
Abstract
Background
Chromosome 6pter-p24 deletion syndrome (OMIM #612582) is a recognized chromosomal disorder. Most of the individuals with this syndrome carry a terminal deletion of the short arm of chromosome 6 (6p) with a breakpoint within the 6p25.3p23 region. An approximately 2.1 Mb terminal region has been reported to be responsible for some major features of the syndrome. The phenotypic contributions of other deleted regions are unknown. Interstitial deletions of the region are uncommon, and reciprocal interstitial duplication in this region is extremely rare.
Case presentation
We present a family carrying an interstitial deletion and its reciprocal duplication within the 6p25.1p24.3 region. The deletion is 5.6 Mb in size and was detected by array comparative genomic hybridization (aCGH) in a 26-month-old female proband who presented speech delay and mild growth delay, bilateral conductive hearing loss and dysmorphic features. Array CGH studies of her family members detected an apparently mosaic deletion of the same region in the proband’s mildly affected mother, but a reciprocal interstitial duplication in her phenotypically normal brother. Further chromosomal and fluorescence in situ hybridization (FISH) analyses revealed that instead of a simple mosaic deletion of 6p25.1p24.3, the mother actually carries three cell populations in her peripheral blood, including a deletion (~70 %), a duplication (~8 %) and a normal (~22 %) populations. Therefore, both the deletion and duplication seen in the siblings were apparently inherited from the mother.
Conclusions
Interstitial deletion within the 6p25.1p24.3 region and its reciprocal duplication may co-exist in the same individual and/or family due to mitotic unequal sister chromatid exchange. While the deletion causes phenotypes reportedly associated with the chromosome 6pter-p24 deletion syndrome, the reciprocal duplication may have no or minimal phenotypic effect, suggesting possible triploinsensitivity of the same region. In addition, the cells with the duplication may compensate the phenotypic effect of the cells with the deletion in the same individual as implied by the maternal karyotype and her mild phenotype. Chromosomal and FISH analyses are essential to verify abnormal cytogenomic array findings.
Similar content being viewed by others

Background
Deletion of the distal part of 6p is a clinically recognized syndrome (chromosome 6pter-p24 deletion syndrome, OMIM #612582) characterized by developmental delay/mental retardation, language impairment, Dandy-Walker malformation, conductive hearing loss, anomalies of the anterior chamber of eyes, cardiac abnormalities, and craniofacial dysmorphism [10]. More than 30 deletion cases have been reported and the vast majority of them involve terminal deletions (0.3 - 14 Mb in size) with breakpoints within the 6p25.3p23 region [11, 12, 17, 21, 24, 25]. Only four interstitial deletions within this region have been reported [7, 14, 21]. An approximately 2.1 Mb commonly deleted terminal region was identified, which is possibly responsible for some of the major features of the syndrome [1, 10]. However, the phenotypic contributions of other deleted regions are unknown. On the other hand, duplications of the 6p25.3p23 region are extremely rare. There are only two cases with eye and cerebellar abnormalities reported in literature [29, 34].
We present here the phenotypic and genomic findings in a family with an affected female proband who carries an interstitial deletion within the 6p25.1p24.3 region, her phenotypically unaffected brother with a reciprocal interstitial duplication of the same region, and their mosaic carrier mother who carries three cell populations with the deletion, the duplication and normal cells, respectively. This rare family provided an excellent opportunity to investigate the genomic etiology and genotype-phenotype correlation of the chromosome 6pter-p24 deletion syndrome.
Case presentation
Proband
The female proband was born to a 33-year-old G2P2 mother at 37-weeks gestation by spontaneous vaginal delivery without complications. Pregnancy was unremarkable except for possible clubfoot noted on ultrasound scan. Birth weight was 2,440 g (20th centile), length 45.5 cm (10th centile), and head circumference 31 cm (10th centile). A hemangioma on the neck was diagnosed at one week after birth. A small patent foramen ovale (PFO, 3 mm x 4 mm) was found at 12 months of age and has been followed without surgical intervention. The proband was referred for clinical genetic evaluation at the age of 26 months for dysmorphic features, speech delay and mild growth delay. She walked at 15–16 months and her fine motor skills were age appropriate. She had four-five words at two years of age. When last reviewed at three years nine months of age, she was able to pronounce words with three syllables and had more than one hundred words. She understood multistep commands and exhibited age-appropriate behavior.
She had mild to slight conductive hearing loss at 500–4000 Hz with a notch or normal hearing at 2000 Hz and she used bilateral hearing aids from age two years 11 months until three years two months, when her 10–15 dB loss had improved. Her teeth were late to erupt and she was missing three primary teeth.
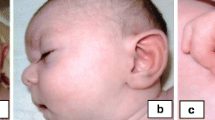
At thee years and nine months of age, height was 90 cm (4rd centile), weight was 13.34 kg (16th centile) and occipitofrontal circumference was 48 cm (19th centile). She showed mild dysmorphic features, including sparse frontal hair with a high anterior hairline, hypertelorism with an interpupillary distance (IPD) measuring 5.8 cm (>97th centile), synophrys, a preauricular pit on the left side, short philtrum with a short columella, downturned corners of the mouth, and small, widely spaced teeth (Fig. 1). She had a resolving hemangioma on the neck that measured five cm, pectus excavatum and a small, reducible umbilical hernia. Her fingers were small with mild fifth finger clinodactyly, but measurements did not show brachydactyly. The second toe overlapped the third toe on right foot.

Facial photograph of the proband at the age of thee years and nine months. The picture shows high anterior hairline with sparse frontal hair, synophrys, upslanting palpebral fissures with hypertelorism and a broad nasal bridge and a short philtrum
Proband’s brother
The proband’s brother was delivered at 39-weeks gestation to the same mother (30 years old, G1P1) without complications. His birth weight, length, and head circumference were 3,650 g, 49.0 cm, and 35 cm, respectively, and all were within the normal range. At age of 5 years, his growth and development were appropriate for age. He had small epicanthic folds and mild clinodactyly of the fifth fingers and toes with mildly small fifth toes, but there were no other findings.
Proband’s mother
The proband’s mother is a 35-year old and typically developed female. She had an embolic stroke at age of 26 years. Investigations with an echocardiogram showed a PFO with an atrial septal aneurysm and the PFO was closed using a transcatheter approach. Her hypercoagulability workup was negative. She had dyslipidemia with a slightly elevated lipoprotein level. Her family history was unremarkable for cardiac disease.
The proband’s father is a normal healthy male.
Methods
Array CGH analysis
Genomic DNA was extracted from peripheral blood using QIAGEN EZ1 kit (QIAGEN). Array CGH was performed using a custom 180 k oligonucleotide-based microarray with an International Standards for Cytogenomic Arrays Consortium V1 Clinical Design complying with the human genome build GRCh37/hg19 (Illumina). Array CGH was set up according to manufacturer’s instruction and the data were analyzed using BlueFuse Multi software (Illumina) and the UCSC genome browser (http://genome.ucsc.edu).
Conventional cytogenetic analysis
Peripheral blood cells were cultured, harvested and banded according to the standard cytogenetic methods [3]. GTG-banded chromosomes were analyzed and imaged using the CytoVision system (Leica Microsystems). Chromosomal abnormalities were described with an International System for Human Cytogenetic Nomenclature 2013 (ISCN 2013) [28].
FISH
FISH was performed to further confirm the aCGH and chromosomal findings. Two directly labeled bacteria artificial chromosome (BAC) clones (Illumina), RP11-339A7 (6p24.3, labeled with green fluorochrome) and RP3-520B18 (6p25.1, labeled with red fluorochrome) were used as FISH probes (Fig. 2c). The hybridization was performed according to the manufacturer’s instruction and the cells were counterstained with DAPI II (Abbott Molecular) after the hybridization. FISH results were analyzed and documented using CytoVision system (Leica Microsystems).

Array CGH and FISH analyses of the family. a. Array CGH detected an interstitial deletion within 6p25.1p24.3 in the proband, a mosaic deletion in her mother, and an interstitial duplication of the same region in her brother. b. Chromosomal analysis revealed normal, duplicated and deleted chromosome 6 s in the mother. c. Metaphase FISH showed a duplication signal pattern in a metaphase cell from the mother using two BAC FISH probes RP3-520B18 and RP11-339A7 (arrow indicates the duplication of 6p). d. Interphase FISH demonstrated normal, duplication, and deletion signal patterns of 6p in the mother
Results
Array CGH findings
Array CGH analysis of the proband revealed a 5.6 Mb interstitial deletion within the 6p25.1p24.3 region: arr[hg19] 6p25.1p24.3(4,745,144-10,384,769)x1 (Fig. 2a). This deleted region carries 17 annotated genes in the database of online Mendelian inheritance in man (OMIM) (CDYL, RPP40, LYRM4, FARS2, NRN1, F13A1, LY86, RREB1, SSR1, CAGE1, DSP, BMP6, BLOC1S5, EEF1E1, SLC35B3, HULC and OFCC1) and 10 other genes (PPP1R3G, LY86-AS1, SNAPC5P1, CNN3P1, BTF3P7, RIOK1, SNRNP48, TXNDC5, BLOC1S5-TXNDC5 and PIP5K1P1).
The mother showed an apparently mosaic loss of the same region seen in the proband (Fig. 2a). The father was normal, in particular without detectable copy changes involving the 6p region (data not shown).
Due to the mother’s carrier status, the proband’s brother was also tested per the family’s request. Surprisingly, instead of the deletion, an interstitial duplication of the same region was detected: arr[hg19] 6p25.1p24.3(4,745,144-10,384,769)x3 (Fig. 2a). This result implied that the duplication might also be derived from the maternal genomic changes that could be more complicated than a simple mosaic deletion.
Chromosome and FISH findings
To further evaluate the unusual array findings, conventional cytogenetic analyses of the siblings and their mother were performed. Indeed, the mother actually has three different cell populations. Of a total of 50 metaphase cells examined, 36 (72.0 %) showed the 6p deletion, 3 (6.0 %) showed the 6p duplication, and the remaining 11 (22.0 %) were normal. Her karyotype is: 46,XX,del(6) (p24p25.1) [36]/46,XX,dup(6) (p24p25.1) [3]/46,XX [11] (Fig. 2b).
The chromosome results were confirmed by both metaphase and interphase FISH studies using two BAC probes located at the proximal and distal ends of the deletion/duplication region, respectively, including RP11-339A7 (6p24.3, labeled with green fluorochrome) and RP3-520B18 (6p25.1, labeled with red fluorochrome) (Fig. 2c). Consistent with the chromosomal findings, all three cell populations were detected in the mother; deletion, duplication and normal signal patterns were detected in approximately 70.0 % (350/500), 8.4 % (42/500) and 21.6 % (108/500) of the interphase cells examined, respectively (Fig. 2d). The metaphase FISH further confirmed the deletion/duplication and provided no evidence of other rearrangements involving the 6p25.1p24.3 region (Fig. 2c). The deletion and duplication in the siblings were also confirmed by FISH (data not shown). These findings demonstrated that the proband and her brother apparently inherited the deletion and duplication from their carrier mother, respectively.
Discussion
The vast majority of deletions seen in the chromosome 6pter-p24 deletion syndrome are terminal deletions. The smallest reported terminal deletion was approximately 2.1 Mb (Fig. 3). This region was suggested to be responsible for some major features of the syndrome [1, 10]. However, the phenotypic contributions of other deleted 6p regions remain unknown. Isolated interstitial deletions within 6p25p24 without involvement of the terminal 2.1 Mb region are uncommon. To our knowledge, only five such deletions, including the one presented in this study, have been reported (Fig. 3) [7, 14, 21]. Individuals with these deletions share many common phenotypic features of the 6pter-p24 deletion syndrome (Table 1, also see the review of DeScipio [10]). Based on the breakpoints of the five overlapping interstitial deletions, nine sub-regions (R1-R9) can be defined along with annotated OMIM genes (Fig. 3 and Table 2). The region R5 (6,100,000-8,330,000 bp) that carries nine OMIM genes appears to be the consensus region of the deletions. Three genes in this region, including RREB1, DSP and BMP6, are of particular interest. The RREB1 gene encodes a zinc finger transcription factor (RREB1 protein) that binds to RAS-responsive elements (RREs). It has been suggested that RREB1 is possibly involved in RAS/RAF-mediated cell differentiation and augments the RAS/RAF-mediated transcriptional response [31]. The RAS/RAF/MEK/ERK-signal transduction pathway is known to be associated with Noonan syndrome (OMIM #163950), Costello syndrome (OMIM #218040) and Cardio-Facial-Cutaneous syndrome (OMIM# 115150) [27, 32]. These syndromes share some of the phenotypic features of the 6p25.1p24.3 deletions, such as cardiac abnormalities, craniofacial dysmorphism and hemangioma [14, 27, 32]. Therefore, we speculate that deletion of the RREB1 gene may underlie some, if not all, of these phenotypes through the RAS/RAF signal pathway. The DSP gene encodes desmoplakin protein that is an obligate component of functional desmosomes (the intercellular junctions that tightly link adjacent cells). Desmoplakin haploinsufficiency was suggested to be associated with keratosis palmoplantaris striata II (PPKS2; OMIM #612908), and recessive mutations in the DSP gene have been associated with skin fragility/woolly hair syndrome (OMIM #607655) and dilated cardiomyopathy (OMIM #605676) [2, 23]. Deletion of DSP could be related to abnormal findings of heart and hair. The BMP6 gene encodes a bone morphogenetic protein 6. Kugimiya et al. and Meynard et al. reported that Bmp6 knockout mice had a delay in ossification, which was strictly confined to sternum [13, 19]. Thus, deletion of the BMP6 gene could be the potential cause of pectus excavatum that was observed in four of the five individuals with the 6p25.1p24.3 deletions.

Reported deletions involving the 6pter-p23 region. Light gray bars represent the deletions involving the terminal 2.1 Mb region; dark gray bars represent the interstitial deletions without the involvement of the terminal 2.1 Mb region; black bar represents the present case of this study. The breakpoints of the five interstitial deletions divide the 6p25.2p24.3 into nine sub-regions, R1-R9. The breakpoint coordinates of Mirza et al. case 5 and case 6 and Davies et al. are estimated based on FISH data [7, 21]
In addition, the region R4 contains the NRN1 gene. The gene’s homologue in mouse encodes neuritin 1 protein that is involved in synaptic plasticity during the brain development possibly through controlling neuronal migration [22, 36]. This implies that deletion of NRN1 may contribute to motor developmental delay. The region R7 contains the OFCC1 gene that has been associated with isolated cleft lip with or without cleft palate (OFC1; OMIM %119530) [8]. One of the three cases with deletion of the OFCC1 gene showed bilateral cleft lips (Table 1, Fig. 3), supporting the association. The region R9 contains the TFAP2A gene, of which haploinsufficiency may be associated with eye anomalies [7, 20]. Consistent with this, the case 3 showed microphthalmia with corneal clouding and it was the only case carried deletion of the region R9 including the TFAP2A gene (Table 2).
Based on the co-existence of phenotype/deletion and reported functions of the genes located in each deletion sub-region, some deletion-related phenotypes could be potentially mapped to the specific deletion sub-regions (Table 2). Such a phenotypic map, although it needs to be further verified, is important for understanding the genomic pathogenesis of the deletions. The phenotypes may not always manifest consistently together with the expected deletion regions. This could be due to incomplete penetrance/expressivity. Also, some phenotypes might not be examined or apparent at the time of reporting. Some shared phenotypes between the individuals with and without the 2.1 Mb terminal deletion could be due to the overlapping deletion regions of the two groups (Fig. 3). It is also possible that some of the phenotypes could be caused by different genomic mechanisms or by multiple genomic alterations.
To our knowledge, only two duplications involving the distal part of 6p have been reported with abnormal clinical findings [29, 34]. However, one of them did not overlap with, and the other was 4.5 Mb (80 %) larger in size (with many additional genes involved) than the present duplication. We also searched DGV (database of genomic variants, http://dgv.tcag.ca/), ClinGen (database of submitted clinical array testing results, http://clinicalgenome.org/) and DECIPHER (database of genomic variation and phenotype in humans using Ensembl resources, https://decipher.sanger.ac.uk/) databases, but no similar duplications were found. The DECIPHER database lists 10 much smaller duplications (0.09 – 2.4 MB) within the 6p region. However, three of them involve multiple additional genomic abnormalities and another three are listed without phenotypes. The phenotypic features associated with the remaining four duplications are not described in detail; in particular, no consistent specific phenotypes are listed. Therefore, the phenotypic features associated with those small duplications could not be determined. In contrast, the proband’s brother who carries a duplication within 6p25.1p24.3 in this study is phenotypically normal at the age of 5 years, suggesting that copy gain of this particular region is likely benign or triploinsensitive. It is noteworthy that approximately 70 % of the blood cells of the proband’s mother carry the deletion, but she apparently does not have most of the phenotypic features seen in the proband except for a mild cardiac anomaly. This implies that dosage compensation from the cells with the duplication might play a role in reducing the phenotypic effect of the deletion, although this might also be due to tissue-specific mosaicism that was not tested in this study. Nevertheless, the findings suggest that the level of phenotypic expression of the 6p25.1p24.3 deletion seems to be related with the overall level of mosaicism of the deletion.
The mosaic reciprocal deletion and duplication in the mother were most likely derived from a mitotic unequal sister chromatid exchange (Fig. 4). When this occurs between two non-allelic loci in a cell during mitosis, it gives rise to two cell populations with a deletion and a reciprocal duplication, respectively, along with an uninvolved normal cell population in an individual. This mitotic non-allelic recombination most likely occurred at an early stage during the maternal embryogenesis, since the percentage of the deletion cell population appeared to be high and the mosaicism was present in multiple tissues, such as blood and gonadal tissues. The mechanism for this unequal sister chromatid exchange is unknown. No segmental duplications, except for a 277-bp Alu repeat, were found in the proximity of the breakpoints in our search using the UCSC genome browser. However, whether this repeat was involved in the unequal exchange needs to be further determined, although Alu repeats have been reported to participate in some homologous recombination events [9, 26].

Unequal sister chromatid exchange involving the 6p25.1p24.3 region
Our study clearly demonstrated the necessity to verify abnormal cytogenomic array findings with cytogenetic methods, including chromosomal analysis when the changes are >3 Mb and metaphase FISH when the changes are <3 Mb. Cytogenetic methods do not only confirm the array findings, but also allow identification of causal structural rearrangements of the array findings and multiple cell populations, as seen in this study, which would otherwise be missed. Such additional information is critical to understand the genomic pathogenesis, to make appropriate decision for clinical management and to provide appropriate counseling to the patient and his/her family.
Conclusions
Our study demonstrated that an interstitial deletion within 6p25.1p24.3 and its reciprocal duplication could occur through mitotic unequal sister chromatid exchange. Both deletion and duplication may be passed independently to the offspring from a parental carrier if his/her gonadal system is affected. Cytogenomic array may detect the deletion or duplication in an individual, but may not detect both in a mosaic carrier. Therefore, the array findings could be misleading in such a mosaic situation. Chromosome and FISH analyses are appropriate methods to identify structural and copy number changes, as well as different cell populations. So, it is essential to verify abnormal cytogenomic array findings with chromosomal and FISH analyses when they are available. In review of other reported interstitial deletions within 6p25.1p24, nine deletion sub-regions with annotated OMIM genes, including a 2.3 Mb consensus region, are defined. Based on the known functions of the annotated genes and the phenotype/deletion co-existence, some phenotypes associated with the deletions can be potentially mapped to specific deletion regions. While the deletion in the proband of this study results in phenotypes reportedly associated with the chromosome 6pter-p24 deletion syndrome due to haploinsufficiency of the region, its reciprocal duplication appears not to have significant phenotypic effect, suggesting possible triploinsensitivity of the same region. In addition, the cells with the duplication may compensate the phenotypic effect of the cells with the deletion in the same individual as implied by the maternal karyotype and her mild phenotype.
Consent
Written informed consent was obtained from the proband’s mother for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Abbreviations
- aCGH:
-
Array comparative genomic hybridization
- BAC:
-
Bacteria artificial chromosome
- DGV:
-
Database of genomic variants
- DECIPHER:
-
Database of genomic variation and phenotype in humans using Ensembl resources
- FISH:
-
Fluorescence in situ hybridization
- ISCN:
-
International system for human cytogenetic nomenclature
- OMIM:
-
Online Mendelian inheritance in man
References
Anderlid BM, Schoumans J, Hallqvist A, Stahl Y, Wallin A, Blennow E, et al. Cryptic subtelomeric 6p deletion in a girl with congenital malformations and severe language impairment. Eur J Hum Genet. 2003;11(1):89–92.
Armstrong DK, McKenna KE, Purkis PE, Green KJ, Eady RA, Leigh IM, et al. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet. 1999;8(1):143–8.
Barch MJ, Knutsen T, Spurbeck KL: AGT Cytogenetics Laboratory Manual. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 1997.
Beby F, Des Portes V, Till M, Mottolese C, Denis P. Chromosome 6p25 deletion syndrome: report of a case with optic disc coloboma and review of published ophthalmic findings. Ophthalmic Genet. 2012;33(4):240–8.
Caluseriu O, Mirza G, Ragoussis J, Chow EW, MacCrimmon D, Bassett AS. Schizophrenia in an adult with 6p25 deletion syndrome. Am J Med Genet A. 2006;140(11):1208–13.
Chen KM, Cherry AM, Hahn JS, Enns GM. Mild developmental delay in terminal chromosome 6p deletion. Am J Med Genet A. 2004;129A(2):201–5.
Davies AF, Mirza G, Flinter F, Ragoussis J. An interstitial deletion of 6p24-p25 proximal to the FKHL7 locus and including AP-2alpha that affects anterior eye chamber development. J Med Genet. 1999;36(9):708–10.
Davies SJ, Wise C, Venkatesh B, Mirza G, Jefferson A, Volpi EV, et al. Mapping of three translocation breakpoints associated with orofacial clefting within 6p24 and identification of new transcripts within the region. Cytogenet Genome Res. 2004;105(1):47–53.
de Smith AJ, Walters RG, Coin LJ, Steinfeld I, Yakhini Z, Sladek R, et al. Small deletion variants have stable breakpoints commonly associated with alu elements. PLoS One. 2008;3(8):e3104.
DeScipio C. The 6p subtelomere deletion syndrome. Am J Med Genet C: Semin Med Genet. 2007;145C(4):377–82.
Descipio C, Schneider L, Young TL, Wasserman N, Yaeger D, Lu F, et al. Subtelomeric deletions of chromosome 6p: molecular and cytogenetic characterization of three new cases with phenotypic overlap with Ritscher-Schinzel (3C) syndrome. Am J Med Genet A. 2005;134A(1):3–11.
Koolen DA, Knoers NV, Nillesen WM, Slabbers GH, Smeets D, de Leeuw N, et al. Partial iris hypoplasia in a patient with an interstitial subtelomeric 6p deletion not including the forkhead transcription factor gene FOXC1. Eur J Hum Genet. 2005;13(11):1169–71.
Kugimiya F, Kawaguchi H, Kamekura S, Chikuda H, Ohba S, Yano F, et al. Involvement of endogenous bone morphogenetic protein (BMP) 2 and BMP6 in bone formation. J Biol Chem. 2005;280(42):35704–12.
Kuipers BC, Vulto-van Silfhout AT, Marcelis C, Pfundt R, de Leeuw N, de Vries BB. Two patients with intellectual disability, overlapping facial features, and overlapping deletions in 6p25.1p24.3. Clin Dysmorphol. 2013;22(1):18–21.
Law CJ, Fisher AM, Temple IK. Distal 6p deletion syndrome: a report of a case with anterior chamber eye anomaly and review of published reports. J Med Genet. 1998;35(8):685–9.
Le Caignec C, De Mas P, Vincent MC, Boceno M, Bourrouillou G, Rival JM, et al. Subtelomeric 6p deletion: clinical, FISH, and array CGH characterization of two cases. Am J Med Genet A. 2005;132A(2):175–80.
Lin RJ, Cherry AM, Chen KC, Lyons M, Hoyme HE, Hudgins L. Terminal deletion of 6p results in a recognizable phenotype. Am J Med Genet A. 2005;136(2):162–8.
Martinet D, Filges I, Besuchet Schmutz N, Morris MA, Gaide AC, Dahoun S, et al. Subtelomeric 6p deletion: clinical and array-CGH characterization in two patients. Am J Med Genet A. 2008;146A(16):2094–102.
Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41(4):478–81.
Milunsky JM, Maher TA, Zhao G, Roberts AE, Stalker HJ, Zori RT, et al. TFAP2A mutations result in branchio-oculo-facial syndrome. Am J Hum Genet. 2008;82(5):1171–7.
Mirza G, Williams RR, Mohammed S, Clark R, Newbury-Ecob R, Baldinger S, et al. Refined genotype-phenotype correlations in cases of chromosome 6p deletion syndromes. Eur J Hum Genet. 2004;12(9):718–28.
Naeve GS, Ramakrishnan M, Kramer R, Hevroni D, Citri Y, Theill LE. Neuritin: a gene induced by neural activity and neurotrophins that promotes neuritogenesis. Proc Natl Acad Sci U S A. 1997;94(6):2648–53.
Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9(18):2761–6.
Palmer CG, Bader P, Slovak ML, Comings DE, Pettenati MJ. Partial deletion of chromosome 6p: delineation of the syndrome. Am J Med Genet. 1991;39(2):155–60.
Plaja A, Vidal R, Soriano D, Bou X, Vendrell T, Mediano C, et al. Terminal deletion of 6p: report of a new case. Ann Genet. 1994;37(4):196–9.
Prak ET, Kazazian Jr HH. Mobile elements and the human genome. Nat Rev Genet. 2000;1(2):134–44.
Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355–69.
Shaffer L.G., McGowan-Jordan J., Schmid M. ISCN 2013: An International System for Human Cytogenetic Nomenclature. Basel: S. Karger; 2013.
Stohler R, Kucharski E, Farrow E, Torres-Martinez W, Delk P, Thurston VC, et al. A case of de novo partial tetrasomy of distal 6p and review of the literature. Am J Med Genet A. 2007;143A(17):1978–83.
Suzuki K, Nakamura M, Amano E, Mokuno K, Shirai S, Terasaki H. Case of chromosome 6p25 terminal deletion associated with Axenfeld-Rieger syndrome and persistent hyperplastic primary vitreous. Am J Med Genet A. 2006;140(5):503–8.
Thiagalingam A, De Bustros A, Borges M, Jasti R, Compton D, Diamond L, et al. RREB-1, a novel zinc finger protein, is involved in the differentiation response to Ras in human medullary thyroid carcinomas. Mol Cell Biol. 1996;16(10):5335–45.
Tidyman WE, Rauen KA. Noonan, Costello and cardio-facio-cutaneous syndromes: dysregulation of the Ras-MAPK pathway. Expert Rev Mol Med. 2008;10:e37.
Topping A, Harris P, Moss AL. The 6p deletion syndrome: a new orofacial clefting syndrome and its implications for antenatal screening. Br J Plast Surg. 2002;55(1):68–72.
Vermeesch JR, Thoelen R, Fryns JP. A familial complex chromosome translocation resulting in duplication of 6p25. Ann Genet. 2004;47(3):275–80.
Vernon HJ, Bytyci Telegrafi A, Batista D, Owegi M, Leigh R. 6p25 Microdeletion: White Matter Abnormalities in an Adult Patient. Am J Med Genet A. 2013;161A(7):1686–9.
Zito A, Cartelli D, Cappelletti G, Cariboni A, Andrews W, Parnavelas J, et al. Neuritin 1 promotes neuronal migration. Brain Struct Funct. 2014;219(1):105–18.
Zurcher VL, Golden WL, Zinn AB. Distal deletion of the short arm of chromosome 6. Am J Med Genet. 1990;35(2):261–5.
Acknowledgements
We wish to express our appreciation to the family for their cooperation in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interest.
Authors’ contributions
ZQ and JY designed the study and wrote the manuscript. JY, LJ and ZQ analyzed and interpreted the aCGH data. JY carried out chromosomal analysis and interpretation. ZQ performed the FISH studies. AS performed the clinical evaluation and genetic counseling. All authors participated in revising the draft and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Qi, Z., Jeng, L.J.B., Slavotinek, A. et al. Haploinsufficiency and triploinsensitivity of the same 6p25.1p24.3 region in a family. BMC Med Genomics 8, 38 (2015). https://doi.org/10.1186/s12920-015-0113-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-015-0113-1