
Abstract
Although most H. pylori infectors are asymptomatic, some may develop serious disease, such as gastric adenocarcinoma, gastric high-grade B cell lymphoma and peptic ulcer disease. Epidemiological and basic studies have provided evidence that infection with H. pylori carrying specific virulence factors can lead to more severe outcome. The virulence factors that are associated with gastric adenocarcinoma development include the presence, expression intensity and types of cytotoxin-associated gene A (CagA, especially EPIYA-D type and multiple copies of EPIYA-C) and type IV secretion system (CagL polymorphism) responsible for its translocation into the host cells, the genotypes of vacuolating cytotoxin A (vacA, s1/i1/m1 type), and expression intensity of blood group antigen binding adhesin (BabA, low-producer or chimeric with BabB). The presence of CagA is also related to gastric high-grade B cell lymphoma occurrence. Peptic ulcer disease is closely associated with cagA-genopositive, vacA s1/m1 genotype, babA2-genopositive (encodes BabA protein), presence of duodenal ulcer promoting gene cluster (dupA cluster) and induced by contact with epithelium gene A1 (iceA1), and expression status of outer inflammatory protein (OipA). The prevalence of these virulence factors is diverse among H. pylori isolated from different geographic areas and ethnic groups, which may explain the differences in disease incidences. For example, in East Asia where gastric cancer incidence is highest worldwide, almost all H. pylori isolates were cagA genopositive, vacA s1/i1/m1 and BabA-expressing. Therefore, selection of appropriate virulence markers and testing methods are important when using them to determine risk of diseases. This review summarizes the evidences of H. pylori virulence factors in relation with gastroduodenal diseases and discusses the geographic differences and appropriate methods of analyzing these virulence markers.
Similar content being viewed by others
Background
H. pylori infection is highly prevalent affecting half of the world’s population. Once infected, H. pylori can be a lifelong infection in the host unless eradicated. Nevertheless, about 85% of the infected patients only have mild asymptomatic gastritis, while 15% of patients can develop to have peptic ulcer disease (PUD), and less than 1% can develop to have gastric cancer [1].
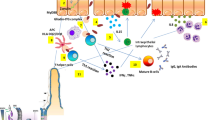
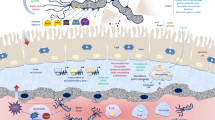
The diverse clinical presentation of H. pylori infection is a result of interaction between bacterial virulence (e.g. CagA, VacA, BabA), host genetic (e.g. IL-1β, IL-10, TNF-α), and environmental factors (e.g. diet, smoke). The virulence factors of H. pylori can be categorized to be related with 3 major pathogenic processes, including colonization, immune escape and disease induction (Table 1). The virulence factors responsible for establishing colonization include urease, flagella, chemotaxis system, and adhesins [2, 3]. Knocking out the urease, flagella or chemotaxis genes all leads to a failure of H. pylori to establish colonization [3]. With increasing antibiotic resistance, these virulence factors provide alternative drug or vaccine targets for H. pylori eradication and prevention [4]. The virulence factors responsible for immune escape help H. pylori escape from host immune clearance and allow its persistence in the human stomach [5]. This review focused on the virulence factors causing more serious clinical outcomes.
Virulence factors associated with gastric adenocarcinoma
Gastric adenocarcinoma is the most deadly disease cause by H. pylori. The virulence factors that are associated with development of gastric adenocarcinoma are summarized in Table 2.
Cytotoxin-associated gene a (CagA) & type IV secretion system (T4SS)
CagA is a well-recognized oncoprotein which is injected into host cells via a pilus structure called type IV secretion system (T4SS) [6]. Successful pilus formation and CagA translocation relies on the binding between CagL on the tip of T4SS and integrin α5β1 receptor on the host cell [7]. The gene locus that encodes CagA and the T4SS is called cag pathogenicity island (cag PAI). After being injected into host cells, CagA alters intracellular signal transduction pathways that facilitates malignant transformation of gastric epithelial cells or activates Lgr5-positive stem cells [8, 9]. Importantly, transgenic mice overexpressing phosphorylation-competent CagA developed gastrointestinal adenocarcinoma, myeloid leukemia and B cell lymphoma, while phosphorylation-resistant CagA could not confer the same pathological changes [10]. These data provided direct evidence that CagA is an oncoprotein and the need of phosphorylation in conferring oncogenesis.
In addition to the above cellular changes, CagA and T4SS also increase gastric inflammation via NFκB signaling and increased IL-8 secretion [11, 12], which predispose to genetic instability and carcinogenesis. CagA can also cause epigenetic changes, such as DNA promoter or histone hypermethylation, which in turn leads to downregulation of tumor suppressor genes (e.g. MGMT) or microRNAs (e.g. let-7) [13, 14]. Alternatively, CagA can attenuate tumor suppressing microRNA expression (e.g. miR-320a, miR-4496) via unknown mechanisms [15, 16]. Therefore, CagA and T4SS can contribute to gastric carcinogenesis via multiple mechanisms.
In concordance with the above-mentioned in vitro and in vivo evidences, several epidemiologic studies also support infection with CagA-positive H. pylori can increase the gastric cancer risk, especially for the non-cardiac location. Most of these studies just tested the serum antibodies against CagA protein to define the status of a CagA-positive H. pylori infection [17,18,19]. Meta-analyses of case-control studies showed CagA-seropositive is associated with 2-fold risk of distal gastric cancer among H. pylori infectors [19], while cagA-genopositive is associated with 2.1–2.4 fold risk of gastric cancer [20]. A cohort study with long-term follow-up also demonstrated that infection with cagA-genopositive strain was associated with greater risk of progression of gastric precancerous lesion (OR = 2.28). However, in East Asia, where nearly all H. pylori strains possess cagA gene [21], presence of serum anti-CagA antibody or cagA gene may not be sensitive enough [22], and CagA subtyping is suggested to identify high risk infectors (Table 2).
Accordingly, the risk of gastric cancer in CagA-positive H. pylori infector can be further stratified by CagA expression level [23, 24], the amount of translocation into host cell [25, 26] and its biological activity [27,28,29]. CagA expression level is higher with the presence of genetic AATAAGATA motif upstream of the translation-starting site, which was associated with greater risk of advanced gastric precancerous lesion [23, 24]. The amount of CagA translocation is greater in strains harboring an amino acid sequence polymorphism (Y58E59) in the CagL of T4SS, which increases its binding affinity with integrin receptor α5β1 on the gastric epithelial cell [26]. Accordingly, gastric cancer risk was increased by 4.6-fold in patients infected by CagL-Y58E59 strain compared with those infected by non-Y58E59 strain [25].
The biological activity of CagA protein is determined by the types and numbers of the EPIYA motifs on its C-terminal region. There are four types of EPIYA motifs based on their distinct conserved flanking sequences, namely EPIYA-A, -B, -C and -D. H. pylori isolates from East Asia where gastric cancer incidence is highest often contains EPIYA A-B-D motif, whereas isolates from Western countries often contains EPIYA A-B-C motif. The A-B-D motif has stronger Src homology 2 phosphatase (SHP-2) binding affinity than the A-B-C motif [27]. A meta-analysis showed 1 EPIYA-D motif was associated with 1.91-fold gastric cancer risk in Asia, compared with 1 EPIYA-C motif [30]. In Western countries, strains harboring multiple EPIYA-C motifs (A-B-C-C or A-B-C-C-C) have higher phosphorylation capacity, SHP-2 binding affinity, and confer higher gastric cancer risk (OR = 3.28) compared with only 1 EPIYA-C motif [30]. Notably, a higher CagA phosphorylation ability was associated with increased risk of gastric precancerous lesions in Taiwan [31]. In addition, amino acid sequence polymorphism within the Western EPIYA-B motif also influences CagA activity, as strains harboring EPIYT-B motif have attenuated ability of inducing hummingbird phenotype and IL-8 in gastric epithelial cells and confer less gastric cancer risk than strains harboring EPIYA-B motifs [29]. Interestingly, EPIYT-B motif was associated with higher duodenal ulcer risk [29].
Vacuolating cytotoxin a (VacA)
VacA was named for its ability to induce vacuole formation in eukaryotic cells. The difference in vacuolating abilities are determined by the variations in the three regions of the vacA gene — the signal (s1 and s2), intermediate (i1 and i2) and middle regions (m1 and m2). A combination of different sequences in the 3 regions leads to multiple alleles and determines the vacuolating activity. Vacuolating activity is high in s1/m1 genotypes, intermediate in s1/m2 genotypes, and absent in s2/m2 genotypes [32]. In clinical isolates, only s1/m2 strain varies in i-type; s1/m1 and s2/m2 strains are exclusively i1 and i2, respectively [33]. The i-type determines vacuolating activity among the s1/m2 strains [33]. Even though the physiologic role of vacuolation is unclear, higher vacuolation activity was linked with more severe clinical outcomes of the infection.
Meta-analysis showed individuals infected with H. pylori harboring vacA s1 or m1 has an increased risk of gastric cancer in Western populations (OR = 2.50–5.32, Table 2) [20]. Moreover, vacA i1 type H. pylori infection is associated with higher gastric cancer risk in the Middle Asia and Middle East area (OR = 10.9–15.0) [34]. However, similar to CagA, the high prevalence of vacA s1/i1/m1 genotype in the East Asian and Southeast Asian population dampens its application as a disease determinant in these areas [35].
Interestingly, the s1/i1/m1 type of vacA is often linked with genopositive cagA [36]. Therefore, neither of the virulence markers can be considered an independent factor for disease outcome [37]. In fact, when multiple virulence factors are present, the risk of severe clinical outcome is greater. For example, in a long-term follow-up cohort, infection with strains that are simultaneously cagA-genopositive and vacA s1/m1 was associated 4.8-fold risk of progression of gastric precancerous lesions compared to those infected with cagA-negative/vacA s2/m2 strains, which was higher than each of the virulence factors considered alone (OR = 2.28–3.38) [38].
Blood group antigen binding adhesin (BabA)
BabA encoded by babA2 gene is a major adhesin on the outer membrane of H. pylori, which recognizes Lewis b (Leb) blood group antigens on the host cells and determines H. pylori colonization density [39, 40]. Two other paralogs of BabA had been found — the BabB and BabC, encoded by babB and babC gene, respectively. The sequence of the 3 bab genes was similar in the 5′ and 3′ region particularly between babA and babB, but the middle region was diverse and likely mediates the binding function. Thus, only BabA has Leb antigen binding ability [41, 42]. The BabA protein expression is mainly regulated by the recombination between babA and babB gene, which forms chimeric proteins (BabA/B or BabB/A) [41, 43, 44]. For example, intra-genomic recombination with babB brings variable number of cytosine-thiamidine (CT) dinucleotide to the 5′-region of the babA sequence, leading to phase variation and affects the expression of BabA [43]. Other mechanisms that regulate BabA expression includes mutation in the coding region of the babA2 gene, or the presence of 4 additional adenines (poly[A] nucleotides) within the − 10 to − 35 region of the babA2 promoter, which diminishes the strength of the promoter [43].
Therefore, using a single pair of PCR primers to determine babA2 genopositivity may not reflect the actual expression status of BabA. This may explain the conflicting results of studies exploring the correlation between babA2 genopositivity and gastric cancer [45]. Fujimoto et al. determined BabA expression level by immunoblotting and classified H. pylori into BabA high producers (BabA-H) with Leb binding activity, BabA low producers (BabA-L) without Leb binding activity, and BabA-negative strain (babA2-genonegative) [46]. Notably, infection with BabA-L strains was associated with highest risk of gastric cancer, followed by infection with BabA-H and BabA-negative strains. In Western countries, infection with BabA-L and BabA-H strain are associated with 54.8-fold and 19.8-fold risk of gastric cancer compared to BabA-negative infectors. Moreover, BabA-L strain infectors had highest gastric H. pylori colonization density, neutrophil infiltration, and mucosal atrophy. However, since all H. pylori isolates from East Asia are either BabA-H or BabA-L, the categorization is not sensitive enough to risk stratify infectors in this area.
In Taiwan, we explored babA and babB recombination using multiple pairs of PCR primers. Four types of babA and babB recombination can be found — the A B genotype without recombination, AB B with babB inserted into babA, A BA with babA inserted into babB, and AB BA with both of the recombination [44]. The isolates from gastric cancer patients had a higher rate of AB BA genotype than those from non-cancer patients (40.0% vs. 9.7%, OR = 6.2, p < 0.05). Interestingly, isolates with AB BA genotype had lower BabA expression level than isolates with A B genotype [44]. Therefore, although babA2-genonegative strain was associated with lowest gastric cancer risk [46], in babA2-genopositive strain, a lower BabA expression level seemed to be associated higher gastric cancer risk [44, 46]. These data suggest multiple pairs of PCR primers should be used to reflect actual BabA status and determine the risk of gastric cancer, especially in East Asia where nearly 100% H. pylori are babA2-genopositive [40, 46].
Notably, H. pylori that simultaneously expresses multiple virulence factors is associated with an even higher risk of severe clinical outcomes. A case-control study showed Infection with strains “triple-positive” for cagA, vacAs1 and babA2 genes significantly correlates to the development of peptic ulcer (p < 0.0001) and adenocarcinoma (p = 0.014) and discriminated adverse disease outcome better than did the dual-positive (cagA and vacA1) classification [47].
High temperature requirement a (HtrA)
H. pylori can secrete proteases as well as induce the expression of host proteases to cleave extracellular matrix and intercellular junctional proteins. Disruption of junctional protein is particularly important for H. pylori to exploit the host receptors located on the basolateral side of the cell membrane, such as integrin [48]. The serine protease and chaperone HtrA is most studied protease expressed by H. pylori. Intracellular HtrA acts as chaperone that refold and degrade misfolded proteins. Thus, HtrA is important for bacterial survival under stressful conditions, such as extreme temperature, pH or salt concentration [49]. All clinical H. pylori isolates possesses htrA gene and suppression of HtrA proteolytic activity is sufficient to kill H. pylori [50]. Therefore, HtrA is a promising target for anti-H. pylori therapy. In addition to the essential role in H. pylori survival, secreted HtrA can cleave E-cadherin and fibronectin [51]. E-cadherin cleavage disrupts cell junctions which exposes basolateral integrin receptors for binding by the H. pylori T4SS, as well as induces epithelial-mesenchymal transition. Since fibronectin has integrin binding motif — RGD, its proteolysis may release integrin receptors on the gastric epithelial cells to interact with H. pylori T4SS, and subsequently facilitate the translocation of CagA [7]. It is novel to assess whether htrA genetic polymorphism is associated with gastric cancer risk, especially in the high gastric cancer incidence area.
Virulence factors associated with gastric B cell lymphoma
Previous studies showed cagA gene was found more frequently (p < 0.05) in the biopsies of gastric high-grade B cell lymphoma (76.7%, 23/30) compared to the gastritis (30.3%, 17/56) and the low-grade lymphoma of the mucosa associated lymphoid tissue (MALToma) cases (37.8%, 14/37) [52]. In addition, the prevalence of serum anti-CagA antibody was higher (p < 0.05) in patients with gastric diffuse large B cell lymphoma (75%, 12/16) than those with low-grade MALToma (44.8%, 13/29) and non-ulcer dyspepsia (43.1%, 22/53) [53]. These data indicates CagA is associated with development of gastric high-grade B cell lymphoma.
In vitro study showed CagA is able to be translocated into human B lymphocytes via T4SS [54]. Once in the cytoplasm, it binds to SHP-2, which stimulates B lymphocyte proliferation and inhibits apoptosis via regulation of intracellular pathways, including activation of endoplasmic reticulum kinases 1 and 2 (ERK 1 and ERK 2) and p38 MAP kinase (MAPK) and increased expression of Bcl-2 and Bcl-xL [54]. Clinical study also showed positive correlations between the expression of CagA and phospho-SHP-2 (p-SHP-2), p-ERK, p-p38 MAPK, Bcl-2 and Bcl-xL in gastric MALToma tissue [55]. Therefore, CagA may promote gastric low-grade MALToma transformation to high-grade B cell lymphoma via the above pathways.
Recently, the genomes of three H. pylori strains isolated from MALToma patients were sequenced. Nine genes were identified to be shared by 3 MALToma strains and absent in the reported 5 gastritis/ulcer strains [56]. Further investigations are needed to clarify the impact of these genes in gastric lymphomagenesis.
Virulence factors associated with peptic ulcer disease (PUD)
The virulence factors that are associated with development of PUD are summarized in Table 3.
Cytotoxin-associated gene a (CagA)
In a large meta-analysis including 44 studies and 17,374 patients from both Eastern and Western regions, CagA-seropositive was associated with a 1.69-fold risk of PUD, which was lower than its association with gastric cancer (OR = 2.44) [20]. However, due to diverse cagA genoprevalence in the various geographic areas, the methods used to identify high risk population for PUD should be different. In Western and Southeast Asian population, where cagA-genopositive rate is lower, cagA-genopositive is sensitive enough to identify high risk infector for PUD [57, 58]. In East Asia, where nearly all H. pylori strains possess cagA gene [21], CagA subtyping is suggested to identify high risk infectors. Accordingly, a meta-analysis showed multiple EPIYA-C motifs is associated with 2.3-fold risk of DU compared with 1 EPIYA-C motif in Asian population [30].
Vacuolating cytotoxin a (VacA)
As mentioned above, the higher vacuolation activity of strains carrying vacA s1, i1 or m1 genotypes were linked with more severe clinical outcomes than the s2, i2 or m2 genotypes [20, 32, 33, 36, 58, 59]. However, similar to cagA genoprevalence, diversity in the prevalence of vacA risky genotypes (s1, i1 and m1) was noted in different geographic regions. Therefore, the use of vacA genotypes to determine PUD risk depends on the prevalence of risky genotypes in each geographic region. In America, Europe, Africa and Middle East where the prevalence of vacA risky genotypes (s1/m1) is lower, individuals infected with vacA s1 or m1 H. pylori strains have an increased risk of PUD compared with those with s2 or m2 strains [20, 32, 36, 59]. In Southeast Asia, vacA m1 is associated with increased risk of PUD [58]. In East Asia, where most strains are vacA s1/i1/s1 genotype, vacA genotypes cannot differentiate high risk infectors, and other virulence markers should be used [35]. The vacA i1 genotype is not associated with risk of PUD in meta-analysis [34].
Blood group antigen binding adhesin (BabA)
Both animal and human studies showed that infection by BabA-expressing strains is associated with higher bacterial density and more severe injury in the gastric mucosa [46, 60]. A meta-analysis of case-control studies showed that babA2 genopositive is associated with an increased risk of PUD (OR = 2.07) in Western countries, but not in Asian countries [45]. As mentioned above, Fujimoto et al. determined BabA expression level by immunoblotting [46]. BabA-L (BabA low producers) and BabA-H (BabA high producers) strains were also associated with higher risk of duodenal ulcer than BabA-negative strains in Western countries (OR = 33.9 and 18.2, respectively) [46]. However, the underlying mechanisms remained to be elucidated. Despite the positive findings of babA2 genopositive and BabA expression intensity in determining peptic ulcer risk in Western countries, these methods are not sensitive enough to differentiate high risk infector in East Asia. Further studies using multiple sets of babA and babB PCR primers [44] are warranted to determine whether babA/B recombination can determine ulcer risk.
Duodenal ulcer promoting gene (DupA)
DupA was initially identified in 2005 and named for its role to increase risk of DU (i.e. duodenal ulcer promoting). The original data showed the presence of dupA gene was associated with increased risk of DU, as well as neutrophil infiltration and IL-8 expression in the antrum [61]. In contrast, its presence was also associated with decreased risk of gastric atrophy, intestinal metaplasia, and gastric cancer [61]. The data are compatible with the findings that antral predominant gastritis often leads to higher gastric acid secretion and duodenal ulcer formation. Nevertheless, although two meta-analyses found a small increase in DU risk (OR = 1.4) by dupA-genopositive strain [62, 63], conflicting results were found [64, 65]. In addition, the association was only found in Asian countries, but not in Western countries [63]. It has been reported that dupA forms T4SS with vir genes around it (called dupA cluster). H. pylori with complete dupA cluster was associated with 2.1-fold risk of DU than that with incomplete dupA cluster or negative dupA [66]. Therefore, merely testing the presence/absence of dupA gene may not reflect the competent function of DupA and analysis of whole dupA cluster may be more accurate to determine DU risk, especially in Western countries [67].
Induced by contact with epithelium gene a (IceA)
The iceA gene was originally identified in 1998 when investigating genes “induced by contact of H. pylori with epithelium” [68]. Two families of iceA have been found, iceA1 and iceA2. Infection with iceA1-genopositive strain is associated with PUD and increased mucosal levels of IL-8 [57, 68, 69]. Meta-analysis showed the presence of iceA1 gene was associated with a small increase of peptic ulcer risk (OR = 1.28) in Western countries [70].
Outer inflammatory protein (OipA)
OipA is an outer membrane protein that functions in adhesion and IL-8 induction. Its functional status (on or off) is regulated by slipped-strand mispairing based on the number of CT dinucleotide repeats in the 5′ region of oipA gene [71]. Infection with oipA “on” strain has been linked with higher H. pylori colonization density, neutrophil infiltration and IL-8 levels in the human stomach [72]. However, the corresponding receptor for OipA has not been identified.
Previous study showed oipA “on” status was closely linked to cagA-positive, vacA s1/m1, and babA2-positive genotypes [73]. Moreover, oipA “on” status was associated with increased risk of DU independent of the other virulence factors [72]. A meta-analysis also reported that the oipA “on”, but not “off”, status is significantly associated with an increased risk of PUD (OR = 3.97) and gastric cancer (OR = 2.43), especially in the Western countries [74]. Importantly, merely investigating the presence/absence of oipA gene would overlook its functional on/off status and may be unreliable to predict risks of PUD or GC [74].
Conclusions
Epidemiological studies had provide evidence that infection with H. pylori carrying specific virulence factors is associated with increased risk of serious clinical outcomes. To identify infectors who are at high risk of serious clinical outcomes, one should select appropriate virulence factors and testing methods according to the epidemiological data of each geographic area and ethnic group.
Abbreviations
- cag PAIc :
-
ag pathogenicity island
- CagA:
-
Cytotoxin-associated gene A
- DU:
-
Duodenal ulcer
- EPIYA:
-
Glu-Pro-Ile-Tyr-Ala
- ERK 1 and ERK 2:
-
Endoplasmic reticulum kinases 1 and 2
- GGT:
-
Gamma-glutamyl transpeptidase
- GU:
-
Gastric ulcer
- HtrA:
-
High temperature requirement A
- LPS:
-
Lipopolysaccharide
- MALToma:
-
B cell lymphoma of the mucosa associated lymphoid tissue
- MGMT:
-
O6-methylguanine DNA methyltransferase
- PUD:
-
Peptic ulcer disease
- RUNX3:
-
Runt-related transcription factor 3
- SHP-2:
-
Src homology 2 phosphatase
- T4SS:
-
Type IV secretion system
- TFF2:
-
Trefoil factor 2
- VacA:
-
Vacuolating cytotoxin A
References
Amieva MR, El-Omar EM. Host-bacterial interactions in helicobacter pylori infection. Gastroenterology. 2008;134(1):306–23.
Sheu BS, Yang HB, Yeh YC, Wu JJ. Helicobacter pylori colonization of the human gastric epithelium: a bug's first step is a novel target for us. J Gastroenterol Hepatol. 2010;25(1):26–32.
Kao CY, Sheu BS, Wu JJ. Helicobacter pylori infection: an overview of bacterial virulence factors and pathogenesis. Biom J. 2016;39(1):14–23.
Menard R, Schoenhofen IC, Tao L, Aubry A, Bouchard P, Reid CW, et al. Small-molecule inhibitors of the pseudaminic acid biosynthetic pathway: targeting motility as a key bacterial virulence factor. Antimicrob Agents Chemother. 2014;58(12):7430–40.
Mejias-Luque R, Gerhard M. Immune evasion strategies and persistence of Helicobacter pylori. Curr Top Microbiol Immunol. 2017;400:53–71.
Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287(5457):1497–500.
Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449(7164):862–6.
Backert S, Blaser MJ. The role of CagA in the gastric biology of Helicobacter pylori. Cancer Res. 2016;76(14):4028–31.
Sigal M, Rothenberg ME, Logan CY, Lee JY, Honaker RW, Cooper RL, et al. Helicobacter pylori activates and expands Lgr5(+) stem cells through direct colonization of the gastric glands. Gastroenterology. 2015;148(7):1392–404. e21
Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A. 2008;105(3):1003–8.
Lamb A, Yang XD, Tsang YH, Li JD, Higashi H, Hatakeyama M, et al. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009;10(11):1242–9.
Gorrell RJ, Guan J, Xin Y, Tafreshi MA, Hutton ML, MA MG, et al. A novel NOD1- and CagA-independent pathway of interleukin-8 induction mediated by the Helicobacter pylori type IV secretion system. Cell Microbiol. 2013;15(4):554–70.
Sepulveda AR, Yao Y, Yan W, Park DI, Kim JJ, Gooding W, et al. CpG methylation and reduced expression of O6-methylguanine DNA methyltransferase is associated with Helicobacter pylori infection. Gastroenterology. 2010;138(5):1836–44.
Hayashi Y, Tsujii M, Wang J, Kondo J, Akasaka T, Jin Y, et al. CagA mediates epigenetic regulation to attenuate let-7 expression in Helicobacter pylori-related carcinogenesis. Gut. 2013;62(11):1536–46.
Kang DW, Yang ES, Noh YN, Hwang WC, Jo SY, Suh YA, et al. MicroRNA-320a and microRNA-4496 attenuate Helicobacter pylori cytotoxin-associated gene a (CagA)-induced cancer-initiating potential and chemoresistance by targeting beta-catenin and ATP-binding cassette, subfamily G, member 2. J Pathol. 2017;241(5):614–25.
Noto JM, Piazuelo MB, Chaturvedi R, Bartel CA, Thatcher EJ, Delgado A, et al. Strain-specific suppression of microRNA-320 by carcinogenic Helicobacter pylori promotes expression of the antiapoptotic protein Mcl-1. Am J Physiol Gastrointest Liver Physiol. 2013;305(11):G786–96.
Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55(10):2111–5.
Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut. 1997;40(3):297–301.
Huang JQ, Zheng GF, Sumanac K, Irvine EJ, Hunt RH. Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology. 2003;125(6):1636–44.
Matos JI, de Sousa HA, Marcos-Pinto R, Dinis-Ribeiro M. Helicobacter pylori CagA and VacA genotypes and gastric phenotype: a meta-analysis. Eur J Gastroenterol Hepatol. 2013;25(12):1431–41.
Yang JC, Wang TH, Wang HJ, Kuo CH, Wang JT, Wang WC. Genetic analysis of the cytotoxin-associated gene and the vacuolating toxin gene in Helicobacter pylori strains isolated from Taiwanese patients. Am J Gastroenterol. 1997;92(8):1316–21.
Zhao Z, Li Y, Liu S, Fu W. Serum Helicobacter pylori CagA antibody may not be used as a tumor marker for diagnosing gastric cancer in east Asian countries. Tumour Biol. 2014;35(12):12217–24.
Loh JT, Shaffer CL, Piazuelo MB, Bravo LE, McClain MS, Correa P, et al. Analysis of cagA in Helicobacter pylori strains from Colombian populations with contrasting gastric cancer risk reveals a biomarker for disease severity. Cancer Epidemiol Biomark Prev. 2011;20(10):2237–49.
Ferreira RM, Pinto-Ribeiro I, Wen X, Marcos-Pinto R, Dinis-Ribeiro M, Carneiro F, et al. Helicobacter pylori cagA promoter region sequences influence CagA expression and interleukin 8 secretion. J Infect Dis. 2016;213(4):669–73.
Yeh YC, Chang WL, Yang HB, Cheng HC, Wu JJ, Sheu BS. H. Pylori cagL amino acid sequence polymorphism Y58E59 induces a corpus shift of gastric integrin alpha5beta1 related with gastric carcinogenesis. Mol Carcinog. 2011;50(10):751–9.
Yeh YC, Cheng HC, Yang HB, Chang WL, Sheu BS. H. pylori CagL-Y58/E59 prime higher integrin alpha5beta1 in adverse pH condition to enhance hypochlorhydria vicious cycle for gastric carcinogenesis. PLoS One. 2013;8(8):e72735.
Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, et al. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A. 2002;99(22):14428–33.
Beltran-Anaya FO, Poblete TM, Roman-Roman A, Reyes S, de Sampedro J, Peralta-Zaragoza O, et al. The EPIYA-ABCC motif pattern in CagA of Helicobacter pylori is associated with peptic ulcer and gastric cancer in Mexican population. BMC Gastroenterol. 2014;14:223.
Zhang XS, Tegtmeyer N, Traube L, Jindal S, Perez-Perez G, Sticht H, et al. A specific A/T polymorphism in Western tyrosine phosphorylation B-motifs regulates Helicobacter pylori CagA epithelial cell interactions. PLoS Pathog. 2015;11(2):e1004621.
Li Q, Liu J, Gong Y, Yuan Y. Association of CagA EPIYA-D or EPIYA-C phosphorylation sites with peptic ulcer and gastric cancer risks: a meta-analysis. Medicine (Baltimore). 2017;96(17):e6620.
Chuang CH, Yang HB, Sheu SM, Hung KH, Wu JJ, Cheng HC, et al. Helicobacter pylori with stronger intensity of CagA phosphorylation lead to an increased risk of gastric intestinal metaplasia and cancer. BMC Microbiol. 2011;11:121.
Atherton JC, Cao P, Peek RM Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270(30):17771–7.
Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M, et al. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology. 2007;133(3):926–36.
Liu X, He B, Cho WC, Pan Y, Chen J, Ying H, et al. A systematic review on the association between the Helicobacter pylori vacA i genotype and gastric disease. FEBS Open Bio. 2016;6(5):409–17.
Ogiwara H, Graham DY, Yamaoka Y. vacA i-region subtyping. Gastroenterology. 2008;134(4):1267. author reply 8
Sugimoto M, Zali MR, Yamaoka Y. The association of vacA genotypes and Helicobacter pylori-related gastroduodenal diseases in the Middle East. Eur J Clin Microbiol Infect Dis. 2009;28(10):1227–36.
Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of helicobacter pylori infection. Clin Microbiol Rev. 2006;19(3):449–90.
Gonzalez CA, Figueiredo C, Lic CB, Ferreira RM, Pardo ML, Ruiz Liso JM, et al. Helicobacter pylori cagA and vacA genotypes as predictors of progression of gastric preneoplastic lesions: a long-term follow-up in a high-risk area in Spain. Am J Gastroenterol. 2011;106(5):867–74.
Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279(5349):373–7.
Sheu BS, Sheu SM, Yang HB, Huang AH, Wu JJ. Host gastric Lewis expression determines the bacterial density of Helicobacter pylori in babA2 genopositive infection. Gut. 2003;52(7):927–32.
Yamaoka Y. Roles of Helicobacter pylori BabA in gastroduodenal pathogenesis. World J Gastroenterol. 2008;14(27):4265–72.
Ansari S, Yamaoka Y. Helicobacter pylori BabA in adaptation for gastric colonization. World J Gastroenterol. 2017;23(23):4158–69.
Backstrom A, Lundberg C, Kersulyte D, Berg DE, Boren T, Arnqvist A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc Natl Acad Sci U S A. 2004;101(48):16923–8.
Sheu SM, Sheu BS, Chiang WC, Kao CY, Wu HM, Yang HB, et al. H pylori clinical isolates have diverse babAB genotype distributions over different topographic sites of stomach with correlation to clinical disease outcomes. BMC Microbiol. 2012;12:89.
Chen MY, He CY, Meng X, Yuan Y. Association of Helicobacter pylori babA2 with peptic ulcer disease and gastric cancer. World J Gastroenterol. 2013;19(26):4242–51.
Fujimoto S, Olaniyi Ojo O, Arnqvist A, Wu JY, Odenbreit S, Haas R, et al. Helicobacter pylori BabA expression, gastric mucosal injury, and clinical outcome. Clin Gastroenterol Hepatol. 2007;5(1):49–58.
Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, et al. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci U S A. 1999;96(22):12778–83.
Posselt G, Crabtree JE, Wessler S. Proteolysis in Helicobacter pylori-induced gastric cancer. Toxins (Basel). 2017;9(4):e134.
Hansen G, Hilgenfeld R. Architecture and regulation of HtrA-family proteins involved in protein quality control and stress response. Cell Mol Life Sci. 2013;70(5):761–75.
Tegtmeyer N, Moodley Y, Yamaoka Y, Pernitzsch SR, Schmidt V, Traverso FR, et al. Characterisation of worldwide Helicobacter pylori strains reveals genetic conservation and essentiality of serine protease HtrA. Mol Microbiol. 2016;99(5):925–44.
Hoy B, Lower M, Weydig C, Carra G, Tegtmeyer N, Geppert T, et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010;11(10):798–804.
Peng H, Ranaldi R, Diss TC, Isaacson PG, Bearzi I, Pan L. High frequency of CagA+ Helicobacter pylori infection in high-grade gastric MALT B-cell lymphomas. J Pathol. 1998;185(4):409–12.
Delchier JC, Lamarque D, Levy M, Tkoub EM, Copie-Bergman C, Deforges I, et al. Helicobacter pylori and gastric lymphoma: high seroprevalence of CagA in diffuse large B-cell lymphoma but not in low-grade lymphoma of mucosa-associated lymphoid tissue type. Am J Gastroenterol. 2001;96(8):2324–8.
Lin WC, Tsai HF, Kuo SH, Wu MS, Lin CW, Hsu P, et al. Translocation of Helicobacter pylori CagA into human B lymphocytes, the origin of mucosa-associated lymphoid tissue lymphoma. Cancer Res. 2010;70(14):5740–8.
Kuo SH, Yeh KH, Chen LT, Lin CW, Hsu PN, Wu MS, et al. Helicobacter pylori CagA translocation is closely associated with the expression of CagA-signaling molecules in low-grade gastric mucosa-associated lymphoid tissue lymphoma. Am J Surg Pathol. 2015;39(6):761–6.
Wang HC, Cheng FC, Wu MS, Shu HY, Sun HS, Wang YC, et al. Genome sequences of three Helicobacter pylori strains from patients with gastric mucosa-associated lymphoid tissue lymphoma. Genome Announc. 2015;3(2):e00229.
van Doorn LJ, Figueiredo C, Sanna R, Plaisier A, Schneeberger P, de Boer W, et al. Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology. 1998;115(1):58–66.
Sahara S, Sugimoto M, Vilaichone RK, Mahachai V, Miyajima H, Furuta T, et al. Role of Helicobacter pylori cagA EPIYA motif and vacA genotypes for the development of gastrointestinal diseases in Southeast Asian countries: a meta-analysis. BMC Infect Dis. 2012;12:223.
Sugimoto M, Yamaoka Y. The association of vacA genotype and Helicobacter pylori-related disease in Latin American and African populations. Clin Microbiol Infect. 2009;15(9):835–42.
Ohno T, Vallstrom A, Rugge M, Ota H, Graham DY, Arnqvist A, et al. Effects of blood group antigen-binding adhesin expression during Helicobacter pylori infection of Mongolian gerbils. J Infect Dis. 2011;203(5):726–35.
Lu H, Hsu PI, Graham DY, Yamaoka Y. Duodenal ulcer promoting gene of helicobacter pylori. Gastroenterology. 2005;128(4):833–48.
Hussein NR. The association of dupA and Helicobacter pylori-related gastroduodenal diseases. Eur J Clin Microbiol Infect Dis. 2010;29(7):817–21.
Shiota S, Matsunari O, Watada M, Hanada K, Yamaoka Y. Systematic review and meta-analysis: the relationship between the Helicobacter pylori dupA gene and clinical outcomes. Gut Pathog. 2010;2(1):13.
Argent RH, Burette A, Miendje Deyi VY, Atherton JC. The presence of dupA in Helicobacter pylori is not significantly associated with duodenal ulceration in Belgium, South Africa, China, or North America. Clin Infect Dis. 2007;45(9):1204–6.
Yeh YC, Cheng HC, Chang WL, Yang HB, Sheu BS. Matrix metalloproteinase-3 promoter polymorphisms but not dupA-H. pylori correlate to duodenal ulcers in H. pylori-infected females. BMC Microbiol. 2010;10:218.
Jung SW, Sugimoto M, Shiota S, Graham DY, Yamaoka Y. The intact dupA cluster is a more reliable Helicobacter pylori virulence marker than dupA alone. Infect Immun. 2012;80(1):381–7.
Talebi Bezmin Abadi A, Perez-Perez G. Role of dupA in virulence of helicobacter pylori. World J Gastroenterol. 2016;22(46):10118–23.
Peek RM Jr, Thompson SA, Donahue JP, Tham KT, Atherton JC, Blaser MJ, et al. Adherence to gastric epithelial cells induces expression of a Helicobacter pylori gene, iceA, that is associated with clinical outcome. Proc Assoc Am Physicians. 1998;110(6):531–44.
Sheu SM, Sheu BS, Yang HB, Li C, Chu TC, Wu JJ. Presence of iceA1 but not cagA, cagC, cagE, cagF, cagN, cagT, or orf13 genes of Helicobacter pylori is associated with more severe gastric inflammation in Taiwanese. J Formos Med Assoc. 2002;101(1):18–23.
Shiota S, Watada M, Matsunari O, Iwatani S, Suzuki R, Yamaoka Y. Helicobacter pylori iceA, clinical outcomes, and correlation with cagA: a meta-analysis. PLoS One. 2012;7(1):e30354.
Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci U S A. 2000;97(13):7533–8.
Yamaoka Y, Kikuchi S, el-Zimaity HM, Gutierrez O, Osato MS, Graham DY. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology. 2002;123(2):414–24.
Dossumbekova A, Prinz C, Mages J, Lang R, Kusters JG, Vliet AH, et al. Helicobacter pylori HopH (OipA) and bacterial pathogenicity: genetic and functional genomic analysis of hopH gene polymorphisms. J Infect Dis. 2006;194(10):1346–55.
Liu J, He C, Chen M, Wang Z, Xing C, Yuan Y. Association of presence/absence and on/off patterns of Helicobacter pylori oipA gene with peptic ulcer disease and gastric cancer risks: a meta-analysis. BMC Infect Dis. 2013;13:555.
Author information
Authors and Affiliations
Contributions
BSS initiated the idea, guided the article structure, and improved the final manuscript. WLC reviewed the published studies and composed the draft of the manuscript. YCY checked and confirmed the analytic methods of each virulence markers. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chang, WL., Yeh, YC. & Sheu, BS. The impacts of H. pylori virulence factors on the development of gastroduodenal diseases. J Biomed Sci 25, 68 (2018). https://doi.org/10.1186/s12929-018-0466-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-018-0466-9