
Abstract
Background
Enterotoxigenic Escherichia coli (ETEC) causes diarrhea in humans, cows, and pigs. The gut microbiota underlies pathology of several infectious diseases yet the role of the gut microbiota in the pathogenesis of ETEC-induced diarrhea is unknown.
Results
By using an ETEC induced diarrheal model in piglet, we profiled the jejunal and fecal microbiota using metagenomics and 16S rRNA sequencing. A jejunal microbiota transplantation experiment was conducted to determine the role of the gut microbiota in ETEC-induced diarrhea. ETEC-induced diarrhea influenced the structure and function of gut microbiota. Diarrheal piglets had lower Bacteroidetes: Firmicutes ratio and microbiota diversity in the jejunum and feces, and lower percentage of Prevotella in the feces, but higher Lactococcus in the jejunum and higher Escherichia-Shigella in the feces. The transplantation of the jejunal microbiota from diarrheal piglets to uninfected piglets leaded to diarrhea after transplantation. Microbiota transplantation experiments also supported the notion that dysbiosis of gut microbiota is involved in the immune responses in ETEC-induced diarrhea.
Conclusion
We conclude that ETEC infection influences the gut microbiota and the dysbiosis of gut microbiota after ETEC infection mediates the immune responses in ETEC infection.
Similar content being viewed by others
Background
The intestinal microbiota is considered as a new “functional organ” as it regulates plentiful physiological functions of host, such as digestion [1], metabolism [2, 3], immunity [4, 5] and so on. Changes in the composition of intestinal microbiota are associated with a series diseases and dysfunctions, including inflammatory bowel disease [6], obesity [7], colorectal cancer [8] and type 2 diabetes [9]. What’s more, changes in the composition of intestinal microbiota also increase the intestinal susceptibility to infection, as the indigenous intestinal microbiota-mediated innate and adaptive defense is disrupted [10]. In contrast, the pathogenic infection in intestine also affects the composition of intestinal microbiota. For example, Salmonella enterica infection, which affects the intestine of poultry and causes intestinal inflammation, increases the relative abundance of Lactobacillaceae in the cecum of chicken [11]. Scores of metabolites are produced by the intestinal microbiota, and certain metabolites play the crucial role in the mediation of host physiological functions. For instance, indolepropionic acid, which produced by Clostridium sporogenes, can reinforce the intestinal barrier by engaging the pregnane X receptor [12]. Thus, changes in the composition of intestinal microbiota may associate with the pathogenesis of several infectious diseases.
Diarrhea in piglets is a typical multifactorial disease in the swine production, and it is also the main cause of piglet death. However, the etiology and epidemiology of diarrhea in piglets is very complicated, and the Enterotoxigenic Escherichia coli (ETEC) is the most common food-borne epidemical pathogen which causes diarrhea [13]. Mechanism of ETEC-induced diarrhea depends on the fimbrial adhesins and enterotoxin, which promotes the pathogen bacteria to adhere on the intestinal epithelial cells of piglets and lead to the fluid-electrolytes disturbance and acid-base imbalance of piglets, respectively [14, 15]. However, not all individuals infected with ETEC suffer from diarrhea [16]. Also, our previous investigation found that about 50% of piglets develop diarrhea after ETEC infection [17]. Whether the intestinal microbiota is related to the susceptibility of individuals and pigs to ETEC infection and development of diarrhea is unknown.
Previous investigations have shown that diarrhea is linked with changes of the intestinal microbiota composition [18,19,20]. However, osmotic diarrhea in humans also induces changes in microbial community structure [21], suggesting the alteration of gut microbiota may be the result of diarrhea. Thus, the cause and effect relationship between the changed intestinal microbiota and infectious diarrhea is unclear. This study was conducted to validate the hypothesis that the intestinal microbiota is changed in ETEC-induced diarrhea, and the intestinal microbiota is involved in the pathogenesis of ETEC-induced diarrhea.
Methods
Bacterial strains
The strain Escherichia coli W25K (O149:K91, K88 ac; LT, STb, EAST) was used in the current study, which was originally isolated from a diarrheal piglet [22].
ETEC infection
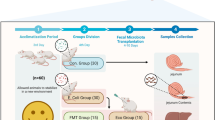
This study was conducted according to the guidelines of College of Animal Science, South China Agricultural University. This study was approved by South China Agricultural University. The Establishment of animal diarrheal model was consistent with our previous study [17, 23]. In brief, fifty-one piglets (Landrace × Yorkshire; 18-days old) were obtained from our partner farm (ZhengDa Co., Chongqing, China). Meanwhile, 41 piglets were randomly selected to receive an oral inoculation with the ETEC W25K (1010 CFUs/ml) and the rest of 10 piglets received the orally inoculation with the same volume LB medium as control. All the piglets experienced a five-day inoculate administration, and the fecal consistency was observed daily. Piglets which challenged with LB medium were defined as control group; piglets which developed watery diarrhea were marked during the whole experiment, and the marked piglets were regarded as recovered piglets if they recovered from diarrhea, otherwise were considered as diarrheal piglets; piglets those challenged with ETEC but not suffered from diarrhea were regarded as resistant piglets. Fresh feces were collected from day 1 to day 5 (post infection) for all kinds of piglets. For diarrheal piglets, their fresh feces were collected before diarrhea after ETEC infection to consider as pre-diarrheal samples. Six control piglets (n = 6), six diarrheal piglets (n = 6), six recovered piglets (n = 6) and six resistant piglets (n = 6) were randomly selected and sacrificed by kalium chloratum injection at day 6. Jejunum contents were collected after ice-cold phosphate buffered saline (PBS; pH = 7.2–7.4) washing. The whole part (including epithelium, mucosa, submucosa, muscular and serosa) of jejunum samples (middle part, about 3 cm) also collected after the ice-cold PBS washing. Luminal contents, feces and jejunum were collected and stored at − 80 °C until processing.
Microbiota transplantation experiment
Jejunal luminal contents from diarrheal piglets and uninfected piglets were collected and used for transplantation. The jejunal luminal contents were collected and mixed with phosphate buffered saline (PBS; pH = 7.2–7.4), and the final volume was adjusted to 50 ml per piglet. The mixed solution was vortexed at maximum speed for 3 min, and then centrifuged for 5 min at 500*g. The donor jejunal solution was orally infused to piglets with oro-gastric tube within 1 h after collection. The jejunal solution from diarrheal piglets was infused to uninfected piglets (n = 12). As controls, the jejunal solution from uninfected piglets was orally infused to uninfected piglets (n = 6). Piglets were orally transplanted for five consecutive days (day 1- day 6) with 20 ml jejunal solution per day. Piglets in control group were defined as control piglets; piglets developed watery diarrhea after transplantation were scarified and defined as transplanted diarrheal piglets; at day 6, piglets without diarrhea after transplantation were scarified and defined as transplanted non-diarrheal piglets. The fresh feces were collected from day 1 to day 5 after transplantation. At day 6, piglets were sacrificed by kalium chloratum injection for sample collection after electrical stunning. The jejunum contents were collected by ice-cold PBS washing and jejunum samples were collected after the ice-cold PBS washing. All the samples were store at − 80 °C for further analysis.
16S rRNA sequencing
The frozen jejunum contents and feces were thawed at the room temperate, and bacterial DNA was extracted by a commercial DNA stool kit (Qiagen, Hilden, Germany) according the manufacturer’s protocols. We measured the DNA concentration and purity with a NanoDrop ND-1000 instrument (NanoDrop Technologies Inc., Wilmington, DE, USA). Equal amounts of DNA from four different piglets were pooled to generate one common sample for each type of treatment. The following 16S rRNA gene amplification and pyrosequencing analysis were entrusted to a commercial company (Biotree, Shanghai, China), and the methodology and procedure were accordance with our previous study [24, 25].
Metagenomics analysis
The DNA extraction was described as above, and equal amounts of DNA from three different piglets were pooled to generate one common sample for each type of treatment (Diarrhea, Recovery, Resistant and Control). The metagenomics analysis of jejunal content was consigned to the commercial company (BGI Life Tech Co., Ltd., Beijing, China). DNA library construction, sequencing, de novo assembly, taxonomic assignment, and gene functional classification were based on their previous work [26, 27]. The total data volume of high-quality reads for our each group was nearly 14 Gbp.
RT-PCR
The mRNA expression of Tlr5, Tlr4 and Lyz-2 was performed by real-time quantitative PCR. Briefly, 100 mg frozen jejunal samples were pulverized in the liquid nitrogen and mixed into 1 ml Trizol (Invitrogen, USA), and the total RNA was extracted following the manufacturer’s protocols. The quality and concentration were detected by a NanoDrop ND-1000 instrument (NanoDrop Technologies Inc., Wilmington, DE, USA). Afterwards, we used the DNase I (Invitrogen, USA) and Superscript II reverse transcriptase (Invitrogen, USA) to produce complementary DNA. To normalise the expression levels of the target genes, β-actin was used as the internal control, and primers used in current study were referred to the previous studies. The RT-PCR was performed as our description in the ref. [28,29,30].
Fecal bacteria analysis using real-time PCR
The protocol and the primers used for feces Bacteroidetes and Firmicutes abundance analysis was conducted as described previously [28, 29].
Statistical analyses
Data in the current study are analyzed by the software Prime 6 and SPSS 22.0, and all the data are presented as means ± standard error of the mean (SEM). The methods of statistical analyses were performed as the previous study [24]. Significant differences were declared when P < 0.05.
Results
ETEC-induced diarrhea was associated with alterations in intestinal microbiota
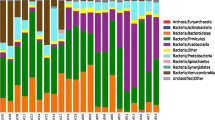
We characterized the jejnunal microbiota in piglets using metagenomics sequencing (Fig. 1a). The two most abundant phyla in diarrheal piglets, accounting for approximately 99% of all assigned sequence reads were Proteobacteria (81%) and Firmicutes (18%). In piglets that recovered from diarrhea, they were Proteobacteria (73%) and Firmicutes (24%). For controls or resistant piglets, they were Proteobacteria (96%) and Firmicutes (2%), and Proteobacteria (96%) and Firmicutes (3%), respectively. At the genus level (Fig. 1b), the percentage of Escherichia (49% vs. 88%) was reduced in diarrheal piglets, while the relative abundance of Lactobacillus (10% vs. 0.6%), Citrobacter (7.1% vs. 0.3%), Klebsiella (6.8% vs. 0.7%), Salmonella (6.2% vs. 1.3%), Enterobacter (6.2% vs. 0.3%), Lactococcus (4.9% vs. 0.008%), and Leuconostoc (1.6% vs. 0.007%) was increased in diarrheal piglets compared to the controls (Fig. 1b). Compared to the resistant piglets, the percentage of Escherichia (49% vs. 86%) was reduced, while the relative abundance of Lactobacillus (10% vs. 1.8%), Citrobacter (7.1% vs. 0.3%), Klebsiella (6.8% vs. 0.8%), Salmonella (6.2% vs. 1.8%), Enterobacter (6.2% vs. 1.0%), Lactococcus (4.9% vs. 0.1%), and Leuconostoc (1.6% vs. 0.06%) was increased in diarrheal piglets (Fig. 1b). For recovered piglets, diarrheal piglets had higher percentage of Escherichia (57% vs. 49%) and Lactobacillus (20% vs. 10%), whereas they had the lower relative abundance of Citrobacter (1.3% vs. 7.1%), Klebsiella (2.8% vs. 6.8%), Salmonella (1.9% vs. 6.2%), Enterobacter (4.4% vs. 6.2%), Lactococcus (0.03% vs. 4.9%), and Leuconostoc (0.1% vs. 1.6%) (Fig. 1b). At the species level, compared to controls, diarrheal piglets had lower relative abundance of Escherichia coli (27% vs. 76%) and Megasphaera elsdeii (0.04% vs. 2.1%), and higher percentage of Lactobacillus reuteri (12% vs. 0.2%), Enterobacter cloacae (3.7% vs. 0.3%), Klebsiella oxytoca (4.2% vs. 0.01%), Lactobacillus johnsonil (4.2% vs. 0.1%), Lactococcus lactis (11% vs. 0.001%) and Citrobacter koseri (7.8% vs. 0.3%) (Fig.1c). Compared to resistant piglets, diarrheal piglets had lower percentage of Escherichia coli (27% vs. 72%), but higher percentage of Lactobacillus reuteri (12% vs. 2.6%), Klebsiella oxytoca (4.2% vs. 0.3%), Lactococcus lactis (11% vs. 0.4%) and Citrobacter koseri (7.8% vs. 0.4%) (Fig.1c). In recovered piglets, the percentage of Escherichia coli (37% vs. 27%) and Megasphaera elsdeii (2% vs. 0.04%) increased, while the percentage of Salmonella enterica (1.9% vs. 4.6%), Klebsiella oxytoca (1.8% vs. 4.2%), Lactobacillus johnsonil (1.6% vs. 4.3%), Lactococcus lactis (0.05% vs. 11%) and Citrobacter koseri (1.2% vs. 7.8%) decreased compared to diarrheal piglets (Fig.1c). Also, compared to diarrheal piglets, recovered piglets had higher relative abundance of Lactobacillus amylovorus (6.3% vs. 0.2%), Lactobacillus acidophilus (2.6% vs. 0.09%) and Lactobacillus crispatus (1.3% vs. 0.2%) (Fig.1c).

Gut microbiota in ETEC induced diarrhea. a The jejunal microbiota at the phylum level among controls, diarrheal piglets, recovered piglets and resistant piglets were analyzed using metagenomics. b The jejunal microbiota at the genus level were analyzed using metagenomics. c The jejunal microbiota at the species level were analyzed using metagenomics. d The jejunal microbiota at the phylum level among controls, diarrheal piglets and resistant piglets were analyzed using 16S rRNA sequencing. e The jejunal microbiota at the genus level were analyzed using 16S rRNA sequencing. f The fecal microbiota at the phylum level among controls, pre-diarrheal piglets, and resistant piglets were analyzed using 16S rRNA sequencing. g Real time-PCR analysis of the relative abundance of Bacteroidetes and Firmicutes in the feces among controls, pre-diarrheal piglets, and resistant piglets (n = 6; *: p < 0.05, one-way ANOVA). h The fecal microbiota at the genus level were analyzed using 16S rRNA sequencing. N = 3 before pooling for a, b, c; and n = 4 before pooling for d, e, f, h
Previous studies have shown that diarrhea may result in a lower Bacteroidetes:Firmicutes ratio because the diarrhea may create a more suitable environment for the survival and growth of Firmicutes as compared with Bacteroidetes [18, 31,32,33]. The Bacteroidetes:Firmicutes ratio was 0.01 in diarrheal piglets, compared with 0.18 in control and 0.10 in resistant piglets. And the Bacteroidetes:Firmicutes ratio was 0.05 in recovered piglets (Table 1).
The microbiota in jejunal luminal content and feces were also analyzed using 16S rRNA sequencing (Table 2). For microbiota in jejunal luminal content, both Shannon and Simpson indices demonstrated that the microbiota diversity in the jejunum of diarrheal piglets was lower than controls or resistant piglets (Table 2). Noticeably, the community richness of microbiota in the jejunum was similar among diarrheal piglets, resistant piglets and controls (Table 2). At the phylum level (Fig. 1d), the three most abundant phyla in jejunal luminal contents were Firmicutes (58%), Proteobacteria (20%) and Bacteroidetes (5%) in diarrheal piglets. For piglets in control or resistant groups, they were Firmicutes (51%), Proteobacteria (16%) and Bacteroidetes (7%), and Firmicutes (49%), Proteobacteria (20%) and Bacteroidetes (6%), respectively. At the genus level (Fig. 1e), the percentage of Streptococcus (35% vs. 13%), Lactococcus (10.5% vs. 4.9%) and Escherichia-Shigella (6.1% vs. 1.9%) were increased, while Weissella (1.1% vs. 13.3%) was decreased in diarrheal piglets, compared to controls. Compared to resistant piglets, the percentage of Streptococcus (35% vs. 21%), Lactococcus (10.5% vs. 2.0%) and Escherichia-Shigella (6.1% vs. 1.4%) were increased in diarrheal piglets (Fig. 1e). The Bacteroidetes:Firmicutes ratio was 0.08 in diarrheal piglets, compared with 0.13 in control and 0.12 in resistant piglets (Table 1).
In feces, the microbiota diversity of diarrheal piglets was lower than pre-diarrheal piglets, resistant piglets and controls, while little difference about the community richness of microbiota was observed among these groups (Table 2). The three most abundant phyla were Firmicutes, Bacteroidetes, and Proteobacteria in all groups (Fig. 1f). Bacteroidetes:Firmicutes ratio in the feces was 0.38 for diarrheal piglets, while it was 0.42 for pre-diarrheal piglets, and 0.50 for control and resistant piglets (Table 1). Real-time PCR data also shown the ratio of Bacteroidetes:Firmicutes was lower (P < 0.05) in diarrheal piglets, compared to the controls and resistant piglets (n = 4) (Fig. 1g). At the genus level (Fig. 1h), from controls, pre-diarrheal piglets to diarrheal piglets, the percentage of Escherichia-Shigella (3.8, 5.5 to 35.3%) increased, while Prevotella (4.2, 1.7 to 0.2%) decreased. Compared to resistant piglets, diarrheal piglets had higher relative abundance of Escherichia-Shigella (35.3% vs. 24.9%), while lower percentage of Prevotella (0.2% vs. 6.7%) (Fig. 1h).
Jejunal microbiota mediates diarrhea
To explore the cause and effect relationship between change in the gut microbiota and diarrhea, we conducted a jejunal microbiota transplantation experiment. As controls, we transplanted the jejunal microbiota from uninfected piglets (n = 4) to uninfected piglets,and we found that no transplanted piglets experienced diarrhea. We then transplanted the jejunal microbiota from diarrheal piglets to uninfected piglets, and 50% of these piglets exhibited diarrhea (n = 5). Compared to the non-diarrheal piglets after transplantation, diarrheal piglets had lower microbiota diversity in the jejunum and feces (Table 2). The relative abundance of Bacteroidetes decreased in diarrheal piglets, but the relative abundance of Firmicutes increased in the jejunum and feces (Fig.2 a-b, Table 3). For microbiota in the jejunum, diarrheal piglets had higher percentage of Lactococcus (45% vs. 23%), Leuconostoc (14% vs. 1.7%), Enterococcus (7% vs. 0.5%) and Lactobacillus (6% vs. 0.7%), but lower Streptococcus (13% vs. 34%) than the non-diarrheal piglets (Fig.2c). In the feces, diarrheal piglets had higher percentage of Escherichia-Shigella (22% vs. 4%) and Erysipelotrichaceae-uncultured (11% vs. 5%), but lower Prevotella (1% vs. 18%) than the non-diarrheal piglets (Fig.2d).

Gut microbiota in transplanted diarrhea. a The jejunal microbiota at the phylum level were analyzed using 16S rRNA sequencing. b Fecal microbiota at the phylum level were analyzed using 16S rRNA sequencing. c The jejunal microbiota at the genus level were analyzed using 16S rRNA sequencing. d Fecal microbiota at the genus level were analyzed using 16S rRNA sequencing. Transplanted-D: recipient piglets that experienced diarrhea after microbiota transplantation from diarrheal piglets. Transplanted-ND: recipient piglets that did not experience diarrhea after microbiota transplantation from diarrheal piglets. N = 4 before pooling for a, b, c, and d
The protein repertoire and pathways impacted by ETEC induced diarrhea
Metagenomic sequences were annotated against the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. At KEGG level one, a total of 6 KEGG entries were identified (Additional file 1: S1), including metabolism, environmental information processing, genetic information processing, cellular processes, human diseases and organismal systems. Diarrheal piglets shows decrease in cellular processes, compared to the control piglets (Table 4). At KEGG level two, a total of 37 KEGG entries were identified (Additional file 1: S2). The six most abundant changed KEGG were cell motility, biosynthesis of other secondary metabolites, excretory system, immune system diseases, immune system and circulatory system (Table 4). ETEC induced diarrhea reduced the cell motility and biosynthesis of other secondary metabolites in jejunal microbiota (Table 4). At KEGG level three, a total of 236 KEGG Orthology (KO) pathways were identified (Additional file 1: S3). Among the 83 most abundant changed KO, ETEC induced diarrhea decreased bacterial invasion of epithelial cells, bacterial motility proteins and flagellar assembly for jejunum microbiota (Table 4). Collectively, these results suggest that ETEC induced diarrhea may influence the function of jejunum microbiota.
Jejunal microbiota mediates the immune inhibition on the jejunum
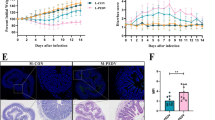
As shown in Table 4, diarrheal piglets suffered the dysbiosis of jejunal microbiota, including decreased cell motility and flagellar assembly (Table 4). This indicates that the inhibition of intestinal immunity in diarrheal piglets may come from the changed intestinal microbiota. To validate this, we transplanted the jejunal microbiota from diarrheal piglets to uninfected piglets, and the controls were transplanted with jejunal microbiota from uninfected piglets. The gene expression of Toll-like receptor (Tlr) 5, Tlr4 and Lyz-2 were analyzed after transplantation because previous study has shown ETEC infection significantly lower the expression of Tlr5, Tlr4 and Lyz-2 in the jejunum [34]. Results found that diarrheal microbiota mediated the mRNA expression of Tlr5 (P < 0.05) (n = 4) (Fig. 3). However, no significant difference was found about the expression of Tlr4 and Lyz-2 (P > 0.05) (n = 4) (Fig. 3). These date suggest that the dysbiosis of jejunal microbiota partly associates with the immune responses in ETEC infection.

mRNA expression of innate immune genes after jejunal microbiota transplantation from normal (control) and diarrheal piglets (transplantation). (n = 4; *: p < 0.05, unpaired t test)
Discussion
Diarrhea and malnutrition are both associated with dysbiosis of the intestinal microbiota [19, 35]. ETEC is an important cause of diarrhea in humans and weaned piglets; however, the role of gut microbiota in ETEC-induced diarrhea is unknown. In current study, with different analysis methods, we found that diarrheal piglets have a dysbiosis of the intestinal microbiota, especially a higher percentage of Lactococcus in the jejunum, and lower Bacteroidetes: Firmicutes ratio in the jejunum and feces. Other interesting findings are that diarrheal piglets have higher percentage of Escherichia-Shigella and lower of Prevotella in the feces, and lower microbiota diversity in the jejunum and feces. There is an obvious difference about the intestinal microbiota between diarrheal piglets and resistant piglets, while little change about the intestinal microbiota is observed between resistant piglets and controls, suggesting the gut microbiota of some individuals or piglets may play a resistant role to diarrhea after exposed to inducers [16, 32, 34, 36]. A previous study found that the gut microbiota in the patients who developed diarrhea are more related to each other than to those did not develop diarrhea [36, 37]. In this study, quite part of piglets didn’t suffer from the diarrhea by the ETEC infection. Thus it seems that a specific, preexisting microbial balance might predispose or protect against diarrhea. However, piglets’ genetic resistance to ETEC has not been tested in the current study, and the jejunal microbiota from diarrheal piglets contains ETEC, it is unclear whether the ETEC in the jejunal transplant material had sufficient levels of the ETEC to cause diarrhea, which leads to cannot fully rule out the direct influence of jejunal ETEC in piglet diarrhea. Thus, the influence of intestinal microbiota on piglet diarrhea needs to further transplantation with synthetic intestinal microbiota without ETEC strain [38]. Also, further investigations are needed to explore the alterations of intestinal microbiota during ETEC induced diarrhea in piglets because this study analyzed the pre-pooled samples. Thus, to overcome the shortages of pooling samples, the intestinal microbiota was analyzed with 16S rRNA sequencing and metagenomics sequencing using different samples. The similarities from both 16S rRNA sequencing and metagenomics sequencing are considered to be the really changes in ETEC induced diarrhea. For example, although there were differences at the phylum level and genus level with each analysis, a consistent finding was that diarrheal piglets have a higher percentage of Lactococcus lactis, compared to the controls. Similarly, in our further experiment using bacterial counting, we found that ETEC infection increases the bacterial load of Lactococcus lactis in the jejunum (manuscript submitted). The discrepancy in results between 16S rRNA sequencing and metagenomics sequencing may come from various determinants, such as species, geography, and host physiology [19, 39, 40]. Indeed, the methods for analysis of intestinal microbiota also highly affect the results [41]. For example, compared to the complete 16S rRNA sequencing, sequencing of individual segments and combinations of segments greatly underestimates the taxonomic diversity [41].
ETEC-induced diarrhea is associated with a decrease in the Bacteroidetes:Firmicutes ratio. Also, a lower ratio of Bacteroidetes:Firmicutes is found in other types of diarrheal models [18, 19, 21, 33]. Thus, diarrhea, regardless of the cause, may establish an environment more suitable for survival and growth of Firmicutes than for Bacteroidetes [18, 19, 21, 33]. Previous study [18] has pointed out that the change in the Bacteroidetes:Firmicutes ratio after diarrhea is not from a change in the abundance of any particular class, but the result of a phylum-level effect. However, higher percentage of Lactococcus (belongs to Firmicutes) in diarrheal piglet jejunum, and lower percentage of Prevotella (belongs to Bacteroidetes) in diarrheal piglet feces may be the reason for lower Bacteroidetes:Firmicutes ratio in ETEC induced diarrhea. The exact roles of Lactococcus and Prevotella in the pathogenesis of ETEC induced diarrhea are unknown. Lactobacillus seems be beneficial for the recovery from ETEC induced diarrhea because Lactobacillus reuteri (11%), Lactobacillus amylovorus (6.3%), Lactobacillus acidophilus (2.6%), Lactobacillus johnsonil (1.6%), and Lactobacillus crispatus (1.3%) are within the top 10 percentages of bacterium in recovered piglet jejunum. Especially, recovered piglets have higher percentage of Lactobacillus amylovorus, Lactobacillus acidophilus, and Lactobacillus crispatus than the diarrheal piglets.
It is unknown why a lower Bacteroidetes:Firmicutes ratio is involved in ETEC-induced diarrhea. One of the possible explanations is the intestinal level of oxygen, which can be diffused from the host tissues into the intestinal lumen [42]. After secretory stimulation (e.g., ETEC infection, Vibrio cholera infection), there is abnormally increase in the intestinal level of oxygen, which inhibits the growth of anaerobic organisms, as well as leads to the accumulation of facultative anaerobes (e.g., Bacilli, member of Firmicutes) to respire oxygen to maintain enteric anoxia [42,43,44]. A decrease in the relative proportion of Bacteroidetes is associated with various diseases, such as obesity [45]. Usually, the difference in function and metabolism between Bacteroidetes and Firmicutes is regarded as the contributor. The changed function (e.g., cell motility and genetic information processing) and metabolism (e.g., xenobiotics biodegradation and metabolism, amino acids and lipid metabolism) of the gut microbiota may be associated with the pathogenesis of diarrhea. A previous study suggested Firmicutes is linked to obesity because Firmicutes can ferment plant polysaccharide to produce short-chain fatty acids (SCFA), providing additional energy for the host [45]. Enhanced production of butyrate (SCFA) could promote the expression of globotriaosylceramide, which is a receptor for the Shiga-like toxin (Stx2), leading to increased bacterial colonization and disease severity in Escherichia coli O157:H7 infection [46]. Thus, the lower Bacteroidetes:Firmicutes ratio possibly could promote the attachment and colonization of pathogens (e.g., ETEC) to the intestine. A previous study found that a fat-rich diet modifies the composition of the conventional intestinal microbiota by increasing the Firmicutes while reducing the Bacteroidetes loads, creating an imbalance that exposes the intestinal epithelial cells to adherence for Campylobacter jejuni [47]. Thus, decreased Bacteroidetes: Firmicutes ratio may also lead to the decrease in colonization resistance against pathogens (e.g., ETEC), which promotes the colonization of pathogens in the intestine.
The gut microbiota is likely to play a pivotal role in the establishment of host-pathogen crosstalk, ultimately shaping the intestinal immune responses after infection [48,49,50]. Previous data have indicated that ETEC-induced diarrhea inhibits intestinal immune responses in the jejunum [34, 51]. Numerous investigations have shown that intestinal pathogens have evolved mechanisms to subvert intestinal immunity by secreting toxins to intestine after colonization [52,53,54]. To colonize to gut mucosal surfaces, pathogens need to inhibit intestinal immunity [53, 55]. In piglets, ETEC induced diarrhea inhibits the activation of the NF-κB pathway and MAPK pathway [34]. ETEC induced diarrhea decreases the expression of innate immune factors, including Tlrs [34, 56]. The inhibition of jejunal immune response in ETEC induced diarrheal piglets might be from the changed jejunal microbiota because they show decreased cell motility and flagellar assembly, which may mean decreased stimulation to the jejunum from jejunum microbiota. Flagellar filament assembly is important for flagellin expressing bacteria, such as α and ε Proteobacteria, to efficiently infect mammalian hosts [57, 58]. TLR5 recognizes bacterial flagellin and activates host inflammatory responses to bacteria [57, 58], thus it is not surprising to found lower expression of Tlr5 in diarrheal piglets compared to the controls [34]. Furthermore, jejunal microbiota transplantation from diarrheal piglets to controls, which changes the gut microbiota to diarrheal situation, also induces lower expression of Tlr5 compared to the controls. This supports the hypotheses that the dysbiosis of gut microbiota mediates the immune responses in ETEC induced diarrhea. Indeed, a previous study also suggested that gut microbiota is necessary for the establishment of host-pathogen crosstalk, ultimately shaping the intestinal immune responses after infection [48, 50]. However, the lack of significant change in the expression of Tlr4 and Lyz-2 after jejunal microbiota transplantation indicates that the immune responses in ETEC induced diarrhea is not fully dependent on the dysbiosis of gut microbiota, but maybe also ETEC. However, the exact function of intestinal microbiota in the immune responses in piglet-ETEC interaction needs further investigations.
Conclusions
In conclusion, ETEC induced diarrhea is associated with the alteration of intestinal microbiota, including lower Bacteroidetes: Firmicutes ratio and microbiota diversity in the jejunum and feces, and lower Prevotella in the feces, but higher percentage of Lactococcus in the jejunum, and Escherichia-Shigella in the feces. Such alteration of intestinal microbiota mediates some aspects of pathogenesis in ETEC induced diarrhea. Our data also suggest there is a specific, preexisting microbial balance that predisposes or protects against ETEC induced diarrhea. It may be fruitful to attempt to treat ETEC induced diarrhea by modulating the gut microbiota.
Abbreviations
- cAMP:
-
Cyclic adenosine monophosphate
- cGMP:
-
Cyclic guanosine monophosphate
- ETEC:
-
Enterotoxigenic Escherichia coli
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LT:
-
Heat-labile toxins
- ST:
-
Heat-stable toxins
- TLR:
-
Toll-like receptor
References
Rafael T, Adolfo S, Marta C, Clara R-G, Abelardo M. Intestinal microbiota in health and disease: role of bifidobacteria in gut homeostasis. World J Gastroenterol. 2014;20(41):15163–76.
Tuddenham S, Sears CL. The intestinal microbiome and health. Curr Opin Infect Dis. 2015;28(5):464.
Nie YF, Hu J, Yan XH. Cross-talk between bile acids and intestinal microbiota in host metabolism and health. Journal of Zhejiang Universityscienceb. 2015;16(6):436.
Brown K, Decoffe D, Molcan E, Gibson DL. Diet-induced Dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients. 2012;4(8):1552–3.
Sjogren YM, Tomicic S, Lundberg A, Bottcher MF, Bjorksten B, Sverremark-Ekstrom E, Jenmalm MC. Influence of early gut microbiota on the maturation of childhood mucosal and systemic immune responses. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2009;39(12):1842–51.
Becker C, Neurath MF, Wirtz S. The intestinal microbiota in inflammatory bowel disease. ILAR J. 2015;56(2):192–204.
Sabatino A, Regolisti G, Cosola C, Gesualdo L, Fiaccadori E. Intestinal microbiota in type 2 diabetes and chronic kidney disease. Current diabetes reports. 2017;17(3):16.
Keku TO, Dulal S, Deveaux A, Jovov B, Han X. The gastrointestinal microbiota and colorectal cancer. Am J Physiol Gastrointest Liver Physiol. 2015;308(5):G351–63.
Yamaguchi Y, Adachi K, Sugiyama T, Shimozato A, Ebi M, Ogasawara N, Funaki Y, Goto C, Sasaki M, Kasugai K. Association of Intestinal Microbiota with metabolic markers and dietary habits in patients with type 2 diabetes. Digestion. 2016;94(2):66–72.
Brown RL, Sequeira RP, Clarke TB. The microbiota protects against respiratory infection via GM-CSF signaling. Nat Commun. 2017;8(1):1512.
Petra V, Frantisek S, Hana H, Marcela F, Ivan R. Influence ofSalmonella entericaserovar Enteritidis infection on the composition of chicken cecal microbiota. BMC Vet Res,9,1(2013-07-15). 2013;9(1):140.
Dodd D, Spitzer MH, Van WT, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, Cowan TM, Nolan GP, Fischbach MA. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature. 2017;3(10):e00438.
Liu G, Ren W, Fang J, C-AA H, Guan G, Al-Dhabi NA, Yin J, Duraipandiyan V, Chen S, Peng Y, et al. L-glutamine and l-arginine protect against enterotoxigenic Escherichia coli infection via intestinal innate immunity in mice. Amino Acids. 2017;49(12):1945–54.
Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, Steinsland H. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes & Infection. 2010;12(2):89–98.
Joffre E, von Mentzer A, Svennerholm AM, Sjoling A. Identification of new heat-stable (STa) enterotoxin allele variants produced by human enterotoxigenic Escherichia coli (ETEC). International journal of medical microbiology : IJMM. 2016;306(7):586–94.
Zhu YH, Li XQ, Zhang W, Zhou D, Liu HY, Wang JF. Dose-dependent effects of lactobacillus rhamnosus on serum interleukin-17 production and intestinal T-cell responses in pigs challenged with Escherichia coli. Appl Environ Microbiol. 2014;80(5):1787–98.
Ren W, Yin J, Chen S, Duan J, Liu G, Li T, Li N, Peng Y, Tan B, Yin Y. Proteome analysis for the global proteins in the jejunum tissues of enterotoxigenicEscherichia coli-infected piglets. Sci Rep. 2016;6:25640.
Costa MO, Chaban B, Harding JC, Hill JE. Characterization of the fecal microbiota of pigs before and after inoculation with "Brachyspira hampsonii". PLoS One. 2014;9(8):e106399.
Pop M, Walker AW, Paulson J, Lindsay B, Antonio M, Hossain MA, Oundo J, Tamboura B, Mai V, Astrovskaya I, et al. Diarrhea in young children from low-income countries leads to large-scale alterations in intestinal microbiota composition. Genome Biol. 2014;15(6):R76.
Ren W, Yin J, Xiao H, Chen S, Liu G, Tan B, Li N, Peng Y, Li T, Zeng B, et al. Intestinal microbiota-derived GABA mediates Interleukin-17 expression during Enterotoxigenic Escherichia coli infection. Front Immunol. 2016;7:685.
Gorkiewicz G, Thallinger GG, Trajanoski S, Lackner S, Stocker G, Hinterleitner T, Gully C, Hogenauer C. Alterations in the colonic microbiota in response to osmotic diarrhea. PLoS One. 2013;8(2):e55817.
Ren W, Liu G, Yin J, Chen S, Li T, Kong X, Peng Y, Yin Y, Hardwidge PR. Draft genome sequence of Enterotoxigenic Escherichia coli strain W25K. Genome Announc. 2014;2(3):675.
Ren W, Yin J, Gao W, Chen S, Duan J, Liu G, Li T, Li N, Peng Y, Yin Y. Metabolomics study of metabolic variations in enterotoxigenic Escherichia coli-infected piglets. RSC Adv. 2015;5(73):59550–5.
Ren W, Wang P, Yan J, Liu G, Zeng B, Hussain T, Peng C, Yin J, Li T, Wei H, et al. Melatonin alleviates weanling stress in mice: involvement of intestinal microbiota. J Pineal Res. 2018;64(2):e12448.
Ren W, Chen S, Zhang L, Liu G, Tarique H, Hao X, Yin J, Duan J, Tan B, Wu G. Interferon tau affects mouse intestinal microbiota and expression of IL-17. Mediat Inflamm. 2016;2016(19):1–9.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65.
Zhu L, Wu Q, Dai J, Zhang S, Wei F. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc Natl Acad Sci U S A. 2011;108(43):17714–9.
Ren W, Chen S, Yin J, Duan J, Li T, Liu G, Feng Z, Tan B, Yin Y, Wu G. Dietary arginine supplementation of mice alters the microbial population and activates intestinal innate immunity. J Nutr. 2014;144(6):988–95.
Ren W, Duan J, Yin J, Liu G, Cao Z, Xiong X, Chen S, Li T, Yin Y, Hou Y, et al. Dietary L-glutamine supplementation modulates microbial community and activates innate immunity in the mouse intestine. Amino Acids. 2014;46(10):2403–13.
Bin P, Liu S, Chen S, Zeng Z, Huang R, Yin Y, Liu G. The effect of aspartate supplementation on the microbial composition and innate immunity on mice. Amino Acids. 2017;49(9):1–7.
Swidsinski A, Loening-Baucke V, Verstraelen H, Osowska S, Doerffel Y. Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology. 2008;135(2):568–79.
Whelan K, Judd PA, Tuohy KM, Gibson GR, Preedy VR, Taylor MA. Fecal microbiota in patients receiving enteral feeding are highly variable and may be altered in those who develop diarrhea. Am J Clin Nutr. 2009;89(1):240–7.
Chaban B, Links MG, Hill JE. A molecular enrichment strategy based on cpn60 for detection of epsilon-proteobacteria in the dog fecal microbiome. Microb Ecol. 2012;63(2):348–57.
Ren W, Yin J, Chen S, Duan J, Liu G, Li T, Li N, Peng Y, Tan B, Yin Y. Proteome analysis for the global proteins in the jejunum tissues of enterotoxigenic Escherichia coli -infected piglets. Sci Rep. 2016;6:25640.
Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature. 2014;510(7505):417–21.
Manichanh C, Varela E, Martinez C, Antolin M, Llopis M, Dore J, Giralt J, Guarner F, Malagelada JR. The gut microbiota predispose to the pathophysiology of acute postradiotherapy diarrhea. Am J Gastroenterol. 2008;103(7):1754–61.
Damman CJ, Surawicz CM. The gut microbiota: a microbial arsenal protecting us from infectious and radiation-induced diarrhea. Gastroenterology. 2009;136(2):722–4.
Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. 2016;167(5):1339–53 e1321.
Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A, Palau F, Nova E, Marcos A, Polanco I, et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut. 2015;64(3):406–17.
Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T, Hanada R, Lipinski S, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487(7408):477–81.
Yarza P, Yilmaz P, Pruesse E, Glockner FO, Ludwig W, Schleifer KH, Whitman WB, Euzeby J, Amann R, Rossello-Mora R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12(9):635–45.
Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, Grunberg S, Baldassano RN, Lewis JD, Li H, et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. 2014;147(5):1055–63 e1058.
David LA, Weil A, Ryan ET, Calderwood SB, Harris JB, Chowdhury F, Begum Y, Qadri F, LaRocque RC, Turnbaugh PJ. Gut microbial succession follows acute secretory diarrhea in humans. mBio. 2015;6(3):e00381–15.
Espey MG. Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radic Biol Med. 2013;55:130–40.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–3.
Zumbrun SD, Melton-Celsa AR, Smith MA, Gilbreath JJ, Merrell DS, O'Brien AD. Dietary choice affects Shiga toxin-producing Escherichia coli (STEC) O157:H7 colonization and disease. Proc Natl Acad Sci U S A. 2013;110(23):E2126–33.
Masanta WO, Heimesaat MM, Bereswill S, Tareen AM, Lugert R, Gross U, Zautner AE. Modification of intestinal microbiota and its consequences for innate immune response in the pathogenesis of campylobacteriosis. Clin Dev Immunol. 2013;2013:526860.
Cantacessi C, Giacomin P, Croese J, Zakrzewski M, Sotillo J, McCann L, Nolan MJ, Mitreva M, Krause L, Loukas A. Impact of experimental hookworm infection on the human gut microbiota. J Infect Dis. 2014;210(9):1431–34.
Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O'Leary CE, Oliver PM, Kolls JK, Weiser JN, Worthen GS. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med. 2014;20(5):524–30.
Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13(5):321–35.
Ren W, Yin J, Duan J, Liu G, Zhu X, Chen S, Li T, Wang S, Tang Y, Hardwidge PR. Mouse intestinal innate immune responses altered by enterotoxigenic Escherichia coli (ETEC) infection. Microbes & Infection. 2014;16(11):954–61.
Wang X, Hardwidge PR. Enterotoxigenic Escherichia coli prevents host NF-kappaB activation by targeting IkappaBalpha polyubiquitination. Infect Immun. 2012;80(12):4417–25.
Wan F, Weaver A, Gao X, Bern M, Hardwidge PR, Lenardo MJ. IKKbeta phosphorylation regulates RPS3 nuclear translocation and NF-kappaB function during infection with Escherichia coli strain O157:H7. Nat Immunol. 2011;12(4):335–43.
de Jong HK, Parry CM, van der Poll T, Wiersinga WJ. Host-pathogen interaction in invasive salmonellosis. PLoS Pathog. 2012;8(10):e1002933.
Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, Raffatellu M. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity. 2014;40(2):262–73.
Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216.
Smith KD, Andersen-Nissen E, Hayashi F, Strobe K, Bergman MA, Barrett SL, Cookson BT, Aderem A. Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat Immunol. 2003;4(12):1247–53.
Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, Aderem A. Evasion of toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A. 2005;102(26):9247–52.
Acknowledgements
The authors thank Prof. Philip Ross Hardwidge from Kansas State University for critical discussion on this study.
Funding
This study was supported by Grant Nos. 2017YFD0500105, 2016YFD0500905 from the National Key Research and Development Program of China, grants from the Chinese National Science Foundation Grant (Nos. 31672579, 30571374, 30771603, 31072136, 31270171, 31873010), and a project founded by the Priority Academic Program of Development Jiangsu High Education Institution. It also grants from the Yangzhou Science and Technology Bureau International Cooperation Project (YZ2018154). The funders supported the charge of experimental animals, 16S rRNA sequencing, metagenomics analysis and related reagents.
Availability of data and materials
The datasets during and/or analyzed during the current study available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
Z.T. and G.Z. conceived and designed the experiments. P.B. and S.C. carried out the animal experiment and sample collection. P.B., S.L. and Y.X contributed to the laboratory experiments and data collection. Z.T., P.B., S.L., S.C. and J.L performed statistical analysis. P.B. and Y.X. wrote the manuscript. G.Z., P.B. S.L. and H.W revised the manuscript critically. All authors read and approve the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
This study was conducted according to the guidelines of College of Animal Science, South China Agricultural University. This study was approved by South China Agricultural University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
S1. A total of 6 KEGG entries were identified at KEGG level one. S2. A total of 37 KEGG entries were identified at KEGG level two. S3. A total of 236 KEGG Orthology (KO) pathways were identified at KEGG level three. (XLSX 70 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bin, P., Tang, Z., Liu, S. et al. Intestinal microbiota mediates Enterotoxigenic Escherichia coli-induced diarrhea in piglets. BMC Vet Res 14, 385 (2018). https://doi.org/10.1186/s12917-018-1704-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-018-1704-9