
Abstract
Neuroblastoma is a tumor arising in the peripheral sympathetic nervous system and is the most common cancer in childhood. Since most of the cellular and molecular mechanisms underlying neuroblastoma onset and progression remain unknown, the generation of new in vivo models might be appropriate to better dissect the peripheral sympathetic nervous system development in both physiological and disease states. This review is focused on the use of zebrafish as a suitable and innovative model to study neuroblastoma development. Here, we briefly summarize the current knowledge about zebrafish peripheral sympathetic nervous system formation, focusing on key genes and cellular pathways that play a crucial role in the differentiation of sympathetic neurons during embryonic development. In addition, we include examples of how genetic changes known to be associated with aggressive neuroblastoma can mimic this malignancy in zebrafish. Thus, we note the value of the zebrafish model in the field of neuroblastoma research, showing how it can improve our current knowledge about genes and biological pathways that contribute to malignant transformation and progression during embryonic life.
Similar content being viewed by others


Background
Neuroblastoma (NB) is the most common extracranial solid tumor arising in the peripheral sympathetic nervous system (PSNS), including spinal sympathetic ganglia and the adrenal medulla [1]. This devastating tumor originates from the sympathoadrenal cell lineage deriving from neural crest cells (NCCs) and is responsible for approximately 15% of childhood cancer-related mortality [2]. NB is characterized by great genetic and biological heterogeneity, and many studies have been performed to understand the complex mechanisms underlying NB origin and progression.
Over the past years, zebrafish has become an attractive alternative tool to the classical mouse model for studying many human diseases, spanning from pediatric to adult tumors and from blood to neurodegenerative disorders (for extended reviews, see [3, 4]). Notably, several experimental techniques that are applied to mice have been extended to zebrafish, and the zebrafish model promises to be extremely useful for development of effective therapeutic strategies for cancer research. For example, zebrafish has emerged as an important tool for the identification of new chemical compounds with potential clinical implications [5], permitting a high-throughput screening of currently available chemical libraries [6].
It is important to highlight that a complete understanding of the biological processes underlying zebrafish PSNS development will be useful for defining the molecular pathways contributing to malignant transformation and NB progression. The biological information obtained with the zebrafish in vivo model might contribute to the discovery of genes and molecular pathways that are dysfunctional during the early stages of NB development.
Neuroblastoma overview
NB presents as either a localized or metastatic disease, and patients with NB have varying survival rates. Patients with a localized tumor have a good outcome, with an overall survival (OS) of more than 99% 5 years after diagnosis. In contrast, the OS of children older than one year with a metastatic tumor is less than 40% 5 years after diagnosis [7, 8].
A large assortment of copy number variations (CNVs), ranging from numerical CNVs in localized tumors to structural CNVs in metastatic tumors, has been observed in NB. The most frequently observed structural CNVs are loss of chromosomes 1p, 3p, 4p, 9p, 11q and 14q and gain of chromosomes 2p and 17q [9–11]. In the last decade, many efforts have been made to identify candidate NB genes. In fact, MYCN oncogene amplification, which is observed in 20% of patients with NB, plays a role in tumor progression and aggressiveness [12]. The genomic amplification of MYCN is frequently observed in patients who are resistant to any therapy and is currently a prognostic marker of risk stratification in patients with NB [13]. Other genes have been identified for NB predisposition. ALK (anaplastic lymphoma kinase) was the first gene to be identified as a familial NB predisposition gene [14]. Several activating mutations have been discovered in the ALK tyrosine kinase domain, although mutations F1174L and R1275Q are the most frequently observed and are associated with a more negative prognosis [15, 16]. Moreover, LMO1 [17], BARD1 [10] and LINC00340 [18] have been identified by genome-wide association studies as other NB predisposition genes. More recently, a massive whole-genome sequencing approach has been used to discover novel key mutations in genes involved in NB onset and progression. Pugh et al. [19] and Molenaar et al. [20] screened more than 200 cases of NB at different clinical stages of the disease and found a low frequency of recurrent mutations. Thus, it seems that NB originates during embryonic life as result of chromosomal instability rather than from a mutation in a single NB gene. There is a general agreement that MYCN gene amplification plays a major role in driving NB tumorigenesis, while ALK gene mutation cooperates with MYCN to enforce tumor aggressiveness [21, 22].
The in vivo models of neuroblastoma
Previous studies have established different in vivo models of NB. Mouse models of NB are clinically relevant tools for studying the growth and metastasis of this aggressive malignancy. Orthotopic and subcutaneous xenograft mouse models have been extensively generated for the preclinical testing of new therapeutic strategies against NB [23–30]. Notably, the xenograft mouse model displays biological features that limit the possibility of obtaining a standardized in vivo system reflecting tumor pathogenesis. These variables include the manipulation of tumor cells prior to their engraftment outside of the natural tumor microenvironment. Due to the imposed and artificial conditions, xenotransplanted mice frequently develop tumors that do not represent the corresponding human cancer that develops in its native environment [31]. Tumor cell manipulation might result in genome and/or transcriptomic changes distinct from those arising in patients’ cancer cells [31]. To overcome these limitations, Weiss and colleagues [32] generated a transgenic NB mouse model through the overexpression of the human MYCN oncogene in neural crest-derived cells driven by the rat tyrosine hydroxylase promoter. These mice spontaneously develop NB exclusively in the sympathoadrenal system with high resemblance to human tumors, demonstrating the involvement of MYCN in NB genesis [33–37].
Other transgenic mouse models have helped to elucidate the cellular mechanisms of NB pathogenesis. In addition to MYCN gene amplification, the in vivo forced expression of genes that are normally involved in sympathoadrenal development during embryonic life (such as ALK and LIN28B) drives NB formation [38, 39]. Thus, these animal models support the prenatal origin of this pediatric cancer. Among the increasing number of models dedicated to recapitulating NB origin, the zebrafish model has emerged as a novel important platform for performing in vivo studies of NB pathogenesis. Zebrafish displays several logistic advantages that make it an attractive alternative for mimicking human diseases: (i) its characteristics of reproduction and transparency will allow the study of the first stages of development in a short amount of time; (ii) the optical transparency of zebrafish embryos and larvae will allow the study of the behavior of engrafted human cancer cells or the expression of fluorescently tagged oncogenes; (iii) its morphology will allow the study of disease phenotypes by following the morphological aberrations that often arise in a short amount of time; (iv) it is possible to have thousands of zebrafish embryos and larvae for tissue specimen collection and high-throughput drug screening; (v) an increasing number of technologies are available for zebrafish genetic manipulation, such as transient/stable gene loss and gain of function as well as high-throughput DNA and RNA sequencing techniques. Thus, the zebrafish model is a powerful tool for dissecting the molecular pathways involved in PSNS development and NB origin. This biological information is important not only for understanding exactly when and where malignant transformations occur but also for more the rapid determination of possible genetic or chemical tumor progression modifiers that might guarantee novel and more effective therapies.
Zebrafish as a model for studying peripheral sympathetic nervous system development
Increasing evidence indicates that NB arises from the NCC lineage during embryonic life (for an extended review, see [40]); thus, it is crucial to understand the characteristics of NCCs under both normal and pathological conditions in order to dissect the initial tumorigenesis steps of this embryonic cancer.
The cells of the sympathetic ganglia and mature adrenal medulla originate from a transient pool of NCCs. These cells pass through four critical developmental phases. First, NCCs undergo an epithelial to mesenchymal transition (EMT) from the most dorsal region of the neural tube and differentiate into sympathoadrenal precursors. Then, sympathoadrenal precursors migrate toward the ventral portion of the notochord near the dorsal aorta. Finally, these cells activate the neuronal differentiation program, giving rise to the mature sympathetic ganglia and chromaffin cells of the adrenal medulla.
Induction of an epithelial to mesenchymal transition in neural crest cells
After neurulation, NCCs undergo an EMT from the roof plate of the neural tube, delaminating from the neuroepithelium and then migrating through the periphery, where they differentiate into several cell types, including cells of the peripheral nervous system and enteric nervous system, pigmented cells, Schwann cells, adrenal medullary cells, and cartilage cells of the craniofacial skeleton [41–43]. The downregulation of cadherins on the cell surface (usually mediated by the upregulation of mesenchymal transcription factors, such as Snail, Slug and Twist), cytoskeleton remodeling and the synthesis of proteases are key events that mediate this transition. These cellular changes are also essential for the development of many tissues during embryogenesis. Interestingly, similar modifications are recapitulated during pathological processes, such as fibrosis, cancer and drug resistance [44, 45].
Recently, in vitro studies have suggested that an EMT might also be involved in NB progression, and Nozato and colleagues [46] found that KRT19, ERBB3, TWIST1 and TCF3 EMT-related genes are differentially expressed in a cohort of patients with NB. Thus, in addition to MYCN gene amplification, the expression level of EMT-related genes might become an additional prognostic marker for patients with NB. Despite these recent discoveries, there is no solid evidence from studies using in vivo models that supports the relationship between EMT and NB pathogenesis. Therefore, additional studies will be necessary to elucidate whether EMT plays a role in tumor development and eventual metastasis in NB. Consequently, the window of time in which EMT impairment might lead to the modulation of tumor-associated signals should be defined.
Neural crest cell migration and differentiation
After an EMT begins, NCCs acquire a polarized morphology and follow two migratory pathways away from the neural tube: the dorsolateral and the ventromedial (Fig. 1). NCC-derived sympathoadrenal precursors ventromedially migrate within the sclerotome, whereas the dorsolateral pathway mostly generates the pigmented cell lineage, including melanocytes [47–49]. Nevertheless, in the trunk region, the route of the NCCs and their migration schedule are not perfectly mirrored in all species but rather show importantly dissimilar patterns among them [50]. Spatially and temporally regulated extrinsic signals from the neural tube, somites, notochord and dorsal aorta are essential for the migration and specification of sympathoadrenal precursors. These signals include many permissive factors, such as the extracellular matrix proteins laminins, versican and fibronectin, which drive the earliest migrating sympathoadrenal precursors into the ventromedial pathway [51–53]. In parallel, ephrins, chondroitin sulfate proteoglycans and other inhibitory molecules block the alternative dorsolateral pathway.

Patterns of NCC migration in zebrafish. Schematic cartoon depicting the two main migratory pathways of NCCs during embryonic development (transverse section of a vertebrate embryo). NCCs (blue) follow the ventromedial pathway (green arrow) and migrate between the neural tube and somites and then bilaterally reach the dorsal portion of the dorsal aorta, where they differentiate into sympathoadrenal progenitors (green). NCCs that migrate dorsolaterally (red arrow) between somites and the overlying ectodermal epithelium give rise to pigment cells (red). NCCs neural crest cells, EE ectodermal epithelium, NT neural tube, NO notochord, DA dorsal aorta, S somite
The molecular development of the peripheral sympathetic nervous system in zebrafish
The development of the PSNS in zebrafish has been well examined [54, 55]. In fact, despite several structural and physiological differences between zebrafish and mammals, the morphogenesis and differentiation of sympathetic neurons in zebrafish are strongly comparable to those of other vertebrates. In zebrafish, trunk sympathoadrenal precursors begin to migrate approximately 16 h post-fertilization (hpf) from somite 7 and follow the ventromedial pathway, indicating that the formation of sympathetic neurons is similar to that of other vertebrates [54, 55]. NCCs are easily identified by the expression of the transcription factor Sox10 [56, 57], and crestin is a multicopy retro-element expressed in pre-migratory and migratory NCCs (Fig. 2) [58]. Moreover, in mammalian embryos, the receptor tyrosine kinase proto-oncogene ErbB3 and its dimerization partner ErbB2 are required for proper trunk NCC migration. Notably, mice mutated in either ErbB display embryonic lethality, limiting their use within early developmental stages [59, 60]. Honjo and colleagues [61] reported that zebrafish mutants lacking ErbB3 or ErbB2 do not form trunk NC-derived sympathetic neurons, whereas cranial NC derivatives appear normal. These data suggest that ErbB2/ErbB3 signaling is required for trunk NCC migration and localization in the embryo within a specific window of time. Moreover, Sox10 increases the expression of ErbB3, describing a positive activating loop from an intrinsic transcriptional factor that can modulate the migration responsiveness of cells [62–64]. Interestingly, the proto-oncogene MYCN appears to be required for the migration, survival and/or differentiation of NCCs that migrate to the dorsal aorta. Accordingly, homozygous mutant mouse embryos display embryonic lethality (at approximately 11.5 days of gestation), and several developing organs are affected, including the dorsal root ganglia and sympathetic ganglia of the PSNS [65–67].

Expression of crestin in neural crest cells (NCCs) during zebrafish development. a Crestin is expressed in premigratory NCCs in wild-type embryos at 19 hpf. b, c Higher magnification images of the embryo in a, showing the streams of migrating NCCs labeled with the crestin probe. d, e At 24 hpf, trunk migratory NCCs labeled by crestin (arrowheads) gradually migrated toward the ventral dorsal aorta. f–h Cross sections through the levels (f, g, h) shown in d. Streams of trunk NCCs defined by crestin expression migrate between the somite and the neural tube. da dorsal aorta; n notochord; s somite; sc spinal cord
When sympathoadrenal precursors reach the dorsal aorta, other molecules, such as N-Cadherin, CXCL12, and Ephrins, play a critical role in the initiation of the segmental organization of sympathetic ganglia in a discrete and metameric pattern [68]. Moreover, the dorsal aorta-derived bone morphogenetic factors (BMPs) are essential for the differentiation of sympathoadrenal precursors into mature sympathoadrenal cells. Later, cells destined to become chromaffin cells of the adrenal medulla retain their responsiveness to BMPs. Consequently, sympathoadrenal precursors continue to migrate toward the BMP4-secreting cortical cells of the adrenal cortex and finally form the adrenal medulla near the anterior pronephros [69]. The sympathoadrenal lineage is specified by a tightly regulated set of transcription factors, and in zebrafish, this process has been characterized in detail. After reaching the proper location near the dorsal aorta, sympathoadrenal precursors begin to express the pan-neuronal marker 16A11, which comprises members of the Hu family of RNA binding proteins [70]. In 2002, An and colleagues [54] demonstrated that sympathoadrenal cells differentiate from the rostral toward the caudal part of the embryo. In fact, cervical neurons develop at 2 days post-fertilization (dpf) and comprise the superior cervical ganglion (SCG), whereas trunk sympathetic neurons express 16A11 several days later (between 4 and 8 dpf). The reason for this delay remains unknown since trunk sympathoadrenal precursors reach the dorsal aorta between 24 and 36 hpf, when the primary source of BMP is still present. Indeed, since NB can develop anywhere along the sympathetic axis beside the adrenal medulla, it may be relevant to investigate how these cells respond differently to BMPs in terms of both space and time [54]. Thus, it is likely that NB may arise from earlier crest derivatives before the development of the sympathoadrenal lineage [1, 71]. This phenomenon could contribute to the heterogeneous histology and pathology found among NBs.
Other transcription factors are required for sympathoadrenal cell development and maintenance, such as phox2b and zash1a, which in turn activate the expression of hand2 and later markers, such as phox2a, GATA2 and AP-2 alpha (tfap2a) [72–75]. For example, the hand2 zebrafish mutant (hands off) fails to reach the complete differentiation of sympathoadrenal cells, even though some early PSNS markers are normally expressed [74]. An analogous phenotype has been observed in mutant embryos lacking the tfap2a gene (mount blanc mutants), in which sympathoadrenal cells fail to differentiate completely [76, 77].
Finally, sympathoadrenal cells undergo catecholaminergic terminal differentiation for the formation of the mature sympathetic chain and the adrenal gland. This last differentiation step requires the expression of tyrosine hydroxylase (TH) and dopamine B-hydroxylase (DBH) genes, which are involved in the conversion of the amino acid l-tyrosine to noradrenalin [78]. In zebrafish, both genes are used to detect fully differentiated sympathetic neurons. The TH protein is detectable in the superior cervical ganglion starting from 2 dpf, whereas its expression in trunk sympathetic neurons becomes detectable at 7 dpf (Fig. 3). In general, by 10 dpf, the majority of sympathetic neurons express TH, and the complete maturation of sympathetic ganglia and chromaffin cells is reached at 28 dpf [54]. Expression protein analysis of DBH during sympathetic neuron development shows that DBH is detectable 1 day after TH expression [54].

Expression of TH by fully differentiated sympathetic neurons in zebrafish. a Dorsal view of a wild-type embryo probed at 48 hpf by in situ hybridization with TH. At this developmental stage, TH mRNA is expressed by dopaminergic neurons of the ventral diencephalon (hatched box), arch-associated catecholaminergic neurons (arrowhead) and neurons constituting the locus coeruleus (arrow). b Higher magnification of dopaminergic neurons stained with the TH probe
Newly developed high-resolution imaging techniques and genetic tools will continue to provide unique insight into the mechanisms controlling cell differentiation during embryogenesis [79]. Accordingly, the identification of new zebrafish mutants affecting PSNS development might contribute to the in vivo analysis of NB onset.
Zebrafish genetic models of neuroblastoma
As previously described, several in vivo strategies have been used to dissect the molecular pathways that normally coordinate sympathetic neuron development. In particular, forward and reverse genetic techniques and transgenic zebrafish models have provide new insight into the function of the genes and pathways involved in either normal vertebrate development or human cancer formation.
The proto-oncogene MYCN: functional interactions with ALK receptor and the tumor suppressor NF1
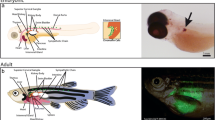
During normal sympathoadrenal development, the proto-oncogene MYCN is highly expressed in the early post-migratory neural crest cells, where it regulates the ventral migration and expansion of NCCs, whereas MYCN protein levels gradually decrease in differentiating sympathetic neurons. This observation suggests that sympathoadrenal maturation requires low or absent MYCN expression [80, 81]. In zebrafish, the persistent expression of the MYCN gene in sympathoadrenal precursors, which mimics the amplification observed in more than 20% of patients with NB, dramatically blocks development toward a chromaffin-like cell phenotype, leading to NB mass formation in the interrenal gland [21, 22]. The tumor formation in the zebrafish interrenal gland recapitulates the adrenal medullary site of origin that is observed in approximately half of the children affected by NB [82]. In contrast, tumors in the murine MYCN transgenic model arise predominately in the sympathetic cervical ganglia complex and the superior cervical ganglia [80, 83]. Tumor masses arising in MYCN-overexpressing zebrafish are histologically, immunohistochemically and ultrastructurally very similar to those arising in humans, supporting the use of this model to investigate NB tumorigenesis. In addition, both the onset and penetrance of this disease are markedly enhanced by the co-expression of the transgene encoding the activated ALK receptor tyrosine kinase, carrying the most common and aggressive activating mutation, F1174L, found in humans [84]. This mutation, although not tumorigenic itself in zebrafish, potentiates the oncogenic power of MYCN by blocking the apoptosis of MYCN-overexpressing sympathoadrenal neuroblasts of the interrenal gland [21]. In fact, this double-transgenic animal model allowed the elucidation of the molecular mechanisms through which MYCN and ALK cooperate in the generation of aggressive tumors. Additional studies using the zebrafish model will be required to determine whether other mutational events can induce NB in a synergistic way, as in the MYCN-ALK model.
More recently, the zebrafish MYCN transgenic model has been used to study which domain of the tumor suppressor protein NF1 is important for inhibiting the growth of NB [85]. When He et al. deleted the gene for the zebrafish version of NF1, the penetrance and the disease onset of MYCN-induced NBs were accelerated and associated with the aberrant activation of the RAS-MAPK signaling. Interestingly, supplying zebrafish with the GTPase-activating protein (GAP)-related domain (GRD) of NF1 was enough to suppress NB growth. In addition, the MEK inhibitor against the RAS-MAPK pathway, trametinib, synergizes with, 13-cis retinoic acid, to suppress nf1-deficient neuroblastoma in vivo. These findings establish the double-transgenic line nf1-deficient/MYCN-overexpressing zebrafish as a valuable tool for testing new therapeutic strategies for NB with mutations affecting the RAS-MAPK pathway, which appear to be a major cause of relapse in patients with NB [85].
The homeobox transcription factor PHOX2B
Previous in vivo models have established PHOX2B as a key regulator of autonomic neuron development. In fact, PHOX2B mutant mice showed embryonic lethality due to a failure of PSNS formation [86]. Moreover, TH-MYCN transgenic mice developed NB in early postnatal sympathetic ganglia predominantly composed of PHOX2B-positive neuronal progenitors [83], and heterozygous germline mutations of PHOX2B are associated with a subset of familial NB [87–90]. These observations indicate a central role of PHOX2B in predisposition to NB.
In 2013, Pei and collaborators [91] reported that morpholino-mediated PHOX2B deletion in zebrafish leads to an impaired sympathetic neuronal differentiation of the PSNS. Similarly, the overexpression of an NB-linked truncation mutation (K155X) [90] or a frameshift mutation (676delG) [85] in the presence of endogenous PHOX2B leads to a similar block in PSNS differentiation, indicating that these variants function in a dominant-negative manner [91]. This study presents an embryonic in vivo model in which aberrant PHOX2B functioning through either allelic deletion or dominant-negative mutations promotes an impaired differentiation of sympathetic neuronal progenitor cells. Future experiments will investigate in more detail the regulatory relationships among the mentioned mutations related to NB.
Aberrant expression of the fgf8 gene in Hagoromo mutants
The histological screening of a collection of zebrafish insertional mutants revealed that four independent lines carrying mutations in the Hagoromo locus develop NB-like tumors with high penetrance [92]. These fish carry viral insertions in the fbxw4 gene that do not affect fbxw4 expression levels but lead to a sustained and aberrant expression of fgf8 after embryogenesis. Notably, tumors arising in Hagoromo mutants are quite different from human NB, since malignant masses develop in cranial ganglia and head of mutant fish. Moreover, tumor cells have a neuronal origin (Hu-positive) but do not express markers of catecholaminergic differentiation, such as TH or DBH, which indicates that these malignant cells originate from a different neural lineage than human NB. Despite these differences, Hagoromo-derived tumors seem to arise through a similar mechanism as in humans [92]. Thus, fgf8 appears to function as an oncogene not only in numerous malignancies, including prostate and breast cancer in mammals [93, 94], but also in NB.
Conclusions
Many cellular and molecular features of NB have been identified over the past decades, and some of these genetic aberrations are now considered strong and powerful predictors of patient outcome. Nevertheless, there is still more complexity behind this pediatric neoplasm, and continued advances are needed to translate this information into more efficient clinical therapy. Although murine and avian models have been used to dissect PSNS development and NB pathogenesis [95], zebrafish displays several advantages, including embryonic optical transparency, small size and external development, making it easier to follow NCC migration and differentiation. Importantly, the generation of new zebrafish models of NB can contribute to the knowledge of NB biology and genetics. Therefore, the generation and use of zebrafish transgenic and mutant lines may provide new opportunities to better understand the biology of NB and to test in vivo potential chemical tumor suppressors through high-throughput drug screening. In addition, zebrafish models of NB crossed with cell signaling reporter fish might be a powerful tool for the in vivo dissection of the signaling cascades and molecular mechanisms involved in tumor development [96, 97].
Abbreviations
- NB:
-
neuroblastoma
- PSNS:
-
peripheral sympathetic nervous system
- NCC:
-
neural crest cell
- OS:
-
overall survival
- CNV:
-
copy number variation
- ALK:
-
anaplastic lymphoma kinase
- EMT:
-
epithelial to mesenchymal transition
- hpf:
-
hours post-fertilization
- BMP:
-
bone morphogenetic factor
- dpf:
-
days post-fertilization
- SCG:
-
superior cervical ganglion
- TH:
-
tyrosine hydroxylase
- DBH:
-
dopamine B-hydroxylase
References
Brodeur GM. Neuroblastoma: biological insight into a clinical enigma. Nat Rev Cancer. 2003;3(3):203–16.
Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010;24(1):65–86. doi:10.1016/j.hoc.2009.11.011.
Goessling W, North TE, Zon LI. New waves of discovery: modeling cancer in zebrafish. J Clin Oncol. 2007;25(17):2473–9.
Stoletov K, Klemke R. Catch of the day: zebrafish as a human cancer model. Oncogene. 2008;27(33):4509–20. doi:10.1038/onc.2008.95.
Li P, Lahvic JL, Binder V, et al. Epoxyeicosatrienoic acids enhance embryonic haematopoiesis and adult marrow engraftment. Nature. 2015;523(7561):468–71. doi:10.1038/nature14569.
Lessman CA. The developing zebrafish (Danio rerio): a vertebrate model for high-throughput screening of chemical libraries. Birth Defects Res C Embryo Today. 2011;93(3):268–80. doi:10.1002/bdrc.20212.
Cotterill SJ, Parker L, More L, et al. Neuroblastoma: changing incidence and survival in young people aged 0–24 years. A report from the North of England Young Persons’ Malignant Disease Registry. Med Pediatr Oncol. 2001;36(1):231–4.
Spix C, Pastore G, Sankila R, et al. Neuroblastoma incidence and survival in European children (1978–1997): report from the Automated Childhood Cancer Information System project. Eur J Cancer. 2006;42(13):2081–91.
Scaruffi P, Coco S, Cifuentes F, et al. Identification and characterization of DNA imbalances in neuroblastoma by high-resolution oligonucleotide array comparative genomic hybridization. Cancer Genetic Cytogenet. 2007;177(1):20–9.
Capasso M, Devoto M, Hou C, et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat Genet. 2009;41(6):718–23. doi:10.1038/ng.374.
Diskin SJ, Hou C, Glessner JT, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009;459(7249):987–91. doi:10.1038/nature08035.
Schwab M. MYCN in neuronal tumours. Cancer Lett. 2004;204(2):179–87.
Maris JM. The biologic basis for neuroblastoma heterogeneity and risk stratification. Curr Opin Pediatr. 2005;17(1):7–13.
Mossé YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–5. doi:10.1038/nature07261.
Passoni L, Longo L, Collini P, et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 2009;69(18):7338–46. doi:10.1158/0008-5472.CAN-08-4419.
George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455(7215):975–8. doi:10.1038/nature07397.
Wang K, Diskin SJ, Zhang H, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. 2011;469(7392):216–20.
Maris JM, Mosse YP, Bradfield JP, et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N Engl J Med. 2008;358(24):2585–93. doi:10.1056/NEJMoa0708698.
Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45(3):279–84. doi:10.1038/ng.2529.
Molenaar JJ, Koster J, Zwijnenburg DA, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483(7391):589–93. doi:10.1038/nature10910.
Zhu S, Lee JS, Guo F, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell. 2012;21(3):362–73. doi:10.1016/j.ccr.2012.02.010.
Zhu S, Look TA. Neuroblastoma and its zebrafish model. Adv Exp Med Biol. 2016;916:451–78. doi:10.1007/978-3-319-30654-4_20.
Coulon A, Flahaut M, Muhlethaler-Mottet A, et al. Functional sphere profiling reveals the complexity of neuroblastoma tumor-initiating cell model. Neoplasia. 2011;13(10):991–1004.
Hansford LM, McKee AE, Zhang L, et al. Neuroblastoma cells isolated from bone marrow metastases contain a naturally enriched tumor-initiating cell. Cancer Res. 2007;67(23):11234–43.
Khanna C, Jaboin JJ, Drakos E, et al. Biologically relevant orthotopic neuroblastoma xenograft models: primary adrenal tumor growth and spontaneous distant metastasis. In vivo. 2002;16(2):77–85.
Patterson DM, Shohet JM, Kim ES. Preclinical models of pediatric solid tumors (neuroblastoma) and their use in drug discovery. Curr Protoc Pharmacol. 2011. Chapter 14:Unit 14.17. doi: 10.1002/0471141755.ph1417s52.
Teitz T, Stanke JJ, Federico S, et al. Preclinical models for neuroblastoma: establishing a baseline for treatment. PLoS ONE. 2011;6(4):e19133. doi:10.1371/journal.pone.0019133.
Bate-Eya LT, Ebus ME, Koster J, et al. Newly-derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur J Cancer. 2014;50(3):628–37. doi:10.1016/j.ejca.2013.11.015.
Joseph JM, Gross N, Lassau N, et al. In vivo ecographic evidence of tumoral vascularization and microenvironment interactions in metastatic orthotopic human neuroblastoma xenografts. Int J Cancer. 2005;113(6):881–90.
Braekeveldt N, Wigerup C, Gisselsson D, et al. Neuroblastoma patient-derived orthotopic xenografts retain metastatic patterns and geno- and phenotypes of patient tumours. Int J Cancer. 2015;136(5):E252–61. doi:10.1002/ijc.29217.
Becher OJ, Holland EC. Genetically engineered models have advantages over zenografts for preclinical studies. Cancer Res. 2006;66(7):3355–8.
Weiss WA, Aldape K, Mohapatra G, et al. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16(11):2985–95.
Moore HC, Wood KM, Jackson MS, et al. Histological profile of tumours from MYCN transgenic mice. J Clin Pathol. 2008;61(10):1098–103.
Weiss WA, Godfrey T, Francisco C, et al. Genome-wide screen for allelic imbalance on a mouse model for neuroblastoma. Cancer Res. 2000;60(9):2483–7.
Hackett CS, Hodgson JG, Law ME, et al. Genome-wide array CGH analysis of murine neuroblastoma reveals distinct genomic aberrations which parallel those in humans tumors. Cancer Res. 2003;63(17):5266–73.
Norris MD, Burkhart CA, Marshall GM, et al. Expression of N-myc and MRP genes and their relationship to N-myc gene disage and tumor formation in a murine neuroblastoma model. Med Pediatr Oncol. 2000;35(6):585–9.
Terrile M, Bryan K, Vaughan L, et al. miRNA expression profiling of the murine TH-MYCN neuroblastoma model reveals similarities with human tumors and identifies novel candidate miRNAs. PLoS ONE. 2011;6(12):e28356. doi:10.1371/journal.pone.0028356.
Molenaar JJ, Domingo-Fernández R, Ebus ME, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet. 2012;44(11):1199–206. doi:10.1038/ng.2436.
Heukamp LC, Thor T, Schramm A, et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci Transl Med. 2012;4(141):141ra91. doi:10.1126/scitranslmed.3003967.
Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277–89. doi:10.1038/nrc3679.
Le Douarin NM, Kalcheim C. The neural crest. Cambridge: Cambridge University Press; 1999.
Lim J, Thiery JP. Epithelial-mesenchymal transitions: insight from development. Development. 2012;139(19):3471–86.
Acloque H, Adams MS, Fishwick K. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119(6):1438–49. doi:10.1172/JCI38019.
Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroad of development and tumor metastasis. Dev Cell. 2008;14(6):818–29. doi:10.1016/j.devcel.2008.05.009.
Pasquier J, Abu-Kaoud N, Al Thani H, et al. Epithelial to mesenchymal transition in a clinical perspective. J Oncol. 2015;2015:792182. doi:10.1155/2015/792182.
Nozato M, Kaneko S, Nakagawara A, et al. Epithelial-mesenchymal transition-related gene expression as a new prognostic marker for neuroblastoma. Int J Oncol. 2013;42(1):134–40. doi:10.3892/ijo.2012.1684.
Kelsh RN, Harris ML, Colanesi S, et al. Stripes and belly-spots—a review of pigment cell morphogenesis in vertebrates. Semin Cell Dev Biol. 2009;20(1):90–104. doi:10.1016/j.semcdb.2008.10.001.
Serbedzija GN, Fraser SE, Bronner-Fraser M. Pathways of trunk neural crest cell migration in the mouse embryo as revealed by vital dye labelling. Development. 1990;108(4):605–12.
Shtukmaster S, Schier MC, Huber K, et al. Sympathetic neurons and chromaffin cells share a common progenitor in the neural crest in vivo. Neural Dev. 2013;8:12. doi:10.1186/1749-8104-8-12.
Theveneau E, Mayor R. Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev Biol. 2012;366(1):34–54. doi:10.1016/j.ydbio.2011.12.041.
Dutt S, Matasci M, Sommer L, et al. Guidance of neural crest cell migration: the inhibitory function of the chondroitin sulfate proteoglycan, versican. ScientificWorldJournal. 2006;6:1114–7.
Perris R, Perissinotto D. Role of the extracellular matrix during neural crest cell migration. Mech Dev. 2000;95(1–2):3–21.
Rorth P. Collective cell migration. Annu Rev Cell Dev Biol. 2009;25:407–29. doi:10.1146/annurev.cellbio.042308.113231.
An M, Luo R, Henion PD. Differentiation and maturation of zebrafish dorsal root and sympathetic ganglion neurons. J Comp Neurol. 2002;446(3):267–75.
Raible DW, Eisen JS. Restriction of neural crest cell fate in the trunk of the embryonic zebrafish. Development. 1994;120(3):495–503.
Dutton KA, Pauliny A, Lopes SS, et al. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development. 2001;128(21):4113–25.
Betters E, Liu Y, Kjaeldgaard A, Sundstrom E, et al. Analysis of early human neural crest development. Dev Biol. 2010;344(2):578–92. doi:10.1016/j.ydbio.2010.05.012.
Rubinstein AL, Lee D, Luo R, et al. Genes dependent on zebrafish cyclops function identified by AFLP differential gene expression screen. Genesis. 2000;26(1):86–97.
Britsch S, Li L, Kirchhoff S, Theuring F, et al. The ErbB2 and ErbB3 receptors and their ligand, neuregulin-1, are essential for development of the sympathetic nervous system. Genes Dev. 1998;12(12):1825–36.
Morris JK, Lin W, Hauser C, et al. Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron. 1999;23(2):273–83.
Honjo Y, Kniss J, Eisen JS. Neuregulin-mediated ErbB3 signaling is required for formation of zebrafish dorsal root ganglion neurons. Development. 2008;135(15):2615–25. doi:10.1242/dev.022178.
Britsch S, Goerich DE, Riethmacher D, et al. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15(1):66–78.
Paratore C, Goerich DE, Suter U, et al. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development. 2001;128(20):3949–61.
Prasad MK, Reed X, Gorkin DU, et al. SOX10 directly modulates ERBB3 transcription via an intronic neural crest enhancer. BMC Dev Biol. 2011;11:40. doi:10.1186/1471-213X-11-40.
Charron J, Malynn BA, Fisher P, et al. Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 1992;6(12A):2248–57.
Sawai S, Shimono A, Wakamatsu Y, et al. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development. 1993;117(4):1445–55.
Stanton BR, Perkins AS, Tessarollo L, et al. Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev. 1992;6(12A):2235–47.
Kasemeier-Kulesa JC, Bradley R, Pasquale EB, Lefcort F, et al. Eph/ephrins and N-cadherin coordinate to control the pattern of sympathetic ganglia. Development. 2006;133(24):4839–47.
Saito D, Takase Y, Murai H, et al. The dorsal aorta initiates a molecular cascade that instructs sympatho-adrenal specification. Science. 2012;336(6088):1578–81.
Yao KM, Samson ML, Reeves R, et al. Gene elav of Drosophila melanogaster: a prototype for neuronal-specific RNA binding protein gene family that is conserved in flies and humans. J Neurobiol. 1993;24(6):723–39.
Maris JM, Hogarty MD, Bagatell R, et al. Neuroblastoma. Lancet. 2007;369(9579):2106–20.
Guillemot F, Joyner AL. Dynamic expression of the murine achaete-scute homologue mash-1 in the developing nervous system. Mech Dev. 1993;42(3):171–85.
Guo S, Brush J, Teraoka H, et al. Development of noradrenergic neurons in the zebrafish hindbrain requires BMP, FGF8, and the homeodomain protein soulless/Phox2a. Neuron. 1999;24(3):555–66.
Lucas ME, Muller F, Rudiger R, et al. The bHLH transcription factor hand2 is essential for noradrenergic differentiation of sympathetic neurons. Development. 2006;133(20):4015–24.
Pattyn A, Morin X, Cremer H, et al. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399(6734):366–70.
Holzschuh J, Barrallo-Gimeno A, Ettl AK, et al. Noradrenergic neurons in the zebrafish hindbrain are induced by retinoic acid and require tfap2a for expression of the neurotransmitter phenotype. Development. 2003;130(23):5741–54.
Knight RD, Nair S, Nelson SS, et al. Lockjaw encodes a zebrafish tfap2a required for early neural crest development. Development. 2003;130(23):5755–68.
Huber K. The sympathoadrenal cell lineage: specification, diversification, and new perspectives. Dev Biol. 2006;298(2):335–43.
Clay MR, Halloran MC. Control of neural crest cell behavior and migration: insights from live imaging. Cell Adh Migr. 2010;4(4):586–94. doi:10.4161/cam.4.4.12902.
Hansford LM, Thomas WD, Keating JM, et al. Mechanisms of embryonal tumor initiation: distinct roles for MycN expression and MYCN amplification. Prot Natl Acad Sci USA. 2004;101(34):12664–9.
Wartiovaara K, Barnabe-Heider F, Miller FD, et al. N-myc promotes survival and induces S-phase entry of postmitotic sympathetic neurons. J Neurosci. 2002;22(3):815–24.
Janoueix-Lerosey I, Schleiermacher G, Delattre O. Molecular pathogenesis of peripheral neuroblastic tumors. Oncogene. 2010;29(11):1566–79. doi:10.1038/onc.2009.518.
Alam G, Cui H, Shi H, et al. MYCN promotes the expansion of Phox2B-positive neuronal progenitors to drive neuroblastoma development. Am J Pathol. 2009;175(2):856–66. doi:10.2353/ajpath.2009.090019.
Chen Y, Takita J, Choi YL, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455(7215):971–4. doi:10.1038/nature07399.
He S, Mansour MR, Zimmerman MW, et al. Synergy between loss of NF1 and overexpression of MYCN in neuroblastoma is mediated by the GAP-related domain. ELife. 2016;5:e14713. doi:10.7554/eLife.14713.
Pattyn A, Morin X, Cremer H, et al. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development. 1997;124:4065–75.
Longo L, Borghini S, Schena F, et al. PHOX2A and PHOX2B genes are highly co-expressed in human neuroblastoma. Int J Oncol. 2008;33(5):985–91.
Perri P, Bachetti T, Longo L, et al. PHOX2B mutations and genetic predisposition to neuroblastoma. Oncogene. 2005;24(18):3050–3.
Mosse YP, Laudenslager M, Khazi D, et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. 2004;75(4):727–30.
Trochet D, Bourdeaut F, Janoueix-Lerosey I, et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. 2004;74(4):761–4.
Pei D, Luther W, Wang W, et al. Distinct neuroblastoma-associated alterations of PHOX2B impair sympathetic neuronal differentiation in zebrafish models. PLoS Genet. 2013;9(6):e1003533. doi:10.1371/journal.pgen.1003533.
Amsterdam A, Lai K, Komisarczuk AZ, et al. Zebrafish Hagoromo mutants up-regulate fgf8 postembryonically and develop neuroblastoma. Mol Cancer Res. 2009;7(6):841–50. doi:10.1158/1541-7786.MCR-08-0555.
Zhong C, Saribekyan G, Liao CP, et al. Cooperation between FGF8b overexpression and PTEN deficiency in prostate tumorigenesis. Cancer Res. 2006;66(4):2188–94. doi:10.1158/0008-5472.CAN-05-3440.
Liu F, You X, Wang Y, et al. The oncoprotein HBXIP enhances angiogenesis and growth of breast cancer through modulating FGF8 and VEGF. Carcinogenesis. 2014;35(5):1144–53. doi:10.1093/carcin/bgu021.
Kasemeier-Kulesa JC, Kulesa PM, Lefcort F. Imaging neural crest dynamics during formation of dorsal root ganglia and sympathetic ganglia. Development. 2005;132:235–45. doi:10.1242/dev.01553.
Moro E, Vettori A, Porazzi P, et al. Generation and application of signaling pathway reporter lines in zebrafish. Mol Genet Genomics. 2013;288(5–6):231–42. doi:10.1007/s00438-013-0750-z.
Schiavone M, Rampazzo E, Casari A, et al. Zebrafish reporter lines reveal in vivo signaling pathways activities involved in pancreatic cancer. Dis Model Mech. 2014;7(7):883–94. doi:10.1242/dmm.014969.
Authors’ contributions
DC designed and drafted the manuscript. SC, MO, SA and GPT were involved in the discussion. GPT revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The present work was supported by Fondazione Italiana per la Lotta al Neuroblastoma. The authors are grateful to Dr. Enrico Moro for the critical reading of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
The present work was funded by Fondazione Italiana per la Lotta al Neuroblastoma.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Corallo, D., Candiani, S., Ori, M. et al. The zebrafish as a model for studying neuroblastoma. Cancer Cell Int 16, 82 (2016). https://doi.org/10.1186/s12935-016-0360-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-016-0360-z