MB-231 cells. The subsequent mesenchymal differentiation contributed to ECM-induced doxorubicin resistance in breast cancer [79]. Ebata et al. showed that Rho/ROCK-associated myosin activation was also involved in ECM stiffness-induced doxorubicin resistance in MCF-7 breast cancer cells [80]. Also, microvesicle-like EVs were shown to carry drug efflux pump proteins from resistant Chronic myelogenous leukemia (CML) cells to sensitive tumor cells, which consequently reduced the intracellular availability of doxorubicin [81]. Resistant breast tumor cell-derived exosomes contain several miRNAs, such as miR-100, miR-17, miR-222, miR-342p and miR-451, among which miR-222 suppressed PTEN expression in recipient drug-sensitive cells to gain resistance to doxorubicin [82].
CAFs seem to play a role in the resistance to other anthracyclines. Genotoxic stress can induce DNA damage in prostate cancer stromal fibroblasts that leads to the expression and secretion of a glial cell-derived neurotrophic factor, which has a paracrine effect on prostate tumor cells resulting in acquired resistance to mitoxantrone [83]. In breast cancer, CAFs activate a novel estrogen/GPER/cAMP/PKA/CREB signaling axis that triggers the switch to aerobic glycolysis, and the production of extra pyruvate and lactate enables tumor cells to survive epirubicin treatment [84].
As anthracyclines are alkaline chemotherapeutic agents, they tend to have limited absorption in the acidic TME. This is due to a phenomenon called ion trapping, which refers to the preference of alkaline chemotherapeutic agents to accumulate in areas of low pH. As tumor cells tend to maintain a neutral pH by overexpressing proton pumps protein, the extracellular pH is more acidic [85, 86]. This pH gradient hinders the absorption of alkaline chemotherapeutic drugs by tumor cells and prevents the chemicals from reaching their site of action [87].
Other cytotoxic drugs
Bortezomib (BTZ), the first proteasome inhibitor approved for clinical use, was usually applied in the treatment of MM and mantle cell lymphoma (MCL). Coculture of MM cells with BMSCs was recently shown to induce BTZ resistance, which was associated with the aberrant expression of MUC-1, a vital factor for BTZ-resistance in MM. Further observation suggested that IL-6 secreted from BMSC upregulated MUC-1 via the JAK2/STAT3 pathway in MM cells [88]. TAMs also participated in the mechanism of BTZ resistance. A study by De Beule and colleagues showed that TAMs might activate STAT3 signaling and reduce the apoptosis of MM cells through the JAK2 pathway upon BTZ treatment. In vivo co-treatment with BTZ and an ATP-competitive JAK2 inhibitor improved the drug sensitivity of MM [89]. Overall, it can be concluded that the STAT3 pathway plays a vital role in BTZ-resistance in MM.
Paclitaxel, also known as Taxol, can interfere with the normal function of microtubules during tumor cell division. The mechanism of paclitaxel resistance may also be associated with TAMs. The Taxol-treated MMTV-PyMT mouse breast cancer model showed the increased abundance of TAMs, which expressed and released proteases such as cathepsins B and S to prevent tumor cells from undergoing Taxol-induced cell death. This protective effect of TAMs on breast ductal carcinoma cells was independent of direct cell-cell contact [90, 91].
Hypoxia is the major TME factor that promotes non-cell-autonomous resistance to etoposide. In HepG2 cells, hypoxia induced a reduction in p53 to protect cells from etoposide-induced apoptosis and promoted the DNA binding activity of c-jun to prevent DNA damage [92, 93]. Interestingly, Dudley et al. showed that tumor stromal cells are less sensitive to etoposide-induced p53 activation, which endows prostate cancer with drug resistance [94]. The expression of acylglycerol-3-phosphate acyltransferase 2 (AGPAT2) under hypoxic conditions increases lipid droplet accumulation in multiple types of cancer cells, leading to etoposide resistance [95]. Besides, non-cell-autonomous mechanisms of etoposide resistance seem to involve cell adhesion. The adhesion of myeloma cells to fibronectin led to G0/G1 cell cycle arrest, which depends on increased p27/kip1 protein levels and the inhibition of Cyclin A-and Cyclin E-associated kinase activity. Disrupting the interaction between fibronectin and tumor cells initiated cell cycle progression into S phase, which reverted MM cells to an etoposide-sensitive phenotype [96].
Tyrosine kinase inhibitors (TKIs)
Human epidermal growth factor receptor (HER2) inhibitors
Overexpression of HER2 plays a crucial role in cancer development due to its function in stimulating cell growth and differentiation. HER2 inhibitors, including monoclonal antibodies and small-molecule TKIs, have been developed for the treatment of diverse types of cancer, especially breast cancer [97,98,99]. Recent studies have shown that TME-driven non-cell-autonomous mechanisms are involved in resistance to anti-HER2 treatment. Breast tumors next to adipose tissue were found to be more resistant to trastuzumab treatment, which could be associated with the adipose tissue-induced failure of antibody-dependent cellular cytotoxicity. Adipocytes reduce the secretion of interferon-γ (IFNγ) by natural killer cells and induced expression of survival genes in breast tumor cells, leading to trastuzumab treatment failure [100]. Acquired resistance to another anti-HER2 therapy lapatinib was demonstrated in esophageal squamous-cell carcinoma cell lines; this resistance could be associated with CAFs-secreted molecules, including HGF and FGF, which activate the HGF/Met and FGF/FGFR pathways to induce significant resistance to lapatinib [101]. Another study suggested that the spatial proximity of breast ductal carcinoma cells to CAFs also influences lapatinib resistance, as the induction of antiapoptotic Bcl-2/Bcl-x, PI3K/AKT ,and JAK/STAT signaling was observed in lapatinib-treated tumor cells, and this induction was associated with CAF-induced protection by HA in the stroma and with intercellular communication between tumor cells and CAFs through JAK/STAT signaling [102]. In addition, ECM components such as laminin may affect breast ductal carcinoma sensitivity to lapatinib. Tumor cells in niches with laminin-enriched ECM express more antiapoptotic Bcl-2 family proteins and exhibited resistance to lapatinib [103]. These previous studies suggest that multiple non-cell-autonomous mechanisms may be involved in the resistance of tumor cells to anti-HER2 treatment.
EGFR inhibitors
EGFR inhibitors are compounds and antibodies that suppress the activity of either wild-type or mutant EGFR and downstream signaling. As an essential growth pathway, EGFR signaling is generally hyperactive in various types of human cancer [104]. Acquired resistance to cetuximab (CTX), a monoclonal antibody that can block the binding of EGF to EGFR and inhibit the activation of downstream pathways AKT and ERK1/2, was found in head and neck squamous cell carcinomas (HNSCC) [105, 106]. Coculture of HNSCC cells with CAFs significantly reduced CTX-induced growth inhibition, which may be associated with increased expression of MMP-1 in both HNSCC cells and CAFs. The elevation of MMP-1 was due to CAF-derived soluble factors, and MMP-1 can cooperate with other MMPs in the ECM to protect tumor cells from CTX-induced growth inhibition [107]. Another study suggested that CAFs produce HGF in an NF-κB-dependent manner and HGF activates Met-dependent signaling in non-small cell lung cancer. The environmental level of lactic acid promoted HGF production by CAFs and acquired resistance to EGFR TKIs [108].
Gefitinib (GFT) and ERL are EGFR TKIs that are usually used to treat non–small cell lung cancer. It was recently shown that most of the non-cell-autonomous mechanisms of GFT and ERL resistance involve the action of CAFs. The HGF/Met signaling pathway in CAFs is involved in GFT resistance in triple-negative breast cancer, and secreted HGF confers resistance by increasing Met phosphorylation in breast cancer cells [109]. A specific population of CAFs expressing podoplanin was found to be associated with GFT resistance. Patients with higher populations of podoplanin-positive CAFs exhibit worse outcomes after GFT treatment; this finding was supported by the observation of increased ERK1/2 pathway activity in GFT-treated cancer cells cocultured with podoplanin-positive CAFs [110]. In cholangiocarcinoma (CCA), the interaction between cancer cells and CAFs mediated by insulin-like growth factor 2 (IGF2), insulin receptor (IR) and IGF1 receptor (IGF1R) was found to regulate ERL resistance. IGF2 expression in activated CAFs initiates IR/IGF1R-mediated proliferation and survival signaling in cancer cells and induces the production of more IGF2 as a positive feedback to promote CAFs proliferation when CCA tumors are exposed to ERL, leading to an adaptive mechanism by which CCA tumors escape death by ERL treatment [111].
B-Raf inhibitors
B-Raf belongs to the Raf family of kinases and is frequently mutated and hyperactive in multiple types of cancers to facilitate uncontrolled cell growth. The B-Raf V600E mutant inhibitor sorafenib was recently developed to treat liver and renal cancers. The involvement of TME factors, including CAFs, TAMs and tumor-associated neutrophils (TANs), was extensively reported in recent studies. Coculture of CAFs with prostate cancer cells induces sorafenib resistance, which can be overcome by a Bcl-2 inhibitor [112]. In hepatocellular carcinoma (HCC), sorafenib treatment induces SDF1α expression in the stroma, which in turn activates hepatic stellate cells (HSCs) and Gr+ myeloid cell infiltration through a CXCR4-dependent pathway. Sorafenib-activated CXCR4 signaling may contribute to the resistance mechanism by inducing the infiltration of TAMs and regulatory T cells into the TME [113, 114]. In particular, the activation of HSCs induces collagen I and transforming growth factor-β (TGF-β) expression, which increases cell-cell contacts in spheroid culture to cause resistance to sorafenib and cisplatin in HCC cells by regulating EMT activation [115, 116]. It was also reported that TANs affect the sensitivity of HCC to sorafenib. The transformation from normal neutrophils into TANs is a response to the sorafenib-induced hypoxic microenvironment in HCC, which activates the HIF-1α/NF-κB pathway to promote CXCL5 expression. Hypoxia was shown to inhibit TAN apoptosis. As a result, TANs secrete CCL2 and CCL17 to recruit inflammatory macrophages and Treg cells, and cancer tissues in an environment with these cells have a substantially increased tumor microvascular density [117]. Another study with AML showed that sorafenib treatment could induce hypoxia, which evoked the upregulation of Tec family kinase (BMX) expression in AML cells, leading to the activation of STAT5-dependent signaling associated with resistance [118]. Another hypoxia-associated mechanism involves metabolic perturbations that are relevant to the initiation of HCC resistance by hydroxyproline augmentation and accumulation. Under hypoxic conditions, hydroxyproline is tightly related to HIF-dependent tumor phenotypes and glutamine-proline conversion in both normal and tumor cells and confers sorafenib resistance in HCC [119]. It was discovered that HSCs are triggered to myofibroblast-like cells by HCC, and enhanced collagen I expression results in sorafenib resistance and HCC cell migration [115]. This is probably due to the expression of TGF-β1 and CTGF in coculture of HSCs and HCC, which promotes an EMT-like transformation and a collective migration [116].
Vemurafenib was developed to target V600E mutant B-Raf in melanoma. Studies have shown that multiple mechanisms are involved in the acquisition of non-cell-autonomous resistance to vemurafenib [120]. The acidosis in TME was reported to promote the constant phosphorylation of AKT in BRAF-mutated melanoma cells, which activates mTOR signaling and confers vemurafenib resistance [121]. The mature microenvironment increases oxidative stress to augments vemurafenib resistance through a factor secreted by CAFs, sFRP2. As a β-catenin inhibitor, sFRP2 suppresses the production of APE1 via MITF inactivation, losing control of ROS reactions [122].
Besides, there are reports of resistance in thyroid cancer to the combination of sorafenib and vemurafenib, which was associated with the presence of pericytes in the TME. B-Raf inhibitor-treated pericytes secrete TSP-1 to activate the TGFβ1 axis and thus recover ERK, AKT and SMAD3 pathway activity in tumor cells, leading to increased survival and cell death refractoriness [123].
VEGF/VEGFR inhibitors
Bevacizumab is a humanized monoclonal antibody against VEGF-A that blocks angiogenesis in tumors. In ovarian cancer, mutual cross-talk between tumor cells and umbilical vein endothelial cells (UVECs) activates AKT-associated signals in both cell types, thereby inducing the secretion of FGF-2 by HUVECs. The activation of AKT and the secretion of FGF2 were shown to contribute to bevacizumab resistance [124]. In GBM cells, bevacizumab seems to enhance the recruitment of myeloid macrophages, which tended to polarize towards an immunosuppressive M2 phenotype. This could be attributed to the downregulation of macrophage migration inhibitory factor (MIF) in GBM upon VEGF inhibition by bevacizumab [125].
Sunitinib is a VEGFR inhibitor that blocks angiogenesis in multiple types of cancer. The exosome-derived lncRNA LNCARSR sponges tumor suppressive miR-34 and miR-449 to encourage sunitinib resistance [126], which in renal cell carcinoma was found to be associated with MDSCs. Sunitinib increases GM-CSF expression in the TME of renal cell carcinoma, which promotes MDSC survival via a STAT5-dependent pathway. The surviving MDSCs then reduce T cell activity and IFN-γ release to escape immune clearance. In addition, the presence of MDSCs in renal cell carcinoma tissues was correlated with increased expression of proangiogenic factors, suggesting that MDSCs may play a role in antiangiogenic treatment failure [127]. The CD11b+Gr1+ MDSC population was shown to promote resistance to anti-VEGF treatment in multiple types of refractory tumors, and inhibition of this cell population significantly promoted the outcome of anti-VEGF treatment [128]. Comparison of the actions of sunitinib and bevacizumab suggested that sunitinib, but not bevacizumab, could quickly activate the recruitment of macrophages and MDSCs due to the rapid formation of hypoxic conditions. The combination of bevacizumab and sunitinib abolished the recruitment of CD11b+/F4/80+/Gr1- myeloid cells and prolonged the survival of GBM patients compared with sunitinib treatment alone [129].
Vatalanib is a VEGFR inhibitor that is selective for VEGFR-2. A study by Achyut et al. suggested that CD68+ myeloid cells may be involved in the main non-cell-autonomous mechanism of vatalanib resistance. These myeloid cells exhibit CSF1R+ characteristics and can promote angiogenesis and inflammation in the TME of GBM through secreting CXCL7 [130]. The inhibitor of CSF1R could significantly improve vatalanib sensitivity in GBM treatment [131].
Other TKIs
Imatinib is a BCR-ABL inhibitor used for the treatment of leukemia. An early study showed that bone marrow cells might protect the CML cell lines K562 and KU812 from imatinib treatment, perhaps due to the activation of Stat5-mediated NF-κB signaling upon an interaction between bone marrow stroma and CML cells [132]. In the K562 cell line, high HO-1 expression in BMSCs was related to imatinib resistance with considerable changes in signaling, including through the PI3K/AKT pathway, Bcl-2 and the CXCL12/CXCR4 axis [133].
Ibrutinib is an inhibitor of Bruton's tyrosine kinase used for the treatment of MCL. The interaction between the TME and lymphoma cells was shown to be mediated by a signaling network centered on the PI3K/AKT pathway. Sustained high levels of AKT phosphorylation ensured stable mTOR signaling, while integrin-β1 increased the TME lymphoma interaction [134]. Another study identified a population of nurse-like cells in the TME of CLL that had a protective effect on ibrutinib-induced tumor cell apoptosis, which could be partially attributed to the inactivation of natural Bcl-2 antagonist in nurse-like cells [135].
Discussion
As the TME was found to drive significant non-cell-autonomous resistance in multiple types of cancers, treatments that target the TME may regulate the efficacy and effectiveness of antineoplastic drugs; this concept has been well studied and reviewed elsewhere [15, 136], and may provide strategies for new combinations of antineoplastic drugs. For example, locoregional delivery of IL-21 initiated macrophage polarization from the M2 to M1 phenotype, which eliminated immunosuppressive TAMs and induced the T cell response [137]. This type of treatment may be considered as adjuvant therapy to antineoplastic drugs that are susceptible to non-cell-autonomous resistance induced by TAMs. The JAK inhibitor tofacitinib was reported to selectively target the bone marrow microenvironment to block JAK/STAT3 signaling in the stroma [138]. This inhibitory effect works in opposition to BTZ resistance as mentioned above. However, safety and potential side effects of new combinations should be critically evaluated before clinical applications.
Diverse mechanisms by which drugs regulate TME-driven resistance have also been discovered, for instance, studies have found that molecules destroy the TME to improve the release of antineoplastic drugs. Quercetin, a natural compound that blocks the initiation of Wnt16-related signaling in CAFs, can improve the delivery and efficacy of cisplatin [139]. Indeed, recent studies on drug delivery via nanoparticles have shown that drug-containing nanoparticles with supportive components on the surface can destroy the TME of gastric carcinoma and breast cancer, and enhance the efficiency of drug delivery in vitro and in vivo [140, 141]. Nonetheless, it is difficult to predict and judge whether this TME destruction causes harm.
There are several technical difficulties in studying the non-cell-autonomous resistance of cancer cells. Due to tumor heterogenicity, the response to antineoplastic agents may vary among individual tumor cells. The presence of a population of naturally resistant tumor cells makes it difficult to distinguish the non-cell-autonomous and autonomous mechanisms of resistance. Precluding the pre-existence of a resistant population in the tumor will be critical for understanding the role of the TME in acquiring resistance to antineoplastic agents. However, most of the current platforms for studying the drug resistance of tumor cells rely on the in vitro selection of resistant populations, which excludes the influence of other cell types in the TME that may be important in the in vivo acquisition of drug resistance; therefore, these platforms are not suitable for the study of non-cell-autonomous mechanisms of drug resistance. Some attempts to mimic the TME in vitro, such as microenvironment-on-chip, ECM-based tumor cell culture, and tumor-stromal cell coculture, have been made and discussed; however, these systems only partially resemble the actual TME. In vivo selection of resistant populations may identify traits important for non-cell-autonomous drug resistance. A recent study performed in vivo selection of TKI-resistant populations in tumor-bearing mice with acquired resistance to TKI treatment. By isolating cancer cells from resistant tumors in the animal, it was possible to identify whether the resistance was gained by tumor cells themselves or by the influence of the TME [108]. This system could help overcome the current technical problems in studying the non-cell-autonomous mechanism of drug resistance in cancer. In addition, recent studies have attempted to establish human organoids from biopsies to better understand cancer biology and further optimize cancer treatment [142, 143]. Human organoids are cultured ex vivo in 3D, primarily from cancer tissues in individual patients, and therefore retain the signature heterogeneity of the TME, the particular tumor phenotype, and the response to antineoplastic treatment [144]. With these features, human organoids have been proposed as a novel ex vivo tool for estimating the human sensitivity to antineoplastic treatment [145], and they may have broad application in understanding the non-cell-autonomous mechanism of drug resistance in future studies.
Conclusion
Recent studies have identified TME-driven non-cell-autonomous resistance as a critical mechanism that causes refractoriness of cancers and failure of antineoplastic treatment failure. Factors in the TME, including pH, oxygen supply, immune surveillance, fibroblasts and ECM, can respond to drugs and initiate signalings to activate resistance-associated pathways in tumor cells, such as the AKT, mTOR, NF-κB, and STAT3 pathways. Acquired resistance to particular antineoplastic agents may occur via specific non-cell-autonomous mechanisms, while several non-cell-autonomous mechanisms may together contribute to the resistance of tumor cells towards one particular drug. Although the clinical application of TME-targeting molecules to treat cancer resistance requires additional effort in evaluating efficacy, selectivity, and safety, understanding the involvement of TME-driven non-cell-autonomous resistance may prompt trials of novel combinations of currently available antineoplastic agents.
Abbreviations
- AML:
-
Acute myelogenous leukemia
- BC-MSCs:
-
Breast cancer cells with tumor tissue-derived MSCs
- BMSC:
-
Bone marrow stromal cell
- BTZ:
-
Bortezomib
- CAFs:
-
Cancer-associated fibroblasts
- CCA:
-
Cholangiocarcinoma
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myelogenous leukemia
- CTX:
-
Cetuximab
- ECM:
-
Extracellular matrix
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-mesenchymal transition
- ERL:
-
Erlotinib
- EVs:
-
Vesicles
- GBM:
-
Glioblastoma multiforme
- GFT:
-
Gefitinib
- HA:
-
Hyaluronic acid
- HCC:
-
Hepatocellular carcinoma
- HER2:
-
Human epidermal growth factor receptor
- HIF:
-
Hypoxia-inducible factor
- HNSCC:
-
Head and neck squamous cell carcinomas
- HSCs:
-
Hepatic stellate cells
- IGF:
-
Insulin-like growth factor
- IGF1R:
-
Insulin-like growth factor 1 receptor
- IR:
-
IGF2-insulin receptor
- MDSCs:
-
Myeloid-derived suppressor cells
- MGMT:
-
O6-methylguanine-DNA methyltransferase
- miRNAs:
-
microRNAs
- MM:
-
Multiple myeloma
- MSCs:
-
Mesenchymal stem cells
- SASP:
-
Senescence-associated secretory phenotype
- TAMs:
-
Tumor-associated macrophages
- TANs:
-
Tumor-associated neutrophils
- TGF-β:
-
Transforming growth factor-β
- TME:
-
Tumor microenvironment
- TMZ:
-
Temozolomide
- VEGF:
-
Vascular endothelial growth factor
References
Dugger SA, Platt A, Goldstein DB. Drug development in the era of precision medicine. Nat Rev Drug Discov. 2018;17:183–96.
Hoelder S, Clarke PA, Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol Oncol. 2012;6:155–76.
Coussens NP, Braisted JC, Peryea T, Sittampalam GS, Simeonov A, Hall MD. Small-Molecule Screens: A Gateway to Cancer Therapeutic Agents with Case Studies of Food and Drug Administration-Approved Drugs. Pharmacol Rev. 2017;69:479–96.
Garraway LA. Jã¤Nne PA: Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discovery. 2012;2:214–26.
Corso S, Giordano S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: from basic research to a clinical perspective. Cancer Discov. 2013;3:978–92.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26.
Gottesman MM, Tito F, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58.
Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Advanced Pharmaceutical Bulletin. 2017;7:339–48.
Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22:7369–75.
Al-Jamal HAN, Jusoh SAM, Ang Cheng Y, Jamaruddin Mat A, Rosline H, Muhammad Farid J. Silencing of suppressor of cytokine signaling-3 due to methylation results in phosphorylation of STAT3 in imatinib resistant BCR-ABL positive chronic myeloid leukemia cells. Asian Pac J Cancer Prev. 2013;15:4555–61.
Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S. Drug Resistance in Cancer: An Overview. Cancers. 2014;6:1769–92.
Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst. 2000;92:1295–302.
Bonanno L, Favaretto A, Rosell R. Platinum Drugs and DNA Repair Mechanisms in Lung Cancer. Anticancer Research. 2014;34:493–501.
Steinbichler TB, Dudas J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol. 2018;53:156–67.
Tan HY, Wang N, Lam W, Guo W, Feng Y, Cheng YC. Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol Cancer. 2018;17:43.
Hanahan D, Coussens L. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell. 2012;21:309–22.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37.
Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015;368:7–13.
Guri Y, Hall MN. mTOR Signaling Confers Resistance to Targeted Cancer Drugs. Trends Cancer. 2016;2:688–97.
Sun Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016;380:205–15.
Najafi M, Goradel NH, Farhood B, Salehi E, Solhjoo S, Toolee H, Kharazinejad E, Mortezaee K. Tumor microenvironment: Interactions and therapy. J Cell Physiol. 2019;234:5700–21.
Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–72.
Allen E, Mieville P, Warren CM, Saghafinia S, Li L, Peng MW, Hanahan D. Metabolic Symbiosis Enables Adaptive Resistance to Anti-angiogenic Therapy that Is Dependent on mTOR Signaling. Cell Rep. 2016;15:1144–60.
Zhang B, Fu D, Xu Q, Cong X, Wu C, Zhong X, Ma Y, Lv Z, Chen F, Han L, et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat Commun. 2018;9:1723.
Krizhanovsky V, Yon M, Dickins R, Hearn SJ, Miething C, Yee H, Zender L, Lowe S. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–67.
Campisi J, di Fagagna FD. Cellular senescence: when bad things happen to good cells. Nat RevMol Cell Biol. 2007;8:729–40.
Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–36.
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74.
Salony SX, Alves CP, Dey-Guha I, Ritsma L, Boukhali M, Lee JH, Chowdhury J, Ross KN, Haas W. AKT Inhibition Promotes Nonautonomous Cancer Cell Survival. Mol Cancer Ther. 2016;15:142–53.
Kim BH, Yi EH, Ye SK. Signal transducer and activator of transcription 3 as a therapeutic target for cancer and the tumor microenvironment. Arch Pharm Res. 2016;39:1085–99.
Samavati L, Rastogi R, Du W, Huttemann M, Fite A, Franchi L. STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol Immunol. 2009;46:1867–77.
Wang LL, Zhang F, Cui JY, Chen L, Chen YT, Liu BW. CAFs enhance paclitaxel resistance by inducing EMT through the IL-6/JAK2/STAT3 pathway. Oncology Reports. 2018;39:2081–90.
Jeong SK, Kim JS, Lee CG, Park YS, Kim SD, Yoon SO, Han DH, Lee KY, Jeong MH, Jo WS. Tumor associated macrophages provide the survival resistance of tumor cells to hypoxic microenvironmental condition through IL-6 receptor-mediated signals. Immunobiology. 2017;222:55–65.
Yin Y, Yao S, Hu Y, Feng Y, Li M, Bian Z, Zhang J, Qin Y, Qi X, Zhou L, et al. The Immune-microenvironment Confers Chemoresistance of Colorectal Cancer through Macrophage-Derived IL6. Clin Cancer Res. 2017;23:7375–87.
Nagathihalli NS, Castellanos JA, Shi CJ, Beesetty Y, Reyzer ML, Caprioli R, Chen X, Walsh AJ, Skala MC, Moses HL, Merchant NB. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology. 2015;149:1932–+.
Dorayappan KDP, Wanner R, Wallbillich JJ, Saini U, Zingarelli R, Suarez AA, Cohn DE, Selvendiran K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: a novel mechanism linking STAT3/Rab proteins. Oncogene. 2018;37:3806–21.
Li MM, Li M, Yin T, Shi HS, Wen Y, Zhang BL, Chen MH, Xu GC, Ren KX, Wei YQ. Targeting of cancer-associated fibroblasts enhances the efficacy of cancer chemotherapy by regulating the tumor microenvironment. Mol Med Rep. 2016;13:2476–84.
Billottet C, Tuefferd M, Gentien D, Rapinat A, Thiery JP, Broet P, Jouanneau J. Modulation of several waves of gene expression during FGF-1 induced epithelial-mesenchymal transition of carcinoma cells. J Cell Biochem. 2008;104:826–39.
Che Y, Wang J, Li Y, Lu Z, Huang J, Sun S, Mao S, Lei Y, Zang R, Sun N, He J. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 2018;9:759.
Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, Orimo A. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107:20009–14.
Castells M, Milhas D, Gandy C, Thibault B, Rafii A, Delord JP, Couderc B. Microenvironment mesenchymal cells protect ovarian cancer cell lines from apoptosis by inhibiting XIAP inactivation. Cell Death Dis. 2013;4:e887.
Xu H, Zhou Y, Li W, Zhang B, Zhang H, Zhao S, Zheng P, Wu H, Yang J. Tumor-derived mesenchymal-stem-cell-secreted IL-6 enhances resistance to cisplatin via the STAT3 pathway in breast cancer. Oncol Lett. 2018;15:9142–50.
Skolekova S, Matuskova M, Bohac M, Toro L, Durinikova E, Tyciakova S, Demkova L, Gursky J, Kucerova L. Cisplatin-induced mesenchymal stromal cells-mediated mechanism contributing to decreased antitumor effect in breast cancer cells. Cell Commun Signal. 2016;14:7.
Samuel P, Mulcahy LA, Furlong F, McCarthy HO, Brooks SA, Fabbri M, Pink RC, Carter DRF. Cisplatin induces the release of extracellular vesicles from ovarian cancer cells that can induce invasiveness and drug resistance in bystander cells. Philos Trans R Soc Lond B Biol Sci. 2018;373:20170065.
Bliss SA, Sinha G, Sandiford OA, Williams LM, Engelberth DJ, Guiro K, Isenalumhe LL, Greco SJ, Ayer S, Bryan M, et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016;76:5832–44.
Sung PL, Jan YH, Lin SC, Huang CC, Lin H, Wen KC, Chao KC, Lai CR, Wang PH, Chuang CM, et al. Periostin in tumor microenvironment is associated with poor prognosis and platinum resistance in epithelial ovarian carcinoma. Oncotarget. 2016;7:4036–47.
Mhawech-Fauceglia P, Yan L, Sharifian M, Ren X, Liu S, Kim G, Gayther SA, Pejovic T, Lawrenson K. Stromal Expression of Fibroblast Activation Protein Alpha (FAP) Predicts Platinum Resistance and Shorter Recurrence in patients with Epithelial Ovarian Cancer. Cancer Microenviron. 2015;8:23–31.
Dijkgraaf EM, Heusinkveld M, Tummers B, Vogelpoel LT, Goedemans R, Jha V, Nortier JW, Welters MJ, Kroep JR, van der Burg SH. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013;73:2480–92.
Perrotta C, Cervia D, Di Renzo I, Moscheni C, Bassi MT, Campana L, Martelli C, Catalani E, Giovarelli M, Zecchini S, et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated With Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front Immunol. 2018;9:1186.
Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, Wu W, James CD, Sarkaria JN. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-oncology. 2009;11:281–91.
Chen WL, Wang CC, Lin YJ, Wu CP, Hsieh CH. Cycling hypoxia induces chemoresistance through the activation of reactive oxygen species-mediated B-cell lymphoma extra-long pathway in glioblastoma multiforme. J Transl Med. 2015;13:389.
Ge X, Pan MH, Wang L, Li W, Jiang C, He J, Abouzid K, Liu LZ, Shi Z, Jiang BH. Hypoxia-mediated mitochondria apoptosis inhibition induces temozolomide treatment resistance through miR-26a/Bad/Bax axis. Cell Death Dis. 2018;9:1128.
Ngo MT, Harley BAC. Perivascular signals alter global gene expression profile of glioblastoma and response to temozolomide in a gelatin hydrogel. Biomaterials. 2019;198:122–34.
Chen W, Wang D, Du X, He Y, Chen S, Shao Q, Ma C, Huang B, Chen A, Zhao P, et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med Oncol. 2015;32:43.
Orimo A, Weinberg RA. Stromal fibroblasts in cancer - A novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601.
Udagawa T, Wood M. Tumor-stromal cell interactions and opportunities for therapeutic intervention. Curr Opin Pharmacol. 2010;10:369–74.
Wei L, Ye H, Li G, Lu Y, Zhou Q, Zheng S, Lin Q, Liu Y, Li Z, Chen R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018;9:1065.
Lee J, Yakubov B, Ivan C, Jones DR, Caperell-Grant A, Fishel M, Cardenas H, Matei D. Tissue transglutaminase activates cancer-associated fibroblasts and contributes to gemcitabine resistance in pancreatic cancer. Neoplasia. 2016;18:689–98.
Binenbaum Y, Fridman E, Yaari Z, Milman N, Schroeder A, Ben David G, Shlomi T, Gil Z. Transfer of miRNA in macrophages-derived exosomes induces drug resistance of pancreatic adenocarcinoma. Cancer Res. 2018;78:5287–99.
Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, Simon B, Ryffel B, Connat JL, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19:57–64.
Mak PY, Mak DH, Mu H, Shi Y, Ruvolo P, Ruvolo V, Jacamo R, Burks JK, Wei W, Huang X, et al. Apoptosis repressor with caspase recruitment domain is regulated by MAPK/PI3K and confers drug resistance and survival advantage to AML. Apoptosis. 2014;19:698–707.
OR E, Dhami SPS, Baev DV, Ortutay C, Halpin-McCormick A, Morrell R, Santocanale C, Samali A, Quinn J, O'Dwyer ME, Szegezdi E. Repression of Mcl-1 expression by the CDC7/CDK9 inhibitor PHA-767491 overcomes bone marrow stroma-mediated drug resistance in AML. Sci Rep. 2018;8:15752.
Piya S, Andreeff M, Borthakur G. Targeting autophagy to overcome chemoresistance in acute myleogenous leukemia. Autophagy. 2017;13:214–5.
Sironi S, Wagner M, Kuett A, Drolle H, Polzer H, Spiekermann K, Rieger C, Fiegl M. Microenvironmental hypoxia regulates FLT3 expression and biology in AML. Sci Rep. 2015;5:17550.
Hoellenriegel J, Zboralski D, Maasch C, Rosin NY, Wierda WG, Keating MJ, Kruschinski A, Burger JA. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood. 2014;123:1032–9.
Rosich L, Saborit-Villarroya I, Lopez-Guerra M, Xargay-Torrent S, Montraveta A, Aymerich M, Villamor N, Campo E, Perez-Galan P, Roue G, Colomer D. The phosphatidylinositol-3-kinase inhibitor NVP-BKM120 overcomes resistance signals derived from microenvironment by regulating the Akt/FoxO3a/Bim axis in chronic lymphocytic leukemia cells. Haematologica. 2013;98:1739–47.
Trimarco V, Ave E, Facco M, Chiodin G, Frezzato F, Martini V, Gattazzo C, Lessi F, Giorgi CA, Visentin A, et al. Cross-talk between chronic lymphocytic leukemia (CLL) tumor B cells and mesenchymal stromal cells (MSCs): implications for neoplastic cell survival. Oncotarget. 2015;6:42130–49.
Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14:276–86.
Tu B, Zhu J, Liu S, Wang L, Fan Q, Hao Y, Fan C, Tang TT. Mesenchymal stem cells promote osteosarcoma cell survival and drug resistance through activation of STAT3. Oncotarget. 2016;7:48296–308.
Yang H, Zheng Y, Zhang Y, Cao Z, Jiang Y. Mesenchymal stem cells derived from multiple myeloma patients protect against chemotherapy through autophagy-dependent activation of NF-κB signaling. Leuk Res. 2017;60:82–8.
Ramachandran IR, Condamine T, Lin C, Herlihy SE, Garfall A, Vogl DT, Gabrilovich DI, Nefedova Y. Bone marrow PMN-MDSCs and neutrophils are functionally similar in protection of multiple myeloma from chemotherapy. Cancer Letters. 2016;371:117–24.
Hsu YL, Hung JY, Tsai EM, Wu CY, Ho YW, Jian SF, Yen MC, Chang WA, Hou MF, Kuo PL. Benzyl butyl phthalate increases the chemoresistance to doxorubicin/cyclophosphamide by increasing breast cancer-associated dendritic cell-derived CXCL1/GROalpha and S100A8/A9. Oncol Rep. 2015;34:2889–900.
Mendoza-Rodriguez M, Arevalo Romero H, Fuentes-Panana EM, Ayala-Sumuano JT, Meza I. IL-1beta induces up-regulation of BIRC3, a gene involved in chemoresistance to doxorubicin in breast cancer cells. Cancer Lett. 2017;390:39–44.
Zhang L, Hannay JA, Liu J, Das P, Zhan M, Nguyen T, Hicklin DJ, Yu D, Pollock RE, Lev D. Vascular endothelial growth factor overexpression by soft tissue sarcoma cells: implications for tumor growth, metastasis, and chemoresistance. Cancer Res. 2006;66:8770–8.
Cheteh EH, Augsten M, Rundqvist H, Bianchi J, Sarne V, Egevad L, Bykov VJ, Östman A, Wiman KG. Human cancer-associated fibroblasts enhance glutathione levels and antagonize drug-induced prostate cancer cell death. Cell Death Dis. 2017;8:e2848.
Zhang D, Shi Z, Li M, Mi J. Hypoxia-induced miR-424 decreases tumor sensitivity to chemotherapy by inhibiting apoptosis. Cell Death Dis. 2014;5:e1301.
Li X, Shepard HM, Cowell JA, Zhao C, Osgood RJ, Rosengren S, Blouw B, Garrovillo SA, Pagel MD, Whatcott CJ, et al. Parallel Accumulation of Tumor Hyaluronan, Collagen and Other Drivers of Tumor Progression. Clin Cancer Res. 2018;24:4798–807.
Gangoda L, Keerthikumar S, Fonseka P, Edgington LE, Ang CS, Ozcitti C, Bogyo M, Parker BS, Mathivanan S. Inhibition of cathepsin proteases attenuates migration and sensitizes aggressive N-Myc amplified human neuroblastoma cells to doxorubicin. Oncotarget. 2015;6:11175–90.
Joyce MH, Lu C, James ER, Hegab R, Allen SC, Suggs LJ, Brock A. Phenotypic basis for matrix stiffness-dependent chemoresistance of breast cancer cells to doxorubicin. Front Oncol. 2018;8:337.
Ebata T, Mitsui Y, Sugimoto W, Maeda M, Araki K, Machiyama H, Harada I, Sawada Y, Fujita H, Hirata H, Kawauchi K. Substrate Stiffness Influences Doxorubicin-Induced p53 Activation via ROCK2 Expression. Biomed Res Int. 2017;2017:5158961.
Lopes-Rodrigues V, Di Luca A, Sousa D, Seca H, Meleady P, Henry M, Lima RT, O'Connor R, Vasconcelos MH. Multidrug resistant tumour cells shed more microvesicle-like EVs and less exosomes than their drug-sensitive counterpart cells. Biochim Biophys Acta. 2016;1860:618–27.
Chen WX, Liu XM, Lv MM, Chen L, Zhao JH, Zhong SL, Ji MH, Hu Q, Luo Z, Wu JZ, Tang JH. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS One. 2014;9:e95240.
Huber RM, Lucas JM, Gomez-Sarosi LA, Coleman I, Zhao S, Coleman R. Nelson PS: DNA damage induces GDNF secretion in the tumor microenvironment with paracrine effects promoting prostate cancer treatment resistance. Oncotarget. 2015;6:2134–47.
Yu T, Yang G, Hou Y, Tang X, Wu C, Wu XA, Guo L, Zhu Q, Luo H, Du YE, et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene. 2017;36:2131–45.
Milito AD, Fais S. Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncology. 2005;1:779–86.
Mahoney BP, Raghunand N, Baggett B, Gillies RJ. Tumor acidity, ion trapping and chemotherapeutics I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem Pharmacol. 2003;66:1207–18.
Fan S, Niu Y, Tan N, Wu Z, Wang Y, You H, Ke R, Song J, Shen Q, Wang W, et al. LASS2 enhances chemosensitivity of breast cancer by counteracting acidic tumor microenvironment through inhibiting activity of V-ATPase proton pump. Oncogene. 2013;32:1682–90.
Bar-Natan M, Stroopinsky D, Luptakova K, Coll MD, Apel A, Rajabi H, Pyzer AR, Palmer K, Reagan MR, Nahas MR, et al. Bone marrow stroma protects myeloma cells from cytotoxic damage via induction of the oncoprotein MUC1. Br J Haematol. 2017;176(6):929–38.
De Beule N, De Veirman K, Maes K, De Bruyne E, Menu E, Breckpot K, De Raeve H, Van Rampelbergh R, Van Ginderachter JA, Schots R, et al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation. J Pathol. 2017;241:534–46.
Santoni M, Massari F, Amantini C, Nabissi M, Maines F, Burattini L, Berardi R, Santoni G, Montironi R, Tortora G, Cascinu S. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol Immunother. 2013;62:1757–68.
Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25:2465–79.
Cosse JP, Ronvaux M, Ninane N, Raes MJ. Michiels C: Hypoxia-induced decrease in p53 protein level and increase in c-jun DNA binding activity results in cancer cell resistance to etoposide. Neoplasia. 2009;11:976–86.
Sermeus A, Rebucci M, Fransolet M, Flamant L, Desmet D, Delaive E, Arnould T, Michiels C. Differential effect of hypoxia on etoposide-induced DNA damage response and p53 regulation in different cell types. J Cell Physiol. 2013;228:2365–76.
Dudley AC, Shih SC, Cliffe AR, Hida K, Klagsbrun M. Attenuated p53 activation in tumour-associated stromal cells accompanies decreased sensitivity to etoposide and vincristine. Br J Cancer. 2008;99:118–25.
Shin K, Klosterhoff BS, Han B. Characterization of Cell-Type-Specific Drug Transport and Resistance of Breast Cancers Using Tumor-Microenvironment-on-Chip. Mol Pharm. 2016;13:2214–23.
Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene. 2000;19:4319–27.
Baselga J. Targeting tyrosine kinases in cancer: The second wave. Science. 2006;312:1175–8.
Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–48.
Park S, Jiang ZJ, Mortenson ED, Deng LF, Radkevich-Brown O, Yang XM, Sattar H, Wang Y, Brown NK, Greene M, et al. The Therapeutic Effect of Anti-HER2/neu Antibody Depends on Both Innate and Adaptive Immunity. Cancer Cell. 2010;18:160–70.
Duong MN, Cleret A, Matera EL, Chettab K, Mathé D, Valsesia-Wittmann S, Clémenceau B, Dumontet C. Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Res. 2015;17:57.
Saito S, Morishima K, Ui T, Hoshino H, Matsubara D, Ishikawa S, Aburatani H, Fukayama M, Hosoya Y, Sata N, et al. The role of HGF/MET and FGF/FGFR in fibroblast-derived growth stimulation and lapatinib-resistance of esophageal squamous cell carcinoma. BMC Cancer. 2015;15:82.
Marusyk A, Tabassum DP, Janiszewska M, Place AE, Trinh A, Rozhok AI, Pyne S, Guerriero JL, Shu S, Ekram M, et al. Spatial Proximity to Fibroblasts Impacts Molecular Features and Therapeutic Sensitivity of Breast Cancer Cells Influencing Clinical Outcomes. Cancer Res. 2016;76(22):6495–506.
Zoeller JJ, Bronson RT, Selfors LM, Mills GB, Brugge JS. Niche-localized tumor cells are protected from HER2-targeted therapy via upregulation of an anti-apoptotic program in vivo. NPJ Breast Cancer. 2017;3:18.
Ono M, Kuwano M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin Cancer Res. 2006;12:7242–51.
Li SQ, Schmitz KR, Jeffrey PD, Wiltzius JJW, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–11.
Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116–27.
Johansson AC, Ansell A, Jerhammar F, Lindh MB, Grénman R, Munck-Wikland E, Östman A, Roberg K. Cancer-associated fibroblasts induce matrix metalloproteinase-mediated cetuximab resistance in head and neck squamous cell carcinoma cells. Mol Cancer Res. 2012;10:1158–68.
Apicella M, Giannoni E, Fiore S, Ferrari KJ, Fernandez-Perez D, Isella C, Granchi C, Minutolo F, Sottile A, Comoglio PM, et al. Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metabolism. 2018;28:848–+
Mueller KL, Madden JM, Zoratti GL, Kuperwasser C, List K, Boerner JL. Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of Met. Breast Cancer Res. 2012;14:R104.
Yoshida T, Ishii G, Goto K, Neri S, Hashimoto H, Yoh K, Niho S, Umemura S, Matsumoto S, Ohmatsu H, et al. Podoplanin-positive cancer-associated fibroblasts in the tumor microenvironment induce primary resistance to EGFR-TKIs in lung adenocarcinoma with EGFR mutation. Clin Cancer Res. 2015;21:642–51.
Vaquero J, Lobe C, Tahraoui S, Claperon A, Mergey M, Merabtene F, Wendum D, Coulouarn C, Housset C, Desbois-Mouthon C, et al. The IGF2/IR/IGF1R Pathway in Tumor Cells and Myofibroblasts Mediates Resistance to EGFR Inhibition in Cholangiocarcinoma. Clin Cancer Res. 2018;24:4282–96.
Kharaziha P, Rodriguez P, Li Q, Rundqvist H, Björklund AC, Augsten M, Ullén A, Egevad L, Wiklund P, Nilsson S, et al. Targeting of distinct signaling cascades and cancer-associated fibroblasts define the efficacy of Sorafenib against prostate cancer cells. Cell Death Dis. 2012;3:e262.
Chen Y, Huang Y, Reiberger T, Duyverman AM, Huang P, Samuel R, Hiddingh L, Roberge S, Koppel C, Lauwers GY, et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59:1435–47.
Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, Ochiai H, Kitahara S, Unan EC, Reddy TP, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61:1591–602.
Song Y, Kim SH, Kim KM, Choi EK, Kim J, Seo HR. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci Rep. 2016;6:36750.
Khawar IA, Park JK, Jung ES, Lee MA, Chang S, Kuh HJ. Three Dimensional Mixed-Cell Spheroids Mimic Stroma-Mediated Chemoresistance and Invasive Migration in hepatocellular carcinoma. Neoplasia. 2018;20:800–12.
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, Fan J, Cao Y, Dai Z, Zhou J. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150:1646–+.
van Oosterwijk JG, Buelow DR, Drenberg CD, Vasilyeva A, Li L, Shi L, Wang YD, Finkelstein D, Shurtleff SA, Janke LJ, et al. Hypoxia-induced upregulation of BMX kinase mediates therapeutic resistance in acute myeloid leukemia. J Clin Invest. 2018;128:369–80.
Tang L, Zeng J, Geng PY, Fang CN, Wang Y, Sun MJ, Wang CS, Wang J, Yin PY, Hu CX, et al. Global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular carcinoma. Clin Cancer Res. 2018;24:474–85.
Xing MZ, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B, Yip L, Mian C, Vianello F, Tuttle RM, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. Jama-J Am Med Assoc. 2013;309:1493–501.
Ruzzolini J, Peppicelli S, Andreucci E, Bianchini F, Margheri F, Laurenzana A, Fibbi G, Pimpinelli N, Calorini L. Everolimus selectively targets vemurafenib resistant BRAF(V600E) melanoma cells adapted to low pH. Cancer Lett. 2017;408:43–54.
Kaur A, Webster MR, Marchbank K, Behera R, Ndoye A, Kugel CH, Dang VM, Appleton J, O'Connell MP, Cheng P, et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature. 2016;532:250–4.
Prete A, Lo AS, Sadow PM, Bhasin SS, Antonello ZA, Vodopivec DM, Ullas S, Sims JN, Clohessy JG, Dvorak AM, et al. Pericytes elicit resistance to vemurafenib and sorafenib therapy in thyroid carcinoma via the TSP-1/TGFβ1 axis. Clin Cancer Res. 2018;24:6078–97.
Guerrouahen BS, Pasquier J, Kaoud NA, Maleki M, Beauchamp MC, Yasmeen A, Ghiabi P, Lis R, Vidal F, Saleh A, et al. Akt-activated endothelium constitutes the niche for residual disease and resistance to bevacizumab in ovarian cancer. Mol Cancer Ther. 2014;13:3123–36.
Castro BA, Flanigan P, Jahangiri A, Hoffman D, Chen W, Kuang R, De Lay M, Yagnik G, Wagner JR, Mascharak S, et al. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene. 2017;36:3749–59.
Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y, Chen W, Liu F, Sun W, Li XF, et al. Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell. 2016;29:653–68.
Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy ☆. Int Immunopharmacol. 2011;11:856–61.
Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–20.
Piao Y, Liang J, Holmes L, Zurita AJ, Henry V, Heymach JV, de Groot JF. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-Oncol. 2012;14:1379–92.
Achyut BR, Shankar A, Iskander AS, Ara R, Angara K, Zeng P, Knight RA, Scicli AG, Arbab AS. Bone marrow derived myeloid cells orchestrate antiangiogenic resistance in glioblastoma through coordinated molecular networks. Cancer Lett. 2015;369:416–26.
Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, Joyce JA. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene. 2017;36:6049–58.
Xu X, Zhang X, Liu Y, Yang L, Huang S, Lu L, Wang S, Guo Q, Zhao L. BM microenvironmental protection of CML cells from imatinib through Stat5/NF-kappa B signaling and reversal by Wogonin. Oncotarget. 2016;7:24436–54.
Liu P, Ma D, Yu Z, Zhe N, Ren M, Wang P, Yu M, Huang J, Fang Q, Wang J. Overexpression of heme oxygenase-1 in bone marrow stromal cells promotes microenvironment-mediated imatinib resistance in chronic myeloid leukemia. Biomed Pharmacother. 2017;91:21–30.
Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, Zhang L, Fu K, Bi C, Li J, et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat Commun. 2017;8:14920.
Boissard F, Fournie JJ, Quillet-Mary A, Ysebaert L, Poupot M. Nurse-like cells mediate ibrutinib resistance in chronic lymphocytic leukemia patients. Blood Cancer J. 2015;5:e355.
Zhang Z, Yu X, Wang Z, Wu P, Huang J. Anthracyclines potentiate anti-tumor immunity: A new opportunity for chemoimmunotherapy. Cancer Lett. 2015;369:331–5.
Xu M, Liu M, Du X, Li S, Li H, Li X, Li Y, Wang Y, Qin Z, Fu YX, Wang S. Intratumoral delivery of IL-21 Overcomes Anti-Her2/Neu resistance through shifting tumor-associated macrophages from M2 to M1 phenotype. J Immunol. 2015;194:4997–5006.
Lam C, Ferguson ID, Mariano MC, Lin YT, Murnane M, Liu H, Smith GA, Wong SW, Taunton J, Liu JO, et al. Repurposing tofacitinib as an anti-myeloma therapeutic to reverse growth-promoting effects of the bone marrow microenvironment. Haematologica. 2018;103:1218–28.
Hu K, Miao L, Goodwin TJ, Li J, Liu Q, Huang L. Quercetin remodels the tumor microenvironment to improve the permeation, retention, and antitumor effects of nanoparticles. ACS Nano. 2017;11:4916–25.
Amini MA, Abbasi AZ, Cai P, Lip H, Gordijo CR, Li J, Chen B, Zhang L, Rauth AM, Wu XY. Combining Tumor Microenvironment Modulating Nanoparticles with Doxorubicin to Enhance Chemotherapeutic Efficacy and Boost Antitumor Immunity. J Natl Cancer Inst. 2019;111.
Fang J, Zhang S, Xue X, Zhu X, Song S, Wang B, Jiang L, Qin M, Liang H, Gao L. Quercetin and doxorubicin co-delivery using mesoporous silica nanoparticles enhance the efficacy of gastric carcinoma chemotherapy. Int J Nanomedicine. 2018;13:5113–26.
Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920–6.
Lampis A, Carotenuto P, Vlachogiannis G, Cascione L, Hedayat S, Burke R, Clarke P, Bosma E, Simbolo M, Scarpa A, et al. MIR21 drives resistance to heat shock protein 90 inhibition in cholangiocarcinoma. Gastroenterology. 2018;154:1066–+.
Papapetrou EP. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat Med. 2016;22:1392–401.
Mazzarella L, Curigliano G. A new approach to assess drug sensitivity in cells for novel drug discovery. Expert Opin Drug Discov. 2018;13:339–46.
Cloosen S, Gratama J, van Leeuwen EBM, Senden-Gijsbers bLMG, Oving EBH, von Mensdorff-Pouilly S, Tarp MA, Mandel U, Clausen H, Germeraad WTV, Bos GMJ. Cancer specific Mucin-1 glycoforms are expressed on multiple myeloma. Br J Haematol. 2006;135:513–6.
Yin L, Ahmad R, Kosugi M, Kufe T, Vasir B, Avigan D, Kharbanda S, Kufe D. Survival of human multiple myeloma cells is dependent on MUC1 C-terminal transmembrane subunit oncoprotein function. Mol Pharmacol. 2010;78:166–74.
Kharbanda A, Rajabi H, Jin C, Tchaicha J, Kikuchi E, Wong K-K, Kufe D. Targeting the oncogenic MUC1-C protein inhibits mutant EGFR-mediated signaling and survival in non-small cell lung cancer cells. Clin Cancer Re. 2014;20:5423–34.
Emeagi PU, Maenhout S, Dang N, Heirman C, Thielemans K, Breckpot K. Downregulation of Stat3 in melanoma: reprogramming the immune microenvironment as an anticancer therapeutic strategy. Gene Therapy. 2013;20:1085–92.
Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J Hematol Oncol. 2011;4:13.
Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T, Stilgenbauer S, Jumaa H, Veelken H, Zirlik K. Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood. 2010;115:4497–506.
Koch H, Wilhelm M, Ruprecht B, Beck S, Frejno M, Klaeger S, Kuster B. Phosphoproteome profiling reveals molecular mechanisms of growth-factor-mediated kinase inhibitor resistance in EGFR-overexpressing cancer cells. J Proteome Res. 2016;15:4490–504.
Murakami A, Takahashi F, Nurwidya F, Kobayashi I, Minakata K, Hashimoto M, Nara T, Kato M, Tajima K, Shimada N, et al. Hypoxia increases gefitinib-resistant lung cancer stem cells through the activation of insulin-like growth factor 1 receptor. Plos One. 2014;9:e86459.
Lee J, Condello S, Yakubov B, Emerson R, Caperell-Grant A, Hitomi K, Xie J, Matei D. Tissue transglutaminase mediated tumor-stroma interaction promotes pancreatic cancer progression. Clin Cancer Res. 2015;21:4482–93.
Guo L, Zheng J, Yu T, Liu Y, Duo L. Elevated expression of SATB1 is involved in pancreatic tumorigenesis and is associated with poor patient survival. Mol Med Rep. 2017;16:8842–8.
Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, Gil Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene. 2014;33:3812–9.
Hamada S, Masamune A, Miura S, Satoh K, Shimosegawa T. MiR-365 induces gemcitabine resistance in pancreatic cancer cells by targeting the adaptor protein SHC1 and pro-apoptotic regulator BAX. Cellular Signalling. 2014;26:179–85.
Grugan KD, Miller CG, Yao Y, Michaylira CZ, Ohashi S, Klein-Szanto AJ, Diehl A, Herlyn M, Han M, Nakagawa H, Rustgi AK. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc Natl Acad Sci U S A. 2010;107:11026–31.
Siddiquee KAZ, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18:254–67.
Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–10.
Zhou S-L, Zhou Z-J, Hu Z-Q, Li X, Huang X-W, Wang Z, Fan J, Dai Z, Zhou J. CXCR2/CXCL5 axis contributes to epithelial–mesenchymal transition of HCC cells through activating PI3K/Akt/GSK-3β/Snail signaling. Cancer Lett. 2015;358:124–35.
Zhou SL, Dai Z, Zhou ZJ, Chen Q, Wang Z, Xiao YS, Hu ZQ, Huang XY, Yang GH, Shi YH, et al. CXCL5 contributes to tumor metastasis and recurrence of intrahepatic cholangiocarcinoma by recruiting infiltrative intratumoral neutrophils. Carcinogenesis. 2014;35:597–605.
Hsieh CH, Lee CH, Liang JA, Yu CY, Shyu WC. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol Rep. 2010;24:1629–36.
Gielen PR, Aftab Q, Ma N, Chen VC, Hong XT, Lozinsky S, Naus CC. Sin WC: Connexin43 confers Temozolomide resistance in human glioma cells by modulating the mitochondrial apoptosis pathway. Neuropharmacology. 2013;75:539–48.
Xu GY, Li JY. Differential expression of PDGFRB and EGFR in microvascular proliferation in glioblastoma. Tumor Biol. 2016;37:10577–86.
Acknowledgements
Not applicable.
Funding
This study was supported by the Special Projects of Cooperation between Jilin University and Jilin Province in China (SXGJSF2017-1), the “13th Five-year” Science and Technology Projects from Education Department in Jilin Province (JJKH20190108KJ), Research Grant Council, HKSAR (Project code: RGC GRF 17152116), Commissioner for Innovation Technology, HKSAR (Project code: ITS/091/16FX) and Health and Medical Research Fund (Project code: 15162961).
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
YDQ, BD and HYT retrieved the data and drafted the manuscript. DW, NW and YBF initiated the study and drafted and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Qu, Y., Dou, B., Tan, H. et al. Tumor microenvironment-driven non-cell-autonomous resistance to antineoplastic treatment. Mol Cancer 18, 69 (2019). https://doi.org/10.1186/s12943-019-0992-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-0992-4