
Abstract
Background
Mitochondrial maternally inherited hearing impairment (HI) appears to be increasing in frequency. The incidence of mitochondrial defects causing HI is estimated to be between 6 and 33% of all hearing deficiencies. Mitochondrial m.1555A > G mutation is the first mtDNA mutation associated with non-syndromic sensorineural deafness and also with aminoglycoside induced HI. Its prevalence varied geographically between different populations.
Methods
We carried out PCR, restriction enzyme based screening, and sequencing of 337 subjects (including 132 patients diagnosed clinically with hereditary deafness) from 54 families from Syria for m.1555A > G mitochondrial mutation.
Results
Mitochondrial m.1555A > G mutation was detected in one of fifty-four families (1.85%), six out of the 132 (4.5%) of all patients with NSHI and one propositus of the 205 individuals with normal hearing (0.48%).
Conclusion
This is the first study to report prelingual deafness causative gene mutations identified by sequencing technology in Syrian families. It is obvious from the results that the testing for the m.1555A > G mutation is useful for diagnosis of hearing loss in Syrian patients and should also be considered prior to treatment with aminoglycosides in predisposed individuals.
Similar content being viewed by others

Background
Hearing impairments (HI) is as one of the most disabling disorders in the world, limiting a person’s ability to communicate with others [1]. It is caused by genetic and/or environmental factors. Genetically caused HI is 70% non-syndromic and 30% syndromic [2], with each form related to specific genes. Non-syndromic hearing impairment (NSHI) is isolated hearing deficit without other medical derangements [3]. It affects about 0.1% of live newborns and 4% of all people below 45 years of age [4]. This number increases dramatically in countries and regions where consanguineous marriages are common, like Syria and other Middle Eastern countries [5,6,7]. Up to 75–80% of cases of non-syndromic hearing impairment have an autosomal recessive cause, 10–15% inherited as autosomal dominant, and few are X-linked or mitochondrial [8].
Mitochondria are intracellular organelles that contain their own DNA (mtDNA). A substitution of A- > G nitrogenous bases in position 1555 of the mitochondrial 12S rRNA gene is the most prevalent mutation in the mitochondrial genome, associated with both late onset and congenital NSHI. It is of particular interest being a key cause of antibiotic-induced HI [9]. This mutation occurs in a highly conserved region of the 12S rRNA gene, where aminoglycosides are known to bind and result in defective ATP production in cochlear cells [10].
The m.1555A > G mutation has been identified in more than 120 families throughout the world with frequencies differing according to the population’s ethnic group [11,12,13,14]. So far, no report exists regarding the involvement of the mtDNA mutations in pathogenesis of deafness in Syria. Syria is a country in the Middle East, along the eastern shore of the Mediterranean Sea. The purpose of this study was to determine the prevalence of m.1555A > G mitochondrial mutation among patients with NSHI from Syria.
Methods
All subjects were recruited from Aleppo, which lies in the northwestern region of Syria, with the help of deafness related schools and institutes. The examined group consisted of 337 subjects including 132 affected patients from 54 Syrian families with 2 or more affected subjects (up to 6) with prelingual, profound, sensorineural, bilateral, non-syndromic hearing loss and 205 unrelated healthy controls.
Patients
In this study 175 females and 162 males were participated, with an age range between two and 72 years (mean age 18.2 years). Detailed family pedigrees were drawn. Information on consanguinity, age at onset of HL, detailed prenatal and perinatal history, use of ototoxic drugs (aminoglycosides), etc. was obtained through a detailed questionnaire. Physical examination was performed to exclude syndromic forms of hearing impairment at Alrazi Hospital (Aleppo), ENT examination such as Tympanometry, and BERA (Brainstem Electric Response Audiometry) were also applied at ENT department of the hospital Alrazi.
Mutation Screening
10 ml of peripheral venous blood was obtained from each affected and healthy family members. Genomic DNA was extracted using Qiagen FlexiGene kits in accordance with the manufacturer’s instructions. Mitochondrial DNA m.1555A > G mutation was detected using PCR-RFLP strategy (m.1555A > G, F: AGAAATGGGCTACATTTTCTACCC; m.1555A > G, R: GTTCGTCCAAGTGCACCTTCCA) as mentioned in [15] followed by BsmAI-digest (site: GTC TCN/) (New England Bio Labs®). Products with m.1555A > G mutation showed one band with 248 bp and wild individual displayed two bands (192 bp and 56 bp) after polyacrylamide gel electrophoresis (6%)(Fig. 1a).

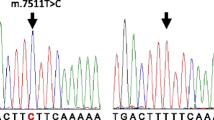
(a): PCR-RFLP analysis of the only Syrian family found with m.1555A > G mutation: a 248 bp PCR fragment is digested with BsmAI. DNA ladder (the first panel). The wild-type mtDNA is cleaved in to tow fragments, 192 and 56 bp in length (the last panel). PCR product containing the m.1555A > G mutation is not cleaved (the other left panels). (b): Partial Sequence chromatograms from a normal hearing individual (down) and affected proband with the m.1555A > G mutation in the mitochondrial 12S rRNA gene (top) with the forward and reverse primers. The small arrows indicate the localization of the change of an Adenine to Guanine nucleotide at position 1555 of the 12S rRNA gene
The presence of the m.1555A > G mutation was verified using standard Sanger sequencing. Detected mutation was confirmed at least two times and compared with the reference sequence (NC_012920).
Results
The m.1555A > G mutation was found in only one out of 54 families with NSHI(Fig. 1a). The family consisted of four generations. The m.1555A > G mutation was detected in six out of the 132 NSHI patients and in one of the 205 individuals with normal hearing. This result was confirmed by direct sequencing of the corresponding PCR product (Fig. 1b).
The family pedigree of patients with m.1555A > G mutation is shown in Fig. 2. Proposita (I: 2, Fig. 2.) is a 72 year old female with age related hearing impairment (presbycusis). She had eight offspring (4 males and 4 females) all with congenital, profound, bilateral and sensorineural deafness since childhood in accordance with mitochondrial inheritance (Fig. 2). Unfortunately drug history of aminoglycoside usage in this family was unavailable. The m.1555A > G mutation in the mitochondrial 12S rRNA gene was found in this mother and all her investigated offspring (Fig. 2).

A graphic map of a four generation large pedigree of a Syrian family found with m.1555A > G mutation in our study. The squares represent males, while the circles represent females. Blue filled shapes are affected individuals (deaf), white filled shapes are unaffected (controls)
Discussion
The mtDNA m.1555A > G mutation is the most prevalent mutation of mitochondrial genome, associated with both late onset and congenital NSHI, with aminoglycoside-induced hearing loss, in people with different ethnic backgrounds [16]. This is the first study to report the prevalence of m.1555A > G mitochondrial mutation in Syria, one of the Middle East countries. We found the m.1555A > G mutation in one out fifty-four families (1.85%), six out of the 132 NSHI patients (4.5%) and one proposita of the 205 individuals with a normal hearing (0.48%). It shows that the average frequency of the m.1555A > G mutation is 1.85% in the Syrian deaf population. Similar results were previously found in Egypt [17] and reports from other Caucasian populations [7, 12, 18], but not other Middle Eastern countries like Qatar [19], and Iran [20] probably due to the ethnic differences between these populations and the population under study.
Even though it has now been clearly shown that the Prevalence this mutation (1.85%) is not so high in familial cases in Syria, it has nevertheless a very important role. Detection of this mutation would delay the onset of the disease or even prevent it through a lifelong strict avoidance of taking aminoglycosides. When indicated, such a genetic analysis prior to the administration of aminoglycosides would be very valuable, since even a single, small parenteral administration may cause bilateral deafness as a serious side effect.. These findings will help the establishment of effective diagnosis for nonsyndromic hearing loss, improve genetic counselling, and serve as a potential therapeutic platform in the future for the affected patients in Syria.
Conclusion
This is the first study to report prelingual deafness causative gene mutations identified by sequencing technology in Syrian families. It is obvious from the results that the testing for the m.1555A > G mutation is useful for diagnosis of hearing loss in Syrian patients and should also be considered prior to treatment with aminoglycosides in predisposed individuals.
Abbreviations
- ATP:
-
Adenosine Tri Phosphate
- ENT:
-
Ear Nose Throat
- HI:
-
Hearing Impairement
- mDNA:
-
mitochondrial Deoxy-ribonucleic Acid
- NSHI:
-
Non-syndromic Hearing Impairment
- PCR:
-
Polymerase Chain Reaction
References
Newborn Hearing Screening: Overview, Prevalence of Hearing Loss, The High-Risk Register [Internet]. [cited 2017 Apr 16]. Available from: http://emedicine.medscape.com/article/836646-overview
Smith RJ, Shearer AE, Hildebrand MS, Van Camp G. Deafness and Hereditary Hearing Loss Overview [Internet]. GeneReviews(®). University of Washington, Seattle; 1993 [cited 2017 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301607
Schrijver I. Hereditary non-syndromic sensorineural hearing loss: transforming silence to sound. J Mol Diagn [Internet]. 2004 Nov [cited 2017 Apr 16];6(4):275–84. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15507665.
Smith RJ, Jones M-KN. Nonsyndromic Hearing Loss and Deafness, DFNB1 [Internet]. GeneReviews(®). University of Washington, Seattle; 1993 [cited 2017 Apr 18]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301449
Othman H, Saadat M. Prevalence of consanguineous marriages in syria. J Biosoc Sci [Internet]. 2009 Sep 12 [cited 2017 Apr 18];41(5):685. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19433003
Essammak BF, Ashour MJ, Sharif FA. Non-Syndromic autosomal recessive deafness in Gaza strip: A study of five GJB2 Gene mutations. Int J Genet Genomics [Internet]. 2014 [cited 2017 Apr 18];2(5):92–96. Available from: http://www.sciencepublishinggroup.com/j/ijgg
Bener A, Mohammad RR. Global distribution of consanguinity and their impact on complex diseases: Genetic disorders from an endogamous population. Egypt J Med Hum Genet [Internet]. 2017 [cited 2017 Apr 18]; Available from: http://www.sciencedirect.com/science/article/pii/S1110863017300174
Bitner-Glindzicz M. Hereditary deafness and phenotyping in humans. Br Med Bull [Internet]. 2002 Oct 1 [cited 2017 Apr 18];63(1):73–94. Available from: https://academic.oup.com/bmb/article-lookup/doi/10.1093/bmb/63.1.73
Perez-Fernandez D, Shcherbakov D, Matt T, Leong NC, Kudyba I, Duscha S, et al. 4′-O-substitutions determine selectivity of aminoglycoside antibiotics. Nat Commun [Internet]. 2014 Jan 28 [cited 2017 Apr 18];5:486–501. Available from: http://www.nature.com/doifinder/10.1038/ncomms4112
Min-Xin G. Mitochondrial DNA Mutations Associated with Aminoglycoside Ototoxicity. J Otol [Internet]. 2006 [cited 2017 Apr 18];1(2):65–75. Available from: http://www.sciencedirect.com/science/article/pii/S1672293006500169
Ouyang XM, Yan D, Yuan HJ, Pu D, Du LL, Han DY, et al. The genetic bases for non-syndromic hearing loss among Chinese. J Hum Genet [Internet]. 2009 Mar [cited 2017 Apr 18];54(3):131–140. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19197336
Bae JW, Kim D-B, Choi JY, Park H-J, Lee JD, Hur DG, et al. Molecular and Clinical Characterization of the Variable Phenotype in Korean Families with Hearing Loss Associated with the Mitochondrial A1555G Mutation. Moran M, editor. PLoS One [Internet]. 2012 Aug 6 [cited 2017 Apr 18];7(8):e42463. Available from: http://dx.plos.org/10.1371/journal.pone.0042463
Estivill X, Govea N, Barceló A, Perelló E, Badenas C, Romero E, et al. Familial Progressive Sensorineural Deafness Is Mainly Due to the mtDNA A1555G Mutation and Is Enhanced by Treatment with Aminoglycosides. Am J Hum Genet. 1998;62(1):27–35.
Giordano C, Pallotti F, Walker WF, Checcarelli N, Musumeci O, Santorelli F, et al. Pathogenesis of the deafness-associated A1555G mitochondrial DNA mutation. Biochem Biophys Res Commun. 2002;293(1):521–9.
Kupka S, Tóth T, Wróbel M, Zeißler U, Szyfter W, Szyfter K, et al. Mutation A1555G in the 12S rRNA gene and its epidemiological importance in German, Hungarian, and Polish patients. Hum Mutat [Internet]. 2002 Mar 1 [cited 2017 Aug 21];19(3):308–309. Available from: http://doi.wiley.com/10.1002/humu.9017
Liu XZ, Angeli S, Ouyang XM, Liu W, Ke XM, Liu YH, et al. Audiological and genetic features of the mtDNA mutations. Acta Otolaryngol [Internet]. 2008 Jul [cited 2017 Apr 18];128(7):732–738. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18568513
Fassad MR, Desouky LM, Asal S, Abdalla EM. Screening for the mitochondrial A1555G mutation among Egyptian patients with non-syndromic, sensorineural hearing loss. Int J Mol Epidemiol Genet [Internet]. 2014 [cited 2017 Aug 21];5(4):200–204. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25755848
Berthomieu B, Menasche M. An enumerative approach for analyzing time Petri nets [Internet]. Proceedings IFIP. 1983:41–6. Available from: http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.11.4063
Khalifa Alkowari M, Girotto G, Abdulhadi K, Dipresa S, Siam R, Najjar N, et al. GJB2 and GJB6 genes and the A1555G mitochondrial mutation are only minor causes of nonsyndromic hearing loss in the Qatari population. Int J Audiol [Internet]. 2012 Mar 21 [cited 2017 Aug 21];51(3):181–185. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22103400
Zohour MM, Tabatabaiefar MA, Dehkordi FA, Farrokhi E, Akbari MT, Chaleshtori MH. Large-Scale Screening of Mitochondrial DNA Mutations Among Iranian Patients with Prelingual Nonsyndromic Hearing Impairment. Genet Test Mol Biomarkers [Internet]. 2012 Apr [cited 2017 Aug 21];16(4):271–278. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22077646
Acknowledgements
This study was supported by the Jürgen ManchotStiftung, we thank all the families, patients and control individuals for their cooperation in this study.
Availability of data and materials
Data generated and/or analysed in this manuscript can obtained from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
HK designed the study, AB, MH and AS collected and analysed the data, MA and YB wrote the manuscript, MK revised and critically appraised it. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Human Ethical committee, University of Aleppo (Syria). Written informed consent was taken from all the family members and legal guardians for patients under 18 before participation in the study.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kaheel, H., Breß, A., Hassan, M.A. et al. Frequency of mitochondrial m.1555A > G mutation in Syrian patients with non-syndromic hearing impairment. BMC Ear Nose Throat Disord 18, 7 (2018). https://doi.org/10.1186/s12901-018-0055-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12901-018-0055-2