
Abstract
Protein tyrosine kinase (PTK) is one of the major signaling enzymes in the process of cell signal transduction, which catalyzes the transfer of ATP-γ-phosphate to the tyrosine residues of the substrate protein, making it phosphorylation, regulating cell growth, differentiation, death and a series of physiological and biochemical processes. Abnormal expression of PTK usually leads to cell proliferation disorders, and is closely related to tumor invasion, metastasis and tumor angiogenesis. At present, a variety of PTKs have been used as targets in the screening of anti-tumor drugs. Tyrosine kinase inhibitors (TKIs) compete with ATP for the ATP binding site of PTK and reduce tyrosine kinase phosphorylation, thereby inhibiting cancer cell proliferation. TKI has made great progress in the treatment of cancer, but the attendant acquired acquired resistance is still inevitable, restricting the treatment of cancer. In this paper, we summarize the role of PTK in cancer, TKI treatment of tumor pathways and TKI acquired resistance mechanisms, which provide some reference for further research on TKI treatment of tumors.
Similar content being viewed by others

Background
Malignant tumors have always been a serious threat to human life. Although the diagnostic and therapeutic levels have improved, many kinds of tumor survival rates have remained low. Anti-tumor research remains a challenging and significant field in the life sciences today. At present, the commonly used anti-tumor drugs are cytotoxic drugs. Cytotoxic cancer drugs are usually of high acute toxicity, which have the disadvantages of poor selectivity, strong side effects and easy to produce drug resistance [1]. In recent years, with the rapid progress of life science research, signal transduction in tumor cells, cell cycle regulation, induction of apoptosis, angiogenesis, the interaction of cells and extracellular matrix and other basic processes are being gradually clarified [2]. In addition, it is pointed out that these drugs may be very specific to certain cellular targets (e.g. DNA, tubulin) present in cancer as well as in normal cells. Using the key enzymes of cell signal transduction pathway associated with tumor cell differentiation and proliferation as drug screening targets, and developing high efficiency, low toxicity and specificity of new anticancer drugs acting on specific targets have become important direction of research and development of antitumor drugs today [3].
Protein tyrosine kinase (PTK) is a class of proteins with tyrosine kinase activity that catalyzes the transfer of phosphate groups on ATP to the tyrosine residues of many important proteins, making proteins phosphorylation, then transferring signal to regulate cell growth, differentiation, death and a series of physiological and biochemical processes [4]. PTK disorders can cause a series of diseases in the body. Previous studies have shown that more than 50% of the proto-oncogene and oncogene products have PTK activities, their abnormal expression will lead to cell proliferation regulation disorders, causing tumorigenesis finally [5]. In addition, PTK abnormal expression is also associated with tumor invasion and metastasis, tumor neovascularization and tumor chemotherapy resistance [6]. Therefore, PTK as a target for drug research and development has become a hot spot for anti-tumor drug research.
By the end of the century, so-called targeted cancer therapy with reduced side effects was made possible by advances such as specific monoclonal antibodies that bound to unique epitopes on the surface of cancerous cells and by small molecules. International major research institutions, pharmaceutical groups have attached great importance to PTK as the target drug research, such as selective tyrosine kinase inhibitors (TKI) that affected specific molecular pathways up-regulated in certain cancers [1]. The pro-spective molecular profiling of cancers to find such ‘driver’ abnormalities became feasible in clinical practice, allowing for routine genotype-directed rather than empiric therapy. In 2001, the first TKI drug imatinib was quickly approved by the FDA and opened up new ideas for cancer treatment. Until 2018 or beyond, a total of more than 20 kinds of TKI approved by the FDA [7,8,9,10,11,12,13], drug-related information as listed in Table 1. These drugs have high selectivity, high efficacy, low side effects, ease of preparation, and have superiority in the treatment of chronic myeloid leukemia(CML), non-small cell lung cancer(NSCLC), renal cell carcinoma(RCC) than traditional cytotoxic antineoplastic agents [14], some have become the first-line drug for the treatment of cancer.
Although TKI has made great strides in the treatment of cancer, it still faces some challenges. Because even in highly sensitive patients with TKI, tumor cells can always be self-adjusting, looking for a way out, to avoid TKI target, acquired resistance and the progress of the disease is still inevitable [15]. The median effective time for TKI therapy was only 5 to 9 months [16]. With our increased understanding of the spectrum of acquired resistance to TKI, major changes in how we conduct clinical research in this setting are now underway. In order to fight against resistance to TKI, the investigators should further study the mechanisms of their resistance and suggest a regimen that prevents or treats their resistance.
PTK and tumor
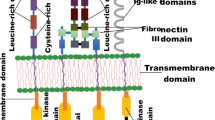
PTK is only found in multicellular animals and is an enzyme that activates and regulates cell proliferation signaling pathways. According to its structure, it can be divided into two categories: Receptor PTK (RTK) and Non-receptor PTK (NRTK). These two types of PTK can be further divided according to their structural homology multiple enzymes. Analysis of human genome data shows that there are 518 kinase genes in the human body, of which 90 have been identified PTK, including RTK 58 species and NRTK 32 species [17].
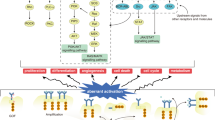
RTK includes epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR) and insulin receptor (InsR) family and so on. They usually have an extracellular domain that binds to a specific ligand, a transmembrane region, and an intracellular kinase domain that selectively binds to and phosphorylates the substrate [18]. RTK can bind to ligands and phosphorylate tyrosine residues of target proteins and transmit information through PI3K/AKT/mTOR; RAS/RAF/MEK/ERK; PLCγ/PKC and other signaling pathways to activate a series of biochemical reactions; or different information combined to cause a comprehensive cellular response (such as cell proliferation) (Fig. 1) [19]. Clinical studies in cancer have shown that these receptors and their ligands are important in many tumors, and many cancers have over-expressed growth factors that cause excessive tyrosine phosphorylation signal into cells [20].

Cell signaling pathways induced by RTK. RTK can bind to ligands and phosphorylate tyrosine residues of target proteins and transmit information through PI3K/AKT/mTOR; RAS/RAF/MEK/ERK; PLCγ/PKC and other signaling pathways to activate a series of biochemical reactions; or different information combined to cause a comprehensive cellular response, including cell proliferation, cell migration and tumor formation
NRTKs generally have no extracellular structure. They are usually coupled to the cell membrane or present in the cytoplasm, including Abl kinase, Src kinase family and so on [21, 22]. NRTK performs signal transduction primarily through cytokine receptors, T-cell receptors and other signaling pathways. T lymphocyte receptors, B lymphocyte receptors, immunoglobulin receptors and so on can recruit NRTK, and then through tyrosine phosphorylation to form signal transduction complex, and then activate the downstream signal transduction, promote cells proliferation, lead to the formation of tumors [23].
Overexpression of the PTK gene enhances the activity of PTK and changes its downstream signaling pathways, causing cell proliferation disorders and eventually leading to tumor formation [5]; mutations in tumor tissue may cause PTK to spontaneously activate in the absence of a stimulus source or appear abnormal growth rate [24]; clinical studies have shown that PTK overexpression or decreased expression can show the biological characteristics of the tumor or predict the response to treatment and survival [25].
EGFR family
The human EGFR gene is localized to the short arm of chromosome 7 (7p12.3-pl2.1), which encodes a product consisting of 1210 amino acids with a molecular weight of about 170 kb. EGFR is a cell surface receptor and plays a pivotal role in regulating survival and apoptosis of epithelial cells and tumors of epithelial cell origin. Overexpression of EGFR and its ligands is present in a variety of epithelial tumor cells such as lung cancer, breast cancer, bladder cancer, prostate cancer and squamous cell carcinoma of the head and neck [26,27,28,29]. It is a member of the ErbB family, a group of four receptor tyrosine kinases sharing similarities in structures and functions: ErbB1 (EGFR or HER1), ErbB2 (HER2), ErbB3 (HER3) andErbB4 (HER4). In breast cancer, overexpression of HER2 is found in approximately 10%–30% of patients and is associated with reduced survival [30]. In addition, EGFR deletion can also be detected in malignant gliomas, NSCLC, breast cancer, medulloblastoma and ovarian cancer [31, 32]. The most common EGFR deletion mutant is EGFR VIII. EGFR VIII lose ligand binding region, but can activate tyrosine kinase itself, stimulate the activation of downstream signaling pathways, and not dependent on its ligand binding region [33]. EGFR overexpression and/or mutation through signal transduction lead to cell growth out of control and malignancy in many tumors. In patients with high expression of EGFR, the degree of malignancy is high, the recurrence interval is short, the recurrence rate is high, the survival time of the patients is short [34].
VEGFR family
VEGFR family members include VEGFR1, VEGFR2 and VEGFR3. The family of receptors has 7 immunoglobulin-like domains in the extracellular domain and a hydrophilic insert sequence in the intracellular tyrosine kinase region [35]. In the malignant growth and metastasis of solid tumors, neovascularization of the tumor plays a very important role, providing the necessary nutrients and oxygen for the tumor growth [36]. VEGF plays an important role in the proliferation, migration, and vascularization of endothelial cells as the most powerful vascular penetrant and endothelium-specific mitotic source [37]. There was a significant positive correlation between the VEGFR expression level and the degree of vascularization and malignancy of tumor tissue.
VEGF is mainly acting on high affinity of the recipient VEGFR1 and VEGFR2 in the vascular endothelial cells and play its biological role, both have different signal transduction pathways [38, 39]. Among them, VEGFR2 is the most important in mediating the biological effect of VEGF, which is closely related to cell chemotaxis, cell division and act in recombination [40]. VEGFR1 has stronger affinity binding to VEGF, and phosphorylation is similar, but the effect of cell division is much smaller [41]. VEGFR3 is highly expressed in the blood vessels of the embryonic vessels, veins and lymphatic vessels, but after the development of the fetus, VEGFR3 only in the lymphoid endothelial cells. In a variety of tumor course, VEGFR3 induced tumor lymph angiogenesis, promoting tumor lymph node invasion and lymph node metastasis. VEGFR3 plays an important role in aiding cellular viability and blocking VEGFR3 signaling hinders this ability, which may induce autophagy [42, 43].
PDGFR family
In addition to PDGFRα and PDGFRβ, members of the PDGFR family also include the colonial stimulating factor-1 receptor (CSF1R), the stem cell growth factor receptor (SCGFR), FLK2/FLK3. The family of receptors has 5 immunoglobulin-like domains in the extracellular domain and a hydrophilic insert sequence in the intracellular tyrosine kinase region [44]. PDGFR is mainly present in fibroblasts, smooth muscle cells, but also expression in the kidney, testis and brain. PDGFR is closely related to tumorigenesis [45]. In most glioblastomas, autocrine loop of PDGF and its receptors is formed. This loop is closely related to the occurrence and development of tumor [46]. In addition, similar loops are also present in melanoma, meningiomas, neuroendocrine tumors, ovarian cancer, prostate cancer, lung cancer and pancreatic cancer [47, 48].
InsR family
InsR family members include INSR, IGF1R and IRR three members. IGF-I and IGF-II have the effect of promoting proliferation and inhibiting apoptosis in breast cancer, cervical cancer, colon cancer and lung cancer [49,50,51]. IGF1R is overexpressed in breast cancer, cervical cancer, and have a great impact on the pathological process of breast cancer [52]. In addition, IGF1R is associated with the metastasis of melanoma at the end of the eye pigment, which is a predictor of this type of tumor metastasis.
Src family
Src is an important member of NRTK, which plays a key role in the regulation of many cells through the extracellular ligand binding to the receptor and the cell adhesion molecule activationin cell cycle specific stage [53,54,55,56]. These include the RAS/RAF/MEK/ERK pathways; the PI3K/AKT/mTOR pathway; and the STAT3 pathway that regulates the expression of c-Myc and Cyclin D1 (Fig. 2).It can affect cell adhesion, mobility, proliferation and angiogenesis. Under normal circumstances, the activity site of Src kinase closed, its expression was inhibited. But under the action of exogenous or endogenous carcinogenic factors, kinase hyperactivated, cell proliferation and differentiation become uncontrolled and lead to tumorigenesis [56, 57].

Cell signaling pathways induced by Src kinases. Src kinases regulate a broad spectrum of cellular events such as cell adhesion, proliferation and mobility. These include the STAT3 pathway that regulates the expression of c-Myc and Cyclin D1; the RAS/RAF/MEK/ERK pathway; and the PI3K/AKT/mTOR pathway
Abl family
Abl family includes two members: c-Abl and Arg. Both proteins can be localized in cytosol, cell membranes, and the actin cytoskeleton. Additionally, c-Abl is also present in the nucleus [58]. In normal cells, c-Abl contributes to actin remodeling, cell adhesion and motility, DNA damage response, and microbial pathogen response. Deregulation and aberrant expression of c-Abl kinases has been implicated in several types of cancer, such as breast cancer [59, 60], colon cancer [61], and NSCLC [62]. Phosporylated c-Abl activates oncogenic signaling pathways by activation of ERK5, Rac/Jnk, and STAT 1/3 pathways (Fig. 3). c-Abl is also known to be important for the genesis of CML, where it forms the oncogenic fusion protein with Bcr after the translocation of a part of chromosome 9 to chromosome 22 [63].

Cell signaling pathways induced by Abl kinases. Phosporylated Abl activates oncogenic signaling pathways by activation of ERK5; Rac/Jnk, and STAT 1/3 pathways. These cascades are required for cancer cell growth and transformation
TKIs biology
TKI can compete ATP binding site of tyrosine kinase with ATP, reduce tyrosine kinase phosphorylation, thereby inhibiting cancer cell proliferation. It has the characteristics of high selectivity, small adverse reaction and convenient oral administration [64]. According to the main targets of different, these TKI can be divided into EGFR inhibitors, VEGFR inhibitors, anaplastic lymphoma kinase (ALK) inhibitors and Bcr-Abl inhibitors. The anti-tumor mechanism of TKI can be achieved by inhibiting the repair of tumor cells, blocking the cell division in G1 phase, inducing and maintaining apoptosis, anti-angiogenesis and so on [65,66,67].
EGFR inhibitors
Gefitinib is a selective EGFR-TKI, which is usually expressed in epithelial-derived solid tumors. Inhibition of EGFR tyrosine kinase activity can prevent tumor growth, metastasis and angiogenesis, and increase tumor cell apoptosis [68, 69]. In vivo, gefitinib extensively inhibited tumor growth of human tumor cell derived lines in nude mice and increased the antitumor activity of chemotherapy, radiotherapy and hormone therapy. It has been shown in clinical trials that gefitinib has antitumor responses to locally advanced or metastatic NSCLC and can improve disease-related symptoms [8, 70].
Lapatinib is a reversible dual inhibitor of EGFR and HER2. Lapatinib can inhibit both EGFR and HER2 tyrosine kinases [12, 71, 72]. Lapatinib inhibits MAPK and PI3K signal transduction in EGFR and HER2 overexpressing tumor cell lines [73, 74]. The response to lapatinib was significantly associated with HER2 overexpression, which inhibited phosphorylation of HER2, RAF, AKT and ERK. Lapatinib has been approved by the FDA in 2007 for the treatment of breast cancer, NSCLC, head and neck cancer and gastric cancer [75].
Erlotinib can inhibit the phosphorylation of intracellular tyrosine kinases associated with EGFR, causing cell growth arrest and/or cell death. This medicine is used for third-line treatment of locally advanced or metastatic NSCLC after a previous failure of at least one chemotherapy regimen [76], combined with gemcitabine for first-line treatment of locally advanced unresectable or metastatic pancreatic cancer [77, 78]. Erlotinib treatment window is very narrow, the recommended dose close to the maximum tolerated dose, more than the recommended dosage may occur unacceptable serious adverse reactions, such as breathing difficulties, cough, diarrhea, rash and so on [79].
VEGFR inhibitors
Sorafenib can inhibit RAF-1, VGFR-2 and VGFR-3 and other RTK activity [80]. It is the first anti-tumor drugs targeting and inhibiting RAF kinase and VEGFR kinase at the same time [81, 82]. It can directly inhibit the proliferation of tumor cells by blocking the cell signaling pathway mediated by RAF/MEK/ERK [83], but also through the action of VEGFR to inhibit the formation of angiogenesis and cut off the nutritional supply of tumor cells to limit the tumor growth [84, 85]. The clinical studies have shown that sorafenib can significantly prolong the progression-free survival of patients with kidney cancer, its major adverse reactions are nausea, diarrhea, rash and high blood pressure [86].
Sunitinib is a multi-target kinase inhibitor that targets VEGFR, PDGFR-α, PDGFR-β, CSF-1R, and the like. It is used to treat inoperable RCC [87] and imatinib-resistant or intolerant gastrointestinal stromal tumors (GIST) [88]. The drug is well tolerated in children with recurrent/refractory gliomas or ependymomas, but specific monotherapy options need further investigation and may be considered in combination with radiotherapy and/or chemotherapy [89].
ALK inhibitor
ALK belongs to the insulin receptor superfamily. The physiological function of ALK in the normal body is not clear, the research suggests that it has a role for nervous system function [90, 91]. Crizotinib is a multi-target tyrosine kinase receptor inhibitor directed against ALK and acts on hepatocyte growth factor receptor (HGFR) in addition to ALK for the treatment of ALK-positive patients with locally advanced or metastatic NSCLC [92]. The study found that crizotinib also has a good anti-tumor effect on patients with NSCLC rearranged gene encoding proto-oncogene receptor (ROS1), the FDA approved in March 2016 its scope of application to broaden to ROS1-positive NSCLC patients [93].
Ceritinib is an oral small-molecule tyrosine kinase inhibitor targeting ALK, IGF-1R, InsR and ROS1, with a highly selective effect on ALK [94]. The main mechanism of action of ceritinib is to inhibit the phosphorylation of ALK itself and ALK-mediated downstream signal proteins, thereby inhibiting the proliferation of ALK-positive cancer cells. It is clinically used to treat ALK positive metastatic NSCLC or NSCLC that is exacerbated and intolerable to be treated with crizotinib [95].
Bcr-Abl kinase inhibitors
Imatinib has three main targets: various Abl, SCGFR and PDGFR, the effect is to inhibit the target-mediated cell events [96]. The molecular mechanism of imatinib is as an ATP inhibitory inhibitor, blocking PTK phosphorylation, inhibiting Bcr-Abl expression, thereby preventing cell proliferation and tumor formation [97, 98]. However, Bcr-Abl products have multiple effect, a single pathway of inhibition cannot completely eliminate the malignant proliferation of tumor cells, so this product is only efficient rather than special effects of anti-cancer drugs [99].
Bosutinib is a dual inhibitor of Abl and Src kinases [100, 101]. Bosutinib has a high anti-proliferative activity, can inhibit the proliferation and survival of CML cells [102]. It can inhibit the activity of CML graft in vivo, making K562 tumor transplant cells subsided in nude mice. The inhibitory activity to Abl kinase is considered to be the main reason for against the proliferation of chronic myeloid leukemia cells [103, 104].
TKI acquired resistance
Most cancer patients can relieve disease after using TKI, but acquired resistance remains a bottleneck in cancer targeted therapy [105]. TKI has a variety of mechanisms for drug resistance, the current researchers in the acquired resistance mechanism and its treatment strategy research has made great progress.
T790M mutation
T790M mutation is the first recognized acquired resistance mechanism after the TKI treatment. T790M mutation is due to EGFR gene 20 exon 790th codon missense mutation, resulting in the product from threonine to methionine [106]. 43%-50% of patients with NSCLC who were resistant to gefitinib or erlotinib were positive for T790M mutations [107]. The cause of resistance may be methionine instead of threonine, a steric hindrance, which affects the formation of hydrogen bonds between tyrosine kinases and TKI, leading to the inability of TKI to bind [108, 109]; Other studies have shown that T790M mutation and EGFR-sensitive mutations results in increased intracellular ATP affinity, whereas the affinity for TKI is reduced, resulting in TKI acquired resistance [110].
More and more studies further support the T790M mutation is an important acquired resistance mechanism in TKI therapy. T790M mutation will increase the tyrosine kinase activity, enhance tumorigenicity [111], but this type of drug-resistant patients still shows the slow progress of the disease trend. After immediate withdrawal, the disease has the possibility of outbreak, and targeted therapy is still effective after treatment interruption, which may be due to drug-resistant tumor cells still exist in a certain proportion of cells sensitive to EGFR-TKI, but the specific mechanism is not clear [112].
In recent years, EGFR irreversible inhibitors have given new hope to patients with failed EGFR-TKI therapy. These drugs act on the ATP binding site of EGFR, covalently bind to the receptor kinase region, and simultaneously inhibit multiple members of the EGFR receptor family [113]. Therefore, theoretically, it can still play an inhibitory effect to the second mutation of T790M EGFR, increase the efficacy and reduce the occurrence of drug resistance [114].
Aftinib (BIBW2992) is a new generation of representative irreversible potent oral inhibitors that simultaneously inhibit EGFR and HER2 targets [115]. BIBW2992 further delays tumor progression by maintaining irreversible binding to EGFR and HER2, maintaining longer activity, suppressing transformation in isogenic cell-based assays, inhibits survival of cancer cell lines and induces tumor regression in xenograft and transgenic lung cancer models, with superior activity over erlotinib [115, 116]. BIBW2992 can benefit clinical patients with refractory NSCLC.
Dacomitinib (PF299) is an oral small molecule that irreversibly inhibits EGFR, HER2 and HER4 tyrosine kinase inhibitors. In vivo and in vitro experiments, it showed potency of T790M mutations and EGFR20 exon insertion mutations, which could overcome EGFR-TKI acquired resistance by inhibition of T790M mutations.
Third generation EGFR TKIs are designed to target EGFR TKI sensitizing mutations and the T790M resistance mutation, thus inhibiting the growth of EGFR T790M-positive tumors. By sparing wild-type EGFR, these compounds are also anticipated to reduce the toxicities that have been associated with first- (e.g. gefitinib; erlotinib) and second-generation (e.g. afatinib) EGFR TKIs. Osimertinib (AZD9291, Tagrisso™), an orally administered, third generation EGFR TKI, has been approved in numerous countries for using in patients with T790M-positive advanced NSCLC [117]. Osimertinib was approved by the FDA, whereas ASP8273 is currently in clinical trials to evaluate the efficacy in patients with T790M-positive EGFR-mutated NSCLC [118].
c-MET gene amplification
Human c-MET gene located in chromosome 7, the coding product is a specific receptor for hepatocyte growth factor. MET occurred amplification, mutations and overexpression in a variety of tumors [119, 120]. After combined with HGF, MET can activate RTK system, promoting cell proliferation and differentiation, inducing epithelial cell migration and inducing angiogenesis. 20% of NSCLC patients with TKI resistance are related to c-MET gene amplification, but its occurrence was not related to the presence of T790M mutation [121]. In the presence of EGFR-TKI, c-MET gene amplification activates ERBB3-PI3K signaling pathway, directly activating EGFR downstream signaling pathway, leading to NSCLC resistance to TKI. Studies have shown that MET may be treatment targets after TKI acquired drug [122].
With the discovery of c-MET gene amplification mechanism, the combination of TKI has become another new idea to overcome the resistance of EGFR-TKI [123, 124]. MetMAb is a unique single-arm antibody that blocks the MET receptor. It inhibits the binding of HGF to the MET receptor and restores its sensitivity to erlotinib [125].
ARQ197 is a novel selective TKI that stabilizes the non-activated conformations of c-MET1 by non-ATP competitive inhibition and inactivates c-MET [126]. In vivo antitumor activity, the antitumor activity of ARQ197 combined with EGFR-TKI was found to be greater than that of ARQ197 and EGFR-TKI mono therapy. At present, ARQ-197 and erlotinib were combined to therapy advanced or metastatic non-small cell lung cancer in the three stages of research [127, 128].
Loss of PTEN expression
The PTEN gene is another tumor suppressor gene that is closely related to tumorigenesis and progression [129]. In the study of PC-9 cell lines resistant to gefitinib, p-AKT in the cell line was significantly up-regulated and the expression of PTEN was reduced. Thus, the expression of PTEN was absent and the tumor cells could find independent on EGFR activation pathway, but effectively activate the PI3K pathway, resulting in resistance to EGFR-TKI treatment [130]. Immunohistochemical staining revealed that 93 NSCLC patients treated with gefitinib had 19 deficient PTEN expression, but this had nothing to do with the objective response rate, the progression of disease, and the overall survival time. This also indicates that EGFR-TKI resistance resulting from loss of PTEN expression is associated with structural changes in EGFR downstream signaling [131].
IGF-1R-mediated EGFR downstream pathway activation
The IGF-1R is overexpressed in many tumors, making the proto-oncogene transcription and translation, and promoting tumor cell growth [132]. IGF-1R activates both RAS/RAF/MAPK and PI3K signaling pathways [133]. In the study of cell lines, IGF-1R leads to EGFR-TKI resistance by regulating the metabolism, proliferation and apoptosis of tumor cells and continuously activating the PI3K-AKT signaling pathway. Studies have found that inhibition of IGF-1R-mediated activation of EGFR downstream pathway can prevent or delay the emergence of drug-resistant after receiving Gefitinib treatment [134, 135].
EGFR pathway downstream signaling molecule PIK3A mutation or/and amplification make ERBB3-mediated PI3K signal transduction pathway activation, PTEN gene deletion or/and mutation can lead to AKT signal activation [136]. Finally, they make resistance to EGFR-TKI. BKM120 is an oral PI3K inhibitor. Preclinical studies have shown it has antitumor activity on malignant tumor with PTEN mutation or/and deletion or PI3K mutation or/and amplification [137, 138].
AKT pathway activation is commonly associated with acquired resistance to EGFR-TKI treatment in NSCLC harboring a diverse array of other, previously identified resistance mechanisms. AKT activation is a convergent feature of acquired EGFR tyrosine kinase inhibitor resistance, across a spectrum of diverse, established upstream resistance mechanisms. Studies have shown that AKT inhibition, specifically, could moreuniformly enhance response and survival in patients with high pAKT levels who are at high risk for AKT-mediated resistance, as this distinct approach has the unique potential to combat the otherwise profound heterogeneity of molecular resistance events that are present in EGFR-mutant NSCLC patients with acquired EGFR-TKI resistance to improve their outcomes [139, 140].
EML4-ALK fusion gene
Echinoderm microtubule associated protein-like 4-anaplastic lymphoma kinase (EML4-ALK) fusion gene is lung cancer-driven gene [141], EML4 and ALK two genes located on human chromosome 2 p21 and p23, intracellular ALK gene and with the N-terminal EML4 inverted fusion induces tyrosine kinase activity by stimulating the PI3K/AKT/MAPK signaling pathway, resulting in the proliferation and differentiation of tumor cells and the inhibition of apoptosis [142]. EML4-ALK fusion gene accounts for 3% to 7% of NSCLC, mostly in non-smoking, young female patients with adenocarcinoma [143]. For the EML4-ALK fusion gene, many highly effective ALK-TKIs have emerged clinically, including the second generation of ceritinib, Brigatinib and the third generation of Loratinib.
Amplification of ALK fusion gene copy number
Amplification of ALK fusion gene copy number is one of the possible mechanisms of crizotinib resistance. In one study, extensive amplification of the ALK fusion gene was detected in 18 patients with lung adenocarcinoma resistant to crizotinib and in acquired drug resistant cell lines with H3122 (including EML4-ALK mutant 1) [144]. ALK signaling pathway is often retained when the ALK fusion gene has a second mutation or increased copy number in the kinase domain and plays a role in tumor survival and drug resistance. Therefore, the use of more effective second- and third-generation ALK inhibitors may be able to overcome the secondary resistance problems caused by these mechanisms.
Activation of signal bypass
ALK belongs to the tyrosine kinase, and its downstream signaling pathways mainly include PI3K/AKT/mTOR, RAS/MEK/ERK and JAK3/STAT3, these signals are related to cell survival and proliferation, crizotinib through its specificity apoptosis was induced by inhibiting the expression of downstream signal of EML4-ALK [145]. When the signal is activated bypasses, the signal transduces around the original target of the inhibitor and activates downstream signals through the signal bypasses, leaving crizotinib not sufficient to suppress tumor growth, leading to drug resistance. These ALK-independent mechanisms of resistance include activation of EGFR, KIT, IGF-1R and other signaling pathways.
Epithelial mesenchymal transformation
The epithelial mesenchymal transformation (EMT) refers to the transformation of epithelial cells into interstitial cells. Through EMT, the epithelial cells lose the polarity of the cells, lose the epithelial phenotype such as the connection with the basement membrane, obtain higher interstitial phenotypes such as migration and invasion, anti-apoptosis and degradation of the extracellular matrix capacity [146, 147]. EMT is an important biological process in which epithelial cell-derived malignant cells acquire the ability to migrate and invade. In recent years, a number of studies have shown EMT is related to tumor stem cell formation, drug resistance and tumor metastasis.
Other possible resistance mechanisms
BRAF gene encoding BRAF protein is the molecular isomer of RAF protein, locates downstream of EGFR signaling pathway. It activated MAPK, promoted cell proliferation and differentiation through the interaction with RAS [148,149,150]. There were about 3% of BRSC gene mutations in NSCLC patients. It has been reported that BRAF gene mutation is one of the resistance mechanisms of EGFR monoclonal antibody in the treatment of colorectal cancer [151]. It has also been reported that mTOR is associated with EGFR resistance, blocking mTOR pathway can interfere with tumor growth [152]. In addition, TKI acquired resistance is also associated with increased VEGF levels, and VEGFR/EGFR dual pathway inhibitors have been shown to have a good therapeutic effect in EGFR-TKI-resistant patients [153].
Conclusions
The mechanism of tumor drug resistance is complicated, and the new emerging mechanism remains to be further studied. On one hand, tumor has a multi-drug resistance mechanism or a escape pathway, combined treatment is possible to block the signal path. In clinical practice, we often need combined application of a number of different drugs to affect the tumor growth [154]. On the other hand, gene therapy technology can inhibit the expression of drug resistance gene mRNA, with a broad clinical application prospects [155, 156]. As the researchers on the tumor resistance mechanism continuously deepening and related treatment technology continuously develop, human can improve the effect of clinical chemotherapy, reverse the resistance of cancer. The potential to truly transform some types of metastatic oncogene-addicted cancers into chronic diseases may now lie within our reach.
Abbreviations
- ALK:
-
Anaplastic lymphoma kinase
- CML:
-
Chronic myeloid leukemia
- CRC:
-
Colon and rectum carcinoma
- CSF1R:
-
Colonial stimulating factor-1 receptor
- DTC:
-
Differential thyroid carcinoma
- EGFR:
-
Epidermal growth factor receptor
- EML4-ALK:
-
Echinoderm microtubule associated protein-like 4-anaplastic lymphoma kinase
- EMT:
-
Epithelial-mesenchymal transition
- GIST:
-
Gastrointestinal stromal tumor
- HCC:
-
Hepatic cellular carcinoma
- HGFR:
-
Hepatocyte growth factor receptor
- InsR:
-
Insulin receptor
- NRTK:
-
Non-receptor PTK
- NSCLC:
-
Non-small cell lung cancer
- PDGFR:
-
Platelet-derived growth factor receptor
- PTK:
-
Protein tyrosine kinase
- RCC:
-
Renal cell carcinoma
- RTK:
-
Receptor PTK
- SCGFR:
-
Stem cell growth factor receptor
- STS:
-
Soft tissue sarcoma
- TKI:
-
Tyrosine kinase inhibitor
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Winkler GC, Barle EL, Galati G, Kluwe WM. Functional differentiation of cytotoxic cancer drugs and targeted cancer therapeutics. Regul Toxicol Pharmacol. 2014;70:46–53.
Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989–5005.
Agarwal E, Brattain MG, Chowdhury S. Cell survival and metastasis regulation by Akt signaling in colorectal cancer. Cell Signal. 2013;25:1711–9.
Wang Z, Cole PA. Catalytic mechanisms and regulation of protein kinases. Methods Enzymol. 2014;548:1–21.
Drake JM, Lee JK, Witte ON. Clinical targeting of mutated and wild-type protein tyrosine kinases in cancer. Mol Cell Biol. 2014;34:1722–32.
Knosel T, Kampmann E, Kirchner T, Altendorf-Hofmann A. [tyrosine kinases in soft tissue tumors]. Pathologe. 2014;35(Suppl 2):198–201.
Tirumani SH, Jagannathan JP, Krajewski KM, Shinagare AB, Jacene H, Ramaiya NH. Imatinib and beyond in gastrointestinal stromal tumors: a radiologist's perspective. AJR Am J Roentgenol. 2013;201:801–10.
Wertheimer C, Siedlecki J, Kook D, Mayer WJ, Wolf A, Klingenstein A, Kampik A, Eibl-Lindner K. EGFR inhibitor Gefitinib attenuates posterior capsule opacification in vitro and in the ex vivo human capsular bag model. Graefes Arch Clin Exp Ophthalmol. 2015;253:409–17.
Burotto M, Manasanch EE, Wilkerson J, Fojo T. Gefitinib and erlotinib in metastatic non-small cell lung cancer: a meta-analysis of toxicity and efficacy of randomized clinical trials. Oncologist. 2015;20:400–10.
Kuczynski EA, Lee CR, Man S, Chen E, Kerbel RS. Effects of Sorafenib dose on acquired reversible resistance and toxicity in Hepatocellular carcinoma. Cancer Res. 2015;75:2510–9.
Cheng AL, Kang YK, Lin DY, Park JW, Kudo M, Qin S, Chung HC, Song X, Xu J, Poggi G, et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol. 2013;31:4067–75.
De Silva N, Schulz L, Paterson A, Qain W, Secrier M, Godfrey E, Cheow H, O'Donovan M, Lao-Sirieix P, Jobanputra M, et al. Molecular effects of Lapatinib in the treatment of HER2 overexpressing oesophago-gastric adenocarcinoma. Br J Cancer. 2015;113:1305–12.
Pusztai L, Moulder S, Altan M, Kwiatkowski D, Valero V, Ueno NT, Esteva FJ, Avritscher R, Qi Y, Strauss L, et al. Gene signature-guided dasatinib therapy in metastatic breast cancer. Clin Cancer Res. 2014;20:5265–71.
Haznedaroglu IC. Monitoring the response to tyrosine Kinase inhibitor (TKI) treatment in chronic myeloid leukemia (CML). Mediterr J Hematol Infect Dis. 2014;6:e2014009.
Togashi Y, Hayashi H, Okamoto K, Fumita S, Terashima M, de Velasco MA, Sakai K, Fujita Y, Tomida S, Nakagawa K, Nishio K. Chronic nicotine exposure mediates resistance to EGFR-TKI in EGFR-mutated lung cancer via an EGFR signal. Lung Cancer. 2015;88:16–23.
OuYang PY, Su Z, Mao YP, Deng W, Xie FY. Combination of EGFR-TKIs and chemotherapy as first-line therapy for advanced NSCLC: a meta-analysis. PLoS One. 2013;8:e79000.
Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 2002;27:514–20.
Jimenez G, Shvartsman SY, Paroush Z. The Capicua repressor--a general sensor of RTK signaling in development and disease. J Cell Sci. 2012;125:1383–91.
Regad T. Targeting RTK signaling pathways in cancer. Cancers (Basel). 2015;7:1758–84.
Annenkov A. Receptor tyrosine kinase (RTK) signalling in the control of neural stem and progenitor cell (NSPC) development. Mol Neurobiol. 2014;49:440–71.
Summy JM, Gallick GE. Treatment for advanced tumors: SRC reclaims center stage. Clin Cancer Res. 2006;12:1398–401.
Schenone S, Manetti F, Botta M. SRC inhibitors and angiogenesis. Curr Pharm Des. 2007;13:2118–28.
Gocek E, Moulas AN, Studzinski GP. Non-receptor protein tyrosine kinases signaling pathways in normal and cancer cells. Crit Rev Clin Lab Sci. 2014;51:125–37.
Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41:1127–32.
Madhusudan S, Ganesan TS. Tyrosine kinase inhibitors in cancer therapy. Clin Biochem. 2004;37:618–35.
Yang CH, Chou HC, Fu YN, Yeh CL, Cheng HW, Chang IC, Liu KJ, Chang GC, Tsai TF, Tsai SF, et al. EGFR over-expression in non-small cell lung cancers harboring EGFR mutations is associated with marked down-regulation of CD82. Biochim Biophys Acta. 2015;1852:1540–9.
Lee HJ, Seo AN, Kim EJ, Jang MH, Kim YJ, Kim JH, Kim SW, Ryu HS, Park IA, Im SA, et al. Prognostic and predictive values of EGFR overexpression and EGFR copy number alteration in HER2-positive breast cancer. Br J Cancer. 2015;112:103–11.
Han YH, Abdul Hamid MR, Telisinghe PU, Haji Hussin JB, Mabruk M. Overexpression of EGFR protein in Bruneian lung cancer patients. Asian Pac J Cancer Prev. 2015;16:233–7.
Park HS, Jang MH, Kim EJ, Kim HJ, Lee HJ, Kim YJ, Kim JH, Kang E, Kim SW, Kim IA, Park SY. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod Pathol. 2014;27:1212–22.
Tong CWS, Wu WKK, Loong HHF, Cho WCS, To KKW. Drug combination approach to overcome resistance to EGFR tyrosine kinase inhibitors in lung cancer. Cancer Lett. 2017;405:100–10.
Ma JY, Yan HJ, Gu W. Association between BIM deletion polymorphism and clinical outcome of EGFR-mutated NSCLC patient with EGFR-TKI therapy: a meta-analysis. J Cancer Res Ther. 2015;11:397–402.
Zhu JY, Xiong Y, Zhang W, Wan J, Wan J. Endophilin B1 regulates EGFR endocytic degradation in prostate cancer cell. Cell Mol Biol (Noisy-le-grand). 2016;62:37–42.
Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, Victor A, Meisen WH, Kim SH, Grandi P, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of Glioblastoma and patient-derived Glioblastoma stem cells. Sci Rep. 2015;5:11483.
Sacher AG, Paweletz C, Dahlberg SE, Alden RS, O'Connell A, Feeney N, Mach SL, Janne PA, Oxnard GR. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2016;2:1014–22.
Shibuya M. VEGFR and type-V RTK activation and signaling. Cold Spring Harb Perspect Biol. 2013;5:a009092.
Gao MN, Li Y. The regulation of VEGFs/VEGFRs in tumor angiogenesis by Wnt/beta-catenin and NF-kappaB signal pathway. Sheng Li Ke Xue Jin Zhan. 2013;44:72–4.
Iacovelli R, Sternberg CN, Porta C, Verzoni E, de Braud F, Escudier B, Procopio G. Inhibition of the VEGF/VEGFR pathway improves survival in advanced kidney cancer: a systematic review and meta-analysis. Curr Drug Targets. 2015;16:164–70.
Ariotti C, Wagner VP, Salvadori G, Carrard VC, Martins MA, da Cunha Filho JJ, Meurer L, Martins MD. VEGFR1 and VEGFR2 in lip carcinogenesis and its association with microvessel density. Tumour Biol. 2015;36:7285–92.
Xu WW, Li B, Lam AK, Tsao SW, Law SY, Chan KW, Yuan QJ, Cheung AL. Targeting VEGFR1- and VEGFR2-expressing non-tumor cells is essential for esophageal cancer therapy. Oncotarget. 2015;6:1790–805.
Pfister NT, Fomin V, Regunath K, Zhou JY, Zhou W, Silwal-Pandit L, Freed-Pastor WA, Laptenko O, Neo SP, Bargonetti J, et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 2015;29:1298–315.
Abbasi O, Mashayekhi F, Mirzajani E, Fakhriyeh Asl S, Mahmoudi T, Saeedi Saedi H. Soluble VEGFR1 concentration in the serum of patients with colorectal cancer. Surg Today. 2015;45:215–20.
Deng Y, Zhang X, Simons M. Molecular controls of lymphatic VEGFR3 signaling. Arterioscler Thromb Vasc Biol. 2015;35:421–9.
Varney ML, Singh RK. VEGF-C-VEGFR3/Flt4 axis regulates mammary tumor growth and metastasis in an autocrine manner. Am J Cancer Res. 2015;5:616–28.
Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med. 2013;19:460–73.
Koschmann C, Zamler D, MacKay A, Robinson D, Wu YM, Doherty R, Marini B, Tran D, Garton H, Muraszko K, et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget. 2016;7:65696–706.
Guha A, Dashner K, Black PM, Wagner JA, Stiles CD. Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int J Cancer. 1995;60:168–73.
Ma Y, Han CC, Li Y, Wang Y, Wei W. Insulin-like growth factor-binding protein-3 inhibits IGF-1-induced proliferation of human hepatocellular carcinoma cells by controlling bFGF and PDGF autocrine/paracrine loops. Biochem Biophys Res Commun. 2016;478:964–9.
Battegay EJ, Raines EW, Seifert RA, Bowen-Pope DF, Ross R. TGF-beta induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell. 1990;63:515–24.
Suh HS, Zhao ML, Derico L, Choi N, Lee SC. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: differential regulation by inflammatory mediators. J Neuroinflammation. 2013;10:37.
Livingstone C. IGF2 and cancer. Endocr Relat Cancer. 2013;20:R321–39.
Chisalita SI, Johansson GS, Liefvendahl E, Back K, Arnqvist HJ. Human aortic smooth muscle cells are insulin resistant at the receptor level but sensitive to IGF1 and IGF2. J Mol Endocrinol. 2009;43:231–9.
Werner H, Sarfstein R. Transcriptional and epigenetic control of IGF1R gene expression: implications in metabolism and cancer. Growth Hormon IGF Res. 2014;24:112–8.
Hunter T. Discovering the first tyrosine kinase. Proc Natl Acad Sci U S A. 2015;112:7877–82.
Roskoski R Jr. Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun. 2004;324:1155–64.
Foda ZH, Shan Y, Kim ET, Shaw DE, Seeliger MA. A dynamically coupled allosteric network underlies binding cooperativity in Src kinase. Nat Commun. 2015;6:5939.
Cobb BS, Parsons JT. Regulation of the cellular src protein tyrosine kinase: interactions of the carboxyl terminal sequences residing between the kinase domain and tyrosine-527. Oncogene. 1993;8:2897–903.
Fan G, Aleem S, Yang M, Miller WT, Tonks NK. Protein-tyrosine Phosphatase and Kinase specificity in regulation of SRC and breast tumor Kinase. J Biol Chem. 2015;290:15934–47.
Wang JY. The capable ABL: what is its biological function? Mol Cell Biol. 2014;34:1188–97.
Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–55.
Sirvent A, Boureux A, Simon V, Leroy C, Roche S. The tyrosine kinase Abl is required for Src-transforming activity in mouse fibroblasts and human breast cancer cells. Oncogene. 2007;26:7313–23.
Chen WS, Kung HJ, Yang WK, Lin W. Comparative tyrosine-kinase profiles in colorectal cancers: enhanced arg expression in carcinoma as compared with adenoma and normal mucosa. Int J Cancer. 1999;83:579–84.
Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–203.
Ross TS, Mgbemena VE. Re-evaluating the role of BCR/ABL in chronic myelogenous leukemia. Mol Cell Oncol. 2014;1:e963450.
Lenihan DJ, Kowey PR. Overview and management of cardiac adverse events associated with tyrosine kinase inhibitors. Oncologist. 2013;18:900–8.
Botting GM, Rastogi I, Chhabra G, Nlend M, Puri N. Mechanism of resistance and novel targets mediating resistance to EGFR and c-met tyrosine Kinase inhibitors in non-small cell lung cancer. PLoS One. 2015;10:e0136155.
Lin Y, Wang X, Jin H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res. 2014;4:411–35.
Liu H, Wang M, Hu K, Xu Y, Ma M, Zhong W, Zhao J, Li L. Wang H: [research progress of the resistance mechanism of non-small cell lung cancer to EGFR-TKIs]. Zhongguo Fei Ai Za Zhi. 2013;16:535–40.
Rahman AF, Korashy HM, Kassem MG. Gefitinib. Profiles Drug Subst Excip Relat Methodol. 2014;39:239–64.
Dhillon S. Gefitinib: a review of its use in adults with advanced non-small cell lung cancer. Target Oncol. 2015;10:153–70.
Chen JC, Su YH, Chiu CF, Chang YW, Yu YH, Tseng CF, Chen HA, Su JL. Suppression of dicer increases sensitivity to gefitinib in human lung cancer cells. Ann Surg Oncol. 2014;21(Suppl 4):S555–63.
D'Amato V, Raimondo L, Formisano L, Giuliano M, De Placido S, Rosa R, Bianco R. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev. 2015;41:877–83.
Dupouy S, Doan VK, Wu Z, Mourra N, Liu J, De Wever O, Llorca FP, Cayre A, Kouchkar A, Gompel A, Forgez P. Activation of EGFR, HER2 and HER3 by neurotensin/neurotensin receptor 1 renders breast tumors aggressive yet highly responsive to lapatinib and metformin in mice. Oncotarget. 2014;5:8235–51.
Hsiao YC, Yeh MH, Chen YJ, Liu JF, Tang CH, Huang WC. Lapatinib increases motility of triple-negative breast cancer cells by decreasing miRNA-7 and inducing Raf-1/MAPK-dependent interleukin-6. Oncotarget. 2015;6:37965–78.
Long XH, Zhang GM, Peng AF, Luo QF, Zhang L, Wen HC, Zhou RP, Gao S, Zhou Y, Liu ZL. Lapatinib alters the malignant phenotype of osteosarcoma cells via downregulation of the activity of the HER2-PI3K/AKT-FASN axis in vitro. Oncol Rep. 2014;31:328–34.
Hicks M, Macrae ER, Abdel-Rasoul M, Layman R, Friedman S, Querry J, Lustberg M, Ramaswamy B, Mrozek E, Shapiro C, Wesolowski R. Neoadjuvant dual HER2-targeted therapy with lapatinib and trastuzumab improves pathologic complete response in patients with early stage HER2-positive breast cancer: a meta-analysis of randomized prospective clinical trials. Oncologist. 2015;20:337–43.
Shi L, Tang J, Tong L, Liu Z. Risk of interstitial lung disease with gefitinib and erlotinib in advanced non-small cell lung cancer: a systematic review and meta-analysis of clinical trials. Lung Cancer. 2014;83:231–9.
Wang Y, Hu GF, Zhang QQ, Tang N, Guo J, Liu LY, Han X, Wang X, Wang ZH. Efficacy and safety of gemcitabine plus erlotinib for locally advanced or metastatic pancreatic cancer: a systematic review and meta-analysis. Drug Des Devel Ther. 2016;10:1961–72.
Philip PA, Lutz MP. Targeting epidermal growth factor receptor-related signaling pathways in pancreatic cancer. Pancreas. 2015;44:1046–52.
D'Arcangelo M, Cappuzzo F. Erlotinib in the first-line treatment of non-small-cell lung cancer. Expert Rev Anticancer Ther. 2013;13:523–33.
Blair HA, Plosker GL. Sorafenib: a review of its use in patients with radioactive iodine-refractory, metastatic differentiated thyroid carcinoma. Target Oncol. 2015;10:171–8.
Nishida N, Kitano M, Sakurai T, Kudo M. Molecular mechanism and prediction of Sorafenib Chemoresistance in human Hepatocellular carcinoma. Dig Dis. 2015;33:771–9.
Nagel C, Armeanu-Ebinger S, Dewerth A, Warmann SW, Fuchs J. Anti-tumor activity of sorafenib in a model of a pediatric hepatocellular carcinoma. Exp Cell Res. 2015;331:97–104.
Mei J, Zhu X, Wang Z, Wang Z. VEGFR, RET, and RAF/MEK/ERK pathway take part in the inhibition of osteosarcoma MG63 cells with sorafenib treatment. Cell Biochem Biophys. 2014;69:151–6.
Cao G, Li X, Qin C, Li J. Prognostic value of VEGF in Hepatocellular carcinoma patients treated with Sorafenib: a meta-analysis. Med Sci Monit. 2015;21:3144–51.
Yildiz C, Kacan T, Akkar OB, Karakus S, Kacan SB, Ozer H, Cetin A. Effects of Pazopanib, Sunitinib, and Sorafenib, anti-VEGF agents, on the growth of experimental endometriosis in rats. Reprod Sci. 2015;22:1445–51.
Bolondi L, Craxi A, Trevisani F, Daniele B, Di Costanzo GG, Fagiuoli S, Camma C, Bruzzi P, Danesi R, Spandonaro F, et al. Refining sorafenib therapy: lessons from clinical practice. Future Oncol. 2015;11:449–65.
Kalra S, Rini BI, Jonasch E. Alternate sunitinib schedules in patients with metastatic renal cell carcinoma. Ann Oncol. 2015;26:1300–4.
Imbulgoda A, Heng DY, Kollmannsberger C. Sunitinib in the treatment of advanced solid tumors. Recent Results Cancer Res. 2014;201:165–84.
Segarra I, Modamio P, Fernandez C, Marino EL. Sunitinib possible sex-divergent therapeutic outcomes. Clin Drug Investig. 2016;36:791–9.
Winkler C, Yao S. The midkine family of growth factors: diverse roles in nervous system formation and maintenance. Br J Pharmacol. 2014;171:905–12.
Allouche M. ALK is a novel dependence receptor: potential implications in development and cancer. Cell Cycle. 2007;6:1533–8.
Dikopf A, Wood K, Salgia R. A safety assessment of crizotinib in the treatment of ALK-positive NSCLC patients. Expert Opin Drug Saf. 2015;14:485–93.
Ye M, Zhang X, Li N, Zhang Y, Jing P, Chang N, Wu J, Ren X, Zhang J. ALK and ROS1 as targeted therapy paradigms and clinical implications to overcome crizotinib resistance. Oncotarget. 2016;7:12289–304.
Dhillon S, Clark M. Ceritinib: first global approval. Drugs. 2014;74:1285–91.
Li S, Qi X, Huang Y, Liu D, Zhou F, Zhou C. Ceritinib (LDK378): a potent alternative to crizotinib for ALK-rearranged non-small-cell lung cancer. Clin Lung Cancer. 2015;16:86–91.
Musumeci F, Schenone S, Grossi G, Brullo C, Sanna M. Analogs, formulations and derivatives of imatinib: a patent review. Expert Opin Ther Pat. 2015;25:1411–21.
Pan X, Dong J, Shao R, Su P, Shi Y, Wang J, He L. Expanding the structural diversity of Bcr-Abl inhibitors: hybrid molecules based on GNF-2 and Imatinib. Bioorg Med Chem Lett. 2015;25:4164–8.
Guo Y, Li Y, Shan Q, He G, Lin J, Gong Y. Curcumin potentiates the anti-leukemia effects of imatinib by downregulation of the AKT/mTOR pathway and BCR/ABL gene expression in ph+ acute lymphoblastic leukemia. Int J Biochem Cell Biol. 2015;65:1–11.
Roy M, Sarkar R, Mukherjee A, Mukherjee S. Inhibition of crosstalk between Bcr-Abl and PKC signaling by PEITC, augments imatinib sensitivity in chronic myelogenous leukemia cells. Chem Biol Interact. 2015;242:195–201.
Keller G, Schafhausen P, Brummendorf TH. Bosutinib. Recent Results Cancer Res. 2010;184:119–27.
Isfort S, Keller-v Amsberg G, Schafhausen P, Koschmieder S, Brummendorf TH. Bosutinib: a novel second-generation tyrosine kinase inhibitor. Recent Results Cancer Res. 2014;201:81–97.
Doan V, Wang A, Prescott H. Bosutinib for the treatment of chronic myeloid leukemia. Am J Health Syst Pharm. 2015;72:439–47.
Levinson NM, Boxer SG. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS One. 2012;7:e29828.
Rassi FE, Khoury HJ. Bosutinib: a SRC-ABL tyrosine kinase inhibitor for treatment of chronic myeloid leukemia. Pharmgenomics Pers Med. 2013;6:57–62.
Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11:473–81.
Watanabe M, Kawaguchi T, Isa S, Ando M, Tamiya A, Kubo A, Saka H, Takeo S, Adachi H, Tagawa T, et al. Ultra-sensitive detection of the pretreatment EGFR T790M mutation in non-small cell lung cancer patients with an EGFR-activating mutation using droplet digital PCR. Clin Cancer Res. 2015;21:3552–60.
Kuiper JL, Heideman DA, Thunnissen E, Paul MA, van Wijk AW, Postmus PE, Smit EF. Incidence of T790M mutation in (sequential) rebiopsies in EGFR-mutated NSCLC-patients. Lung Cancer. 2014;85:19–24.
Hata A, Katakami N, Yoshioka H, Takeshita J, Tanaka K, Nanjo S, Fujita S, Kaji R, Imai Y, Monden K, et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: comparison between T790M mutation-positive and mutation-negative populations. Cancer. 2013;119:4325–32.
Denis MG, Vallee A, Theoleyre S. EGFR T790M resistance mutation in non small-cell lung carcinoma. Clin Chim Acta. 2015;444:81–5.
Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–5.
Kim Y, Ko J, Cui Z, Abolhoda A, Ahn JS, Ou SH, Ahn MJ, Park K. The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor. Mol Cancer Ther. 2012;11:784–91.
Suda K, Onozato R, Yatabe Y, Mitsudomi T. EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol. 2009;4:1–4.
Bordi P, Tiseo M, Bortesi B, Naldi N, Buti S, Ardizzoni A. Overcoming T790M-driven acquired resistance to EGFR-TKIs in NSCLC with afatinib: a case report. Tumori. 2014;100:e20–3.
Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34.
Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–11.
Takezawa K, Okamoto I, Tanizaki J, Kuwata K, Yamaguchi H, Fukuoka M, Nishio K, Nakagawa K. Enhanced anticancer effect of the combination of BIBW2992 and thymidylate synthase-targeted agents in non-small cell lung cancer with the T790M mutation of epidermal growth factor receptor. Mol Cancer Ther. 2010;9:1647–56.
Lamb YN, Scott LJ. Osimertinib: a review in T790M-positive advanced non-small cell lung cancer. Target Oncol. 2017;12:555–62.
Watanabe S, Yoshida T, Kawakami H, Takegawa N, Tanizaki J, Hayashi H, Takeda M, Yonesaka K, Tsurutani J, Nakagawa K. T790M-selective EGFR-TKI combined with dasatinib as an optimal strategy for overcoming EGFR-TKI resistance in T790M-positive non-small cell lung cancer. Mol Cancer Ther. 2017;16(11):2563–71.
Kwak Y, Kim SI, Park CK, Paek SH, Lee ST, Park SH. C-MET overexpression and amplification in gliomas. Int J Clin Exp Pathol. 2015;8:14932–8.
Chen D, Xu C, Wu J, Zhang Y, Fang M. A comparison of consistency of detecting c-MET gene amplification in peripheral blood and tumor tissue of nonsmall cell lung cancer patients. J Cancer Res Ther. 2015;11(Suppl 1):C63–7.
Presutti D, Santini S, Cardinali B, Papoff G, Lalli C, Samperna S, Fustaino V, Giannini G, Ruberti G. MET gene amplification and MET receptor activation are not sufficient to predict efficacy of combined MET and EGFR inhibitors in EGFR TKI-resistant NSCLC cells. PLoS One. 2015;10:e0143333.
Kawakami H, Okamoto I, Arao T, Okamoto W, Matsumoto K, Taniguchi H, Kuwata K, Yamaguchi H, Nishio K, Nakagawa K, Yamada Y. MET amplification as a potential therapeutic target in gastric cancer. Oncotarget. 2013;4:9–17.
Watermann I, Schmitt B, Stellmacher F, Muller J, Gaber R, Kugler C, Reinmuth N, Huber RM, Thomas M, Zabel P, et al. Improved diagnostics targeting c-MET in non-small cell lung cancer: expression, amplification and activation? Diagn Pathol. 2015;10:130.
Janbabai G, Oladi Z, Farazmandfar T, Taghvaei T, Naghshvar F. The prognostic impact of EGFR, ErbB2 and MET gene amplification in human gastric carcinomas as measured by quantitative real-time PCR. J Cancer Res Clin Oncol. 2015;141:1945–52.
Xiang H, Bender BC, Reyes AE 2nd, Merchant M, Jumbe NL, Romero M, Davancaze T, Nijem I, Mai E, Young J, et al. Onartuzumab (MetMAb): using nonclinical pharmacokinetic and concentration-effect data to support clinical development. Clin Cancer Res. 2013;19:5068–78.
Porta C, Giglione P, Ferrari A, Reversi F, Liguigli W, Imarisio I, Ganini C. Tivantinib (ARQ197) in hepatocellular carcinoma. Expert Rev Anticancer Ther. 2015;15:615–22.
Basilico C, Pennacchietti S, Vigna E, Chiriaco C, Arena S, Bardelli A, Valdembri D, Serini G, Michieli P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin Cancer Res. 2013;19:2381–92.
Michieli P, Basilico C, Pennacchietti S. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET--response. Clin Cancer Res. 2013;19:4291.
Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: multiple functions in human malignant tumors. Front Oncol. 2015;5:24.
Chen J, Bi H, Hou J, Zhang X, Zhang C, Yue L, Wen X, Liu D, Shi H, Yuan J, et al. Atorvastatin overcomes gefitinib resistance in KRAS mutant human non-small cell lung carcinoma cells. Cell Death Dis. 2013;4:e814.
Shen H, Zhu F, Liu J, Xu T, Pei D, Wang R, Qian Y, Li Q, Wang L, Shi Z, et al. Alteration in Mir-21/PTEN expression modulates gefitinib resistance in non-small cell lung cancer. PLoS One. 2014;9:e103305.
Cai W, Sakaguchi M, Kleinridders A, Gonzalez-Del Pino G, Dreyfuss JM, O'Neill BT, Ramirez AK, Pan H, Winnay JN, Boucher J, et al. Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat Commun. 2017;8:14892.
Mills K, Gusscott S, Jenkins CE, Lam SH, Giambra V, Pollak M, Weng AP. IGF1R derived PI3K/AKT signaling maintains growth in a subset of human T-cell acute lymphoblastic Leukemias. PLoS One. 2016;11:e0161158.
Ma Y, Tang N, Thompson RC, Mobley BC, Clark SW, Sarkaria JN, Wang J. InsR/IGF1R pathway mediates resistance to EGFR inhibitors in Glioblastoma. Clin Cancer Res. 2016;22:1767–76.
Park E, Park SY, Kim H, Sun PL, Jin Y, Cho SK, Kim K, Lee CT, Chung JH. Membranous insulin-like growth Factor-1 receptor (IGF1R) expression is predictive of poor prognosis in patients with epidermal growth factor receptor (EGFR)-mutant lung Adenocarcinoma. J Pathol Transl Med. 2015;49:382–8.
Davis NM, Sokolosky M, Stadelman K, Abrams SL, Libra M, Candido S, Nicoletti F, Polesel J, Maestro R, D'Assoro A, et al. Deregulation of the EGFR/PI3K/PTEN/Akt/mTORC1 pathway in breast cancer: possibilities for therapeutic intervention. Oncotarget. 2014;5:4603–50.
Liu WL, Gao M, Tzen KY, Tsai CL, Hsu FM, Cheng AL, Cheng JC. Targeting Phosphatidylinositide3-Kinase/Akt pathway by BKM120 for radiosensitization in hepatocellular carcinoma. Oncotarget. 2014;5:3662–72.
Martin SK, Gan ZY, Fitter S, To LB, Zannettino AC. The effect of the PI3K inhibitor BKM120 on tumour growth and osteolytic bone disease in multiple myeloma. Leuk Res. 2015;39:380–7.
Jacobsen K, Bertran-Alamillo J, Molina MA, Teixido C, Karachaliou N, Pedersen MH, Castellvi J, Garzon M, Codony-Servat C, Codony-Servat J, et al. Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer. Nat Commun. 2017;8:410.
Fang L, He J, Xia J, Dong L, Zhang X, Chai Y, Li Y, Niu M, Hang T, Li S. Resistance to epithelial growth factor receptor tyrosine kinase inhibitors in a patient with transformation from lung adenocarcinoma to small cell lung cancer: a case report. Oncol Lett. 2017;14:593–8.
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6.
Mano H. The EML4-ALK oncogene: targeting an essential growth driver in human cancer. Proc Jpn Acad Ser B Phys Biol Sci. 2015;91:193–201.
Han W, Bi M. Clinical meaning of EML4-ALK fusion gene in non-small cell lung cancer. Zhongguo Fei Ai Za Zhi. 2013;16:97–101.
Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra117.
Cappuzzo F, Moro-Sibilot D, Gautschi O, Boleti E, Felip E, Groen HJ, Germonpre P, Meldgaard P, Arriola E, Steele N, et al. Management of crizotinib therapy for ALK-rearranged non-small cell lung carcinoma: an expert consensus. Lung Cancer. 2015;87:89–95.
Li D, Zhang L, Zhou J, Chen H. Cigarette smoke extract exposure induces EGFR-TKI resistance in EGFR-mutated NSCLC via mediating Src activation and EMT. Lung Cancer. 2016;93:35–42.
Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 2015;5:390–401.
Li DD, Zhang YF, Xu HX, Zhang XP. The role of BRAF in the pathogenesis of thyroid carcinoma. Front Biosci (Landmark Ed). 2015;20:1068–78.
Luk PP, Yu B, Ng CC, Mercorella B, Selinger C, Lum T, Kao S, O'Toole SA, Cooper WA. BRAF mutations in non-small cell lung cancer. Transl Lung Cancer Res. 2015;4:142–8.
Nguyen-Ngoc T, Bouchaab H, Adjei AA, Peters S. BRAF alterations as therapeutic targets in non-small-cell lung cancer. J Thorac Oncol. 2015;10:1396–403.
Corcoran RB. New therapeutic strategies for BRAF mutant colorectal cancers. J Gastrointest Oncol. 2015;6:650–9.
Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA. Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit Rev Oncog. 2012;17:69–95.
van Cruijsen H, van der Veldt A, Hoekman K. Tyrosine kinase inhibitors of VEGF receptors: clinical issues and remaining questions. Front Biosci (Landmark Ed). 2009;14:2248–68.
Benedetti R, Conte M, Iside C, Altucci L. Epigenetic-based therapy: from single- to multi-target approaches. Int J Biochem Cell Biol. 2015;69:121–31.
Pazarentzos E, Mazarakis ND. Anticancer gene transfer for cancer gene therapy. Adv Exp Med Biol. 2014;818:255–80.
Ajith TA. Strategies used in the clinical trials of gene therapy for cancer. J Exp Ther Oncol. 2015;11:33–9.
Acknowledgements
Not applicable.
Funding
This work was supported by the China Post doctoral International Exchange Program2015, National Natural Science Foundation of China (no. 81402193), Postdoctoral Innovation Project of Shandong Province (no. 147751), and Postdoctoral Science Foundation of China (no. 2015 M570597) to Y. Wang; by National Natural Science Foundation of China (no. 81500029), Natural Science Foundation of Shandong Province (no. BS2015YY05) to Q. Wang.
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Author information
Authors and Affiliations
Contributions
QLJ, QW, YSW designed and drafted the manuscript. LB, YDR, SLS discussed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This is not applicable for this review.
Consent for publication
This is not applicable for this review.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jiao, Q., Bi, L., Ren, Y. et al. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer 17, 36 (2018). https://doi.org/10.1186/s12943-018-0801-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-018-0801-5