
Abstract
Background
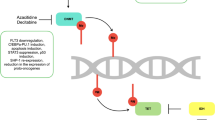
Tumor-suppressor genes are inactivated by methylation in several cancers including acute myeloid leukemia (AML). Src homology-2 (SH2)-containing protein-tyrosine phosphatase 1 (SHP-1) is a negative regulator of the JAK/STAT pathway. Transcriptional silencing of SHP-1 plays a critical role in the development and progression of cancers through STAT3 activation. 5-Azacytidine (5-Aza) is a DNA methyltransferase inhibitor that causes DNA demethylation resulting in re-expression of silenced SHP-1. Lestaurtinib (CEP-701) is a multi-targeted tyrosine kinase inhibitor that potently inhibits FLT3 tyrosine kinase and induces hematological remission in AML patients harboring the internal tandem duplication of the FLT3 gene (FLT3-ITD). However, the majority of patients in clinical trials developed resistance to CEP-701. Therefore, the aim of this study, was to assess the effect of re-expression of SHP-1 on sensitivity to CEP-701 in resistant AML cells.
Methods
Resistant cells harboring the FLT3-ITD were developed by overexposure of MV4-11 to CEP-701, and the effects of 5-Aza treatment were investigated. Apoptosis and cytotoxicity of CEP-701 were determined using Annexin V and MTS assays, respectively. Gene expression was performed by quantitative real-time PCR. STATs activity was examined by western blotting and the methylation profile of SHP-1 was studied using MS-PCR and pyrosequencing analysis. Repeated-measures ANOVA and Kruskal–Wallis tests were used for statistical analysis.
Results
The cytotoxic dose of CEP-701 on resistant cells was significantly higher in comparison with parental and MV4-11R-cep + 5-Aza cells (p = 0.004). The resistant cells showed a significant higher viability and lower apoptosis compared with other cells (p < 0.001). Expression of SHP-1 was 7-fold higher in MV4-11R-cep + 5-Aza cells compared to parental and resistant cells (p = 0.011). STAT3 was activated in resistant cells. Methylation of SHP-1 was significantly decreased in MV4-11R-cep + 5-Aza cells (p = 0.002).
Conclusions
The restoration of SHP-1 expression induces sensitivity towards CEP-701 and could serve as a target in the treatment of AML. Our findings support the hypothesis that, the tumor-suppressor effect of SHP-1 is lost due to epigenetic silencing and its re-expression might play an important role in re-inducing sensitivity to TKIs. Thus, SHP-1 is a plausible candidate for a role in the development of CEP-701 resistance in FLT3-ITD+ AML patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Acute myeloid leukemia (AML) is a hematological malignancy that occurs as a result of genetic aberrations in hematopoietic progenitor cells [1, 2]. Epigenetic silencing due to DNA hypermethylation is a frequent mechanism of inactivation of tumor suppressor genes (TSG) in variety of human cancers including AML [3]. 5-Azacytidine (5-Aza) is a chemotherapeutic agent that induces DNA demethylation by inhibition of DNA methyltransferase (DNMT) enzymes [4, 5]. The suppression of DNMTs in cancer cell lines induces hypomethylation resulting in decreased viability [6]. CEP-701 is a tyrosine kinase inhibitor (TKI) that potentially inhibits FLT3 tyrosine kinase and induces hematological remission in patients with AML. However, the majority of AML patients have only moderate and transient responses to tyrosine kinase inhibitors (TKIs) [7–9].
SHP-1 is a non-transmembrane protein tyrosine phosphatase expressed primarily in hematopoietic stem cells [10–12]. SHP-1 is a TSG that, in normal cells, negatively regulates Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling. The loss of SHP-1 suppressor function results in JAK or STAT activation in cancer cells [13–20]. The JAK/STAT signaling pathway is one of the most important signaling cascades that regulate various cell biological activities including immune response, cell growth, and differentiation [21].
Transcriptional silencing of SHP-1 due to promoter methylation has been reported in lymphoma and leukemia as well as in many hematopoietic cell lines [12, 22, 23]. Epigenetic silencing of SHP-1 in myeloproliferative neoplasms and K562 cells results in constitutive activation of JAK/STAT signaling [24]. The restoration of SHP-1 expression by a demethylating agent such as 5-Aza-2-deoxycytidine (5-Aza2dc) resulted in decreased JAK3, p-JAK3, and p-STAT3 but not STAT3 protein [25]. STAT3 and STAT5 are constitutively activated in myeloid tumors [26]. Resistance to imatinib in chronic myeloid leukemia is conferred by the activation of STAT3 signaling, and the sensitivity is restored by STAT3 inactivation [27].
We hypothesized that JAK/STAT negative regulators may lose their tumor suppression function in TKI-resistant AML cells due to epigenetic silencing, and the re-expression of these genes could re-induce sensitivity to CEP-701. Therefore, gene expression and methylation profiling of SHP-1 and its downstream targets were studied in FLT3-ITD positive AML cells resistant to CEP-701 before and after treatment with 5-Aza.
Methods
Lestaurtinib (CEP-701)
CEP-701 was purchased from LC Laboratories (Woburn, MA, USA) and dissolved in DMSO before use. The stock preparation was 1 mM, which was stored at −20 °C according to the manufacturer’s protocol.
Development of resistant cells
MV4-11, an AML cell line with FLT3-ITD, was obtained from the Department of Haematology, Universiti Sains Malaysia (USM), having originally been purchased from American Type Culture Collection (ATCC). The cells were cultured with RPMI 1640 (Life Technologies, Grand Island, NY, USA) supplemented with 10 % fetal bovine serum (FBS; Life Technologies, Grand Island, NY, USA) at a density of 5 × 104 cells/mL in a humid incubator with 5 % CO2 at 37 °C. A subclone of this line that was resistant to CEP-701, termed MV4-11R-cep, was developed according to the protocol described previously [28]. Briefly, log phase growing MV4-11 cells were co-cultured at a starting dose of 20 nM CEP-701 followed by a step-wise increase in concentration of 10–20 nM for 12 months until the cells were able to survive at the IC50 dose of CEP-701 on parental MV4-11. The resistant cell lines were grown in normal medium without CEP-701 for at least 48 h before starting the experiments.
5-Azacytidine treatment
5-Azacytidine (5-Aza; Sigma-Aldrich Corp., MO, USA) was dissolved in RPMI-1640 and the stocks at 500 μM were prepared for immediate use or stored at −20 °C, to be used within 2–3 days. Resistant cells were sub-cultured in working solution (5 μM) and incubated in a humidified incubator with 5 % CO2 at 37 °C for 4–5 days until confluent. MV4-11R-cep + 5-Aza cells were then sub-cultured in normal medium without treatment for at least one passage before re-treatment with CEP-701.
Growth inhibition assay
MV4-11 cells were seeded in 96-well culture plates at a density of 1 × 104 viable cells/100 μL/well in triplicates, and were treated with CEP-701. Colorimetric CellTiter 96 AQueous One Solution Cell Proliferation assay (MTS assay; Promega, Madison, WI, USA) was used to determine the cytotoxicity. The IC50 values were calculated using GraphPad Prism 3.02 (San Diego, California, USA). Each experiment was performed in triplicate.
Apoptosis assay
Annexin V–FITC binding assay (BD Pharmingen, San Diego, California, USA) was used as recommended by the manufacturer and analyzed by flow cytometry (BD FACSCanto™, San Jose, California, USA). Analysis was performed with Diva software (FACS Diva, 6.1.2, San Jose, California, USA). Each experiment was performed in triplicate.
RNA extraction
Total RNA was extracted from MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza cells using the Rneasy® Mini Kit (Qiagen, Valencia, California, USA), the purity and concentration was measured with a NanoDrop ND-1000 spectrophotometer V3.3.0 (NanoDrop Technologies, Berlin, Germany).
Quantitative reverse transcriptase PCR (RT-qPCR)
High Capacity RNA-to-cDNA kit (Applied Biosystem, Foster City, California, USA) was used to synthesize cDNA according to the manufacturer’s protocol. TaqMan Gene Expression assays (Applied Biosystems) were performed on an Applied Biosystem 7500 Fast Real-Time PCR System according to the manufacturer’s protocol. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. ABI 7500 software v2.0.6 (Applied Biosystem) was used to perform relative quantification of 5 target genes, SHP-1, SOCS-1, SOCS-3, STAT5a, and JAK2 using the comparative threshold cycle (Ct) method.
DNA extraction
DNA was extracted from MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza cells using the NucleoSpin® Tissue kit (Macherey-Nagel, Düren, Germany) following the manufacturer’s instructions. The concentration and purity of DNA were measured by NanoDrop.
Methylation-specific polymerase chain reactions (MS-PCR)
One microgram of DNA was treated with bisulfite using the EZ DNA Methylation-Gold™ Kit (Zymo Research, Irvine, NY, USA) according to the manufacturer’s instructions. MS-PCR was performed as described previously [29] and modified DNA was subjected to two separate PCRs. MS-PCR primers were designed to amplify the methylated (M) or unmethylated (U) alleles. SHP-1 (GeneBank: NM_002831) was amplified using previous designed primers [22]. Universal methylated DNA (Zymo Research, Irvine, NY, USA) was used as a positive control. The 50-μL PCR reaction contained 200 ng of bisulfite-treated DNA, ReddyMix PCR master mix (Bioline Ltd., London, UK) and 0.2 μM of each primer. PCRs were performed in a thermal cycler (PTC-200, Alameda, California, USA). The amplified PCR products were denatured for 2 min at 95 °C followed by 40 cycles: 95 °C for 25 s, 59 °C for 35 s, 52 °C and 72 °C for 65 s, and extension at 72 °C for 5 min. PCR products were electrophoresed on 2 % agarose gels, and visualized by ethidium bromide staining under ultraviolet transillumination. Results from triplicate experiments were used to determine methylation status.
Pyrosequencing analysis
Twenty microliters (1 μg) of purified DNA from each sample were sent to EpigenDx (Hopkinton, MA, USA) for pyrosequencing analysis. The assay was designed to target six CpG islands in the promoter regions of the SHP-1 gene.
Western blot analysis
Protein from MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza cells was extracted by RIPA buffer (Sigma-Aldrich, MO, USA). The three cell lines were incubated with 300 nM CEP-701 for 3 days before protein extraction. BioRad protein dye (BioRad, Hercules, California, USA) and a spectrophotometer (BioPhotometer Plus, Eppendorf, Germany) were employed for the measurement of protein concentrations. Preparation of immunoblotting was performed as described previously [30]. Antibodies used were anti-STAT1, anti-p-STAT1, anti-STAT3, anti-p-STAT3, anti-STAT5, anti-p-STAT5, and anti-β-actin (Thermo Scientific, Waltham, MA, USA).
Statistical and bioinformatics analysis
Repeated-measures ANOVA and Kruskal–Wallis tests were employed for statistical analyses. All statistical analyses were performed using the SPSS software package (Version 20, SPSS, Armonk, NY, USA) and a p value <0.05 was considered as significant.
Results
Long-term co-culture of MV4-11 cells with low doses of CEP-701 resulted in resistant cells
To verify the resistance of MV4-11R-cep cells to CEP-701, we determined the cytotoxicity and apoptosis of CEP-701 on MV4-11R-cep and parental MV4-11 cells. MV4-11 cells were inhibited by 290 nM CEP-701 whereas the resistant MV4-11R-cep cells were only inhibited by a higher dose (3340 nM). The IC50 of CEP-701 on MV4-11R-cep was more than 10-fold higher than that on MV4-11 (p = 0.004) (Fig. 1-a). There was a significant decrease in the percentage of apoptotic cells in MV4-11R-cep compared with parental MV4-11 based on incubation of parental and resistant cells in serial concentrations of CEP-701 (p < 0.001) (Fig. 1-b).

Cell growth inhibition, profile plot of apoptotic cells, and western blot analysis. a Cell growth inhibition by CEP-701 in MV4-11 (IC50 = 290 nM), MV4-11R-cep (IC50 = 3340 nM), and MV4-11R-cep + 5-Aza cells (IC50 = 200 nM). The cells were exposed to serial concentrations of CEP-701 for 72 h, and quantified by cell proliferation assay. Each result is presented as the median percentage of proliferation to unexposed control cells. b Repeated-measures ANOVA between groups based on concentrations was applied. The profile plot shows the adjusted mean (estimated marginal means) of apoptotic cells for all concentrations of CEP-701 (0, 100, 200, and 300 nM). Although the mean percentages of apoptotic cells before treatment with CEP-701 were almost equal for parental, resistant, and MV4-11R-cep + 5-Aza cells, there was a clear increase in the percentages of apoptotic cells in MV4-11 and MV4-11R-cep + 5-Aza cells with increasing concentration of CEP-701, reaching 58 and 65 %, respectively, at 300 nM. In contrast, the increase of apoptosis in the resistant cells was only 21 % at 300 nM PKC-412 (p < 0.001). c The phosphorylation of STAT1, STAT3, and STAT5 in MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza cells was assessed by western blotting. STAT3 was activated in MV4-11R-cep cells but not in MV4-11 and MV4-11R-cep + 5-Aza cells. However, STAT1 and STAT5 showed no phosphorylation in all cells; a, b, c, d, e and f indicate other cell lines not included in the present study but they are in agreement with the findings of this study
Higher sensitivity to CEP-701 in MV4-11R-cep treated with 5-Aza
The MTS assay showed a decrease in the IC50 of CEP-701 on MV4-11R-cep + 5-Aza cells compared with that of MV4-11R-cep cells (p = 0.011) (Fig. 1-a). The IC50 on MV4-11R-cep + 5-Aza was also lower than that of MV4-11 cells, however this difference was not significant (p = 0.099).
Induction of apoptosis in MV4-11 and MV4-11R-cep + 5-Aza in response to CEP-701
The vitality and fraction of apoptotic and necrotic cells of MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza cells after various incubations with CEP-701 are shown in Fig. 2. Upon incubation of cells in the presence of 300 nM CEP-701, a significant reduction of cell viability of 90 % down to 37 and 33 % was detected in MV4-11 and MV4-11R-cep + 5-Aza cells, respectively. In contrast, the resistant cell line MV4-11R-cep still showed 77 % viable cells after treatment (Fig. 2). Figure 3 depicts the course of apoptotic (Q2 and Q4) and necrotic (Q1) cells over 72 h measured by Annexin V/FITC-FACS analysis in MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza cells after addition of 100, 200, and 300 nM of CEP-701. The resistant cells showed a significant increase in the viability with obvious decrease in apoptosis after incubation with CEP-701 compared with the parental and MV4-11R-cep + 5-Aza cell lines. Despite the mean percentage of apoptotic cells before incubations with CEP-701 being almost equal for MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza, there was a sharp increase in apoptotic MV4-11 and MV4-11R-cep + 5-Aza cells with increased drug concentration, reaching 58 and 65 % apoptosis, respectively, at 300 nM (Fig. 1-b). In contrast, there was no significant increase in the apoptotic cells in MV4-11R-cep with increased drug concentrations, with only 21 % apoptotic cells at 300 nM (p < 0.001).

The vitality and fraction of apoptotic cells in MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza cells. Apoptotic cells increased significantly in (a) MV4-11 and (c) MV4-11R-cep + 5-Aza cells compared with (b) MV4-11R-cep cells by increasing concentrations of CEP-701. There was a significant reduction of cell viability from 90% down to 37 and 33% in MV4-11 and MV4-11R-cep + 5-Aza cells, in association with 58 and 65% apoptotic cells, respectively. In contrast, we observed only 21% apoptotic cells in MV4-11R-cep cells at the highest CEP-701 concentration with 77% viable cells remaining (p < 0.001)

CEP-701 induced decrease of vitality in MV4-11 and MV4-11R-cep + 5-Aza cells. Flow cytometric scatterplots of MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza cells after addition of various concentrations of PKC-412 at 72 h. The data depict the course of apoptotic (Q2 and Q4), live (Q3), and necrotic (Q1) cells. The decrease of vitality induced by CEP-701 was concentration dependent and was greater in MV4-11 and MV4-11R-cep + 5-Aza cells compared with MV4-11R-cep
Activated STAT3 in resistant acute myeloid leukemia cells
The activation status of STAT1, STAT3, and STAT5 proteins were studied using western blot. Although these proteins were expressed in all cell lines, STAT3 was only activated in the resistant cells and not the parental and MV4-11R-cep + 5-Aza cells. In contrast, there was no differences in the activity of STAT1 and STAT5 between resistant and parental or MV4-11R-cep + 5-Aza cells (Fig. 1-c).
Restoration of SHP-1 gene expression in MV4-11R-cep + 5-Aza cells
To investigate the correlation between re-expression of SHP-1 and demethylation, gene expression by RT-qPCR was performed on MV4-11, MV4-11R-cep, and MV4-11R-cep + 5-Aza cells. The results showed a significant up-regulation of SHP-1 in MV4-11R-cep + 5-Aza cells compared with MV4-11 and MV4-11R-cep cells (p = 0.011 and p = 0.002, respectively; Fig. 4-a).

Real-time quantitative PCR (RQ-PCR) and methylation status of SHP-1 in all cells. a The relative normalized ratio of RQ-PCR revealed that, SHP-1 is re-expressed in MV4-11R-cep + 5-Aza cells 7-fold higher compared with that in MV4-11 and MV4-11R-cep cells (p = 0.011). b Methylation-specific polymerase chain reaction (MS-PCR) showed methylation of SHP-1 in MV4-11 and MV4-11R-cep cells but not in MV4-11R-cep + 5-Aza cells. c Pyrosequencing analysis revealed low methylation levels of the CpG islands in the promoter region of SHP-1 in MV4-11R-cep + 5-Aza cells. The Kruskal–Wallis test was applied followed by the Multiple Mann–Whitney Test with Bonferroni correction. The box blot showed a significant lower (p = 0.023) of methylation in the CpG islands of SHP-1 gene in MV4-11R-cep + 5-Aza cells compared with that in MV4-11 and MV4-11R-cep cells. However, there was no significant difference in the methylation levels of CpG islands in the same region of SHP-1 genes between MV4-11 and MV4-11R-cep cells (p = 0.200)
Low methylation of SHP-1 gene in MV4-11R-cep + 5-Aza cells
To relate the expression of SHP-1 with methylation status, MS-PCR and pyrosequencing analysis were performed on DNA from the three cell lines as well as positive and negative controls. MS-PCR revealed methylated SHP-1 in parental MV4-11 and resistant MV4-11R-cep cells but not MV4-11R-cep + 5-Aza cells (Fig. 4-b). The methylation status was confirmed by pyrosequencing analysis, which revealed a significantly lower level of methylation in CpG islands in the promoter region of the SHP-1 gene when treated with 5-Aza compared with untreated MV4-11 and MV4-11R-cep cells (p = 0.023). However, there was no significant difference in the methylation of CpG islands in the promoter region of SHP-1 in MV4-11 cells compared with MV4-11R-cep cells (p = 0.200; Table 1 and Fig. 4-c).
Discussion
Resistance to TKIs remains a challenge in the treatment of AML patients. The mechanism of acquired resistance to TKIs has been studied in vitro and in vivo [7, 31–35] but is still not fully understood. Aberrant methylation of tumor suppressor genes (TSG) such as SOCS-1, SOCS-3, SHP-1, and PRG2 has been documented in a variety of cancers including hematological malignancies [3, 36–42], and is a plausible means by which cells can acquire therapy resistance.
In the present study, FLT3-ITD+ AML cells resistant to CEP-701 were developed by overexposure of parental cells to the drug. Acquired resistance was confirmed by cytotoxicity and apoptosis assays that showed significant differences in parental compared with resistant cells. The IC50 of CEP-701 on resistant MV-11R-cep was more than 10-fold higher than that of parental MV4-11 cells (p = 0.004) and acquired resistance was associated with low apoptosis (p < 0.001). This is in agreement with previously reported studies on the development of resistant cell lines to ABT-869 and PKC-412 [7, 31].
Gene expression analysis revealed low expression of SHP-1 and PRG2 in MV4-11 and MV4-11R-cep cells (data not shown for PRG2 gene). However, after treatment of MV4-11R-cep cells with 5-Aza, we observed re-expression of SHP-1 and PRG2 that was associated with inhibition of STAT3 activity. Moreover, the transcriptional silencing of SHP-1 and PRG2 genes was due to hyermethylation of CpG islands in the promoter regions of both genes in MV4-11 and MV4-11R-cep cells. Transcriptional silencing of TSGs is mediated by DNMTs in tumor cells [43–46]. The gene expression analysis also showed significant up-regulation of DNMT1, DNMT3a, and DNMT3b (data not shown), which have been reported to regulate the expression of TSGs through methylation of CpG islands in the promoter regions [47, 48]. Methylation profiling revealed hypermethylation of CpG islands in the promoter regions of SHP-1 and PRG2 in MV4-11 and MV4-11R-cep cells (data not shown for PRG2 gene). These findings suggest that, the up-regulated DNMTs in the parental and resistant cells methylate the CpG islands of SHP-1 and PRG2 genes, resulting in their transcriptional silencing. Our findings are consistent with previous reports that revealed hypermethylation of SHP-1 and PRG2 in leukemic cell lines [22, 38, 42, 49].
In hematological malignancies and leukemic cell lines, the tumor-suppressing function of SHP-1 is lost because of promoter methylation, resulting in constitutive activation of JAK/STAT signaling [22, 24, 39, 50, 51]. Epigenetic silencing of one of the JAK/STAT negative regulators is sufficient for activation of STAT signaling [40]. Methylation of SHP1 is involved in the constitutive activation of STAT3 [50], and a low level of SHP-1 is not sufficient to inhibit activated STAT3 [25]. Transcriptional silencing of SHP-1 also plays a role in the development of resistance to imatinib in BCR-ABL1-positive CML cells [51]. Similarly, constitutive activation of STAT3 and STAT5 are common events in myeloid leukemia and have previously been implicated in resistance to TKIs [7, 26, 52]. Bewry, et al. [27] suggested that, the activation of STAT3 is an important mechanism of imatinib resistance. Likewise, overexpression of PRG2 in myeloid cells blocked G-CSF-dependent proliferation and increased apoptosis [53]. However, epigenetic silencing of PRG2 is associated with higher proliferation and lowered apoptosis in pancreatic cancer cells [54] and leukemic cells [42].
In the present study, we observed activation of STAT3 in MV4-11R-cep cells but not in MV4-11 cells. The findings suggest that, activated STAT3 could be involved in the acquisition of resistance to CEP-701 in MV4-11R-cep cells, which is consistent with previous reports in which, the activation of STAT3 was associated with acquired resistance to ABT-869 in AML [7], and to imatinib in CML [27, 51]. After treatment of MV4-11R-cep with 5-Aza, STAT3 was inactivated and cells showed higher sensitivity to CEP-701. This finding is in accordance with other reports [7, 25, 51]. Our data suggest a crucial role for STAT3 in the development of resistance to TKIs and inhibition of STAT3 phosphorylation provides an effective means of re-inducing sensitivity.
STAT3 is negatively regulated by TSGs such as SHP-1, SOCS-1, and SOCS-3 [7, 41, 51, 55, 56]. Re-expression of these genes by 5-Aza or 5-Aza2dc results in inactivation of STAT3 [7, 25, 51, 57, 58]. In addition, inactivation of STAT3 enhances apoptosis and restores sensitivity towards TKIs [27, 59]. Similarly, we found that the re-expression of SHP-1 and PRG2 is associated with inactivation of STAT3 in 5-Aza-treated cells. Moreover, the sensitivity of MV4-11 and MV4-11R-cep + 5-Aza cells towards CEP-701 was significantly higher with low IC50, at only 200 nM compared with 3340 nM in MV4-11R-cep (p = 0.011). The findings suggest that, the restoration of expression of SHP-1 and PRG2 could induce sensitivity towards CEP-701 through inactivation of STAT3. Our observations are supported by previous reports [7, 25, 27, 31, 51, 59].
We also found that, increasing CEP-701 concentration caused a significant increase of apoptosis in 5-Aza-treated resistant cells compared with the untreated resistant cells (p < 0.001) In addition, we observed no significant difference when CEP-701 concentration was increased by 100 nM in untreated resistant cells whereas the 5-Aza-treated resistant cells showed significant differences at each incremental increase in TKI concentration. Taken together, our results indicate that, re-expression of SHP-1 and suppression of STAT3 are associated with induction of apoptosis in TKI-resistant FLT3-ITD+ AML cells. These findings are similar to those previously reported [7, 60–62]. Enhanced re-expression of SHP-1 could therefore play role in the management of CEP-701-resistant patients. Further studies are needed to clarify the correlation between the re-expression of the SHP-1 gene with STAT3 inhibition and to confirm the clinical utility of this approach.
Conclusion
The epigenetic silencing of SHP-1 results in loss of its tumor suppressor function and re-expression of SHP-1 by 5-Aza may enhance sensitivity to CEP-701 through inactivation of STAT3.
References
Hart S, Goh KC, Novotny-Diermayr V, Tan YC, Madan B, Amalini C, et al. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J. 2011;1:e44.
Yan-Fang T, Jian N, Jun L, Na W, Pei-Fang X, Wen-Li Z, et al. The promoter of miR-663 is hypermethylated in Chinese pediatric acute myeloid leukemia (AML). BMC Med Genet. 2013;14:74.
Leone G, Voso MT, Teofili L, Lubbert M. Inhibitors of DNA methylation in the treatment of hematological malignancies and MDS. Clin Immunol. 2003;109:89–102.
Garcia-Manero G. Demethylating agents in myeloid malignancies. Curr Opin Oncol. 2008;20:705–10.
Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol. 2005;23:3971–93.
Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15:3938–46.
Zhou J, Bi C, Janakakumara JV, Liu SC, Chng WJ, Tay KG, et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009;113:4052–62.
Small D. FLT3 mutations: biology and treatment. Hematology. 2006;2006:178–84.
Fathi A, Levis M. FLT3 inhibitors: a story of the old and the new. Curr Opin Hematol. 2011;18:71.
Yi TL, Cleveland JL, Ihle JN. Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13. Mol Cell Biol. 1992;12:836–46.
Ulyanova T, Blasioli J, Thomas ML. Regulation of cell signaling by the protein tyrosine phosphatases, CD45 and SHP-1. Immunol Res. 1997;16:101–13.
Oka T, Yoshino T, Hayashi K, Ohara N, Nakanishi T, Yamaai Y, et al. Reduction of hematopoietic cell-specific tyrosine phosphatase SHP-1 gene expression in natural killer cell lymphoma and various types of lymphomas/leukemias : combination analysis with cDNA expression array and tissue microarray. Am J Pathol. 2001;159:1495–505.
Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102:276–83.
Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, et al. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73:1445–54.
Jiao H, Berrada K, Yang W, Tabrizi M, Platanias LC, Yi T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol Cell Biol. 1996;16:6985–92.
Wu C, Sun M, Liu L, Zhou GW. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene. 2003;306:1–12.
Wu C, Guan Q, Wang Y, Zhao ZJ, Zhou GW. SHP-1 suppresses cancer cell growth by promoting degradation of JAK kinases. J Cell Biochem. 2003;90:1026–37.
Chim CS, Wong AS, Kwong YL. Epigenetic dysregulation of the Jak/STAT pathway by frequent aberrant methylation of SHP1 but not SOCS1 in acute leukaemias. Ann Hematol. 2004;83:527–32.
Khoury JD, Rassidakis GZ, Medeiros LJ, Amin HM, Lai R. Methylation of SHP1 gene and loss of SHP1 protein expression are frequent in systemic anaplastic large cell lymphoma. Blood. 2004;104:1580–1.
Tassidis H, Culig Z, Wingren AG, Harkonen P. Role of the protein tyrosine phosphatase SHP-1 in Interleukin-6 regulation of prostate cancer cells. Prostate. 2010;70:1491–500.
Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, et al. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005;24:6406–17.
Oka T, Ouchida M, Koyama M, Ogama Y, Takada S, Nakatani Y, et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002;62:6390–4.
Chim CS, Wong KY, Loong F, Srivastava G. SOCS1 and SHP1 hypermethylation in mantle cell lymphoma and follicular lymphoma: implications for epigenetic activation of the Jak/STAT pathway. Leukemia. 2004;18:356–8.
Zhang MY, Fung TK, Chen FY, Chim CS. Methylation profiling of SOCS1, SOCS2, SOCS3, CISH and SHP1 in Philadelphia-negative myeloproliferative neoplasm. J Cell Mol Med. 2013;17:1282–90.
Han Y, Amin HM, Frantz C, Franko B, Lee J, Lin Q, et al. Restoration of shp1 expression by 5-AZA-2'-deoxycytidine is associated with downregulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell lymphoma. Leukemia. 2006;20:1602–9.
Bar-Natan M, Nelson EA, Xiang M, Frank DA. STAT signaling in the pathogenesis and treatment of myeloid malignancies. Jak-Stat. 2012;1:55–64.
Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther. 2008;7:3169–75.
Coley HM. Development of drug-resistant models. Methods Mol Med. 2004;88:267–73.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6.
Frohling S, Scholl C, Levine RL, Loriaux M, Boggon TJ, Bernard OA, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell. 2007;12:501–13.
Stolzel F, Steudel C, Oelschlagel U, Mohr B, Koch S, Ehninger G, et al. Mechanisms of resistance against PKC412 in resistant FLT3-ITD positive human acute myeloid leukemia cells. Ann Hematol. 2010;89:653–62.
Weisberg E, Barrett R, Liu Q, Stone R, Gray N, Griffin JD. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist Updat. 2009;12:81–9.
Villuendas R, Steegmann JL, Pollan M, Tracey L, Granda A, Fernandez-Ruiz E, et al. Identification of genes involved in imatinib resistance in CML: a gene-expression profiling approach. Leukemia. 2006;20:1047–54.
Binato R, Mencalha A, Pizzatti L, Scholl V, Zalcberg I, Abdelhay E. RUNX1T1 is overexpressed in imatinib mesylate-resistant cells. Mol Med Rep. 2009;2:657–61.
Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood. 2007;109:1643–52.
Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97:1172–9.
Hatirnaz O, Ure U, Ar C, Akyerli C, Soysal T, Ferhanoglu B, et al. The SOCS-1 gene methylation in chronic myeloid leukemia patients. Am J Hematol. 2007;82:729–30.
Uhm KO, Lee ES, Lee YM, Park JS, Kim SJ, Kim BS, et al. Differential methylation pattern of ID4, SFRP1, and SHP1 between acute myeloid leukemia and chronic myeloid leukemia. J Korean Med Sci. 2009;24:493–7.
Li Y, Yang L, Pan Y, Yang J, Shang Y, Luo J. Methylation and decreased expression of SHP-1 are related to disease progression in chronic myelogenous leukemia. Oncol Rep. 2014;31:2438–46.
Johan M, Bowen D, Frew M, Goodeve A, Reilly J. Aberrant methylation of the negative regulators RASSFIA, SHP 1 and SOCS 1 in myelodysplastic syndromes and acute myeloid leukaemia. Br J Haematol. 2005;129:60–5.
Al-Jamal HA, Jusoh SA, Yong AC, Asan JM, Hassan R, Johan MF. Silencing of suppressor of cytokine signaling-3 due to methylation results in phosphorylation of STAT3 in imatinib resistant BCR-ABL positive chronic myeloid leukemia cells. Asian Pac J Cancer Prevent. 2014;15:4555–61.
Al-jamal H, Jusoh S, Sidek M, Hassan R, Johan M. Restoration of PRG2 Expression by 5-Azacytidine Involves in Sensitivity of PKC-412 (Midostaurin) Resistant FLT3-ITD Positive Acute Myeloid Leukaemia Cells. J Hematol Thrombo Dis. 2015;3:2.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33.
Song J, Rechkoblit O, Bestor TH, Patel DJ. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011;331:1036–40.
Ding YB, Long CL, Liu XQ, Chen XM, Guo LR, Xia YY, et al. 5-aza-2'-deoxycytidine leads to reduced embryo implantation and reduced expression of DNA methyltransferases and essential endometrial genes. PLoS One. 2012;7:e45364.
Gao J, Wang L, Xu J, Zheng J, Man X, Wu H, et al. Aberrant DNA methyltransferase expression in pancreatic ductal adenocarcinoma development and progression. J Exp Clin Cancer Res. 2013;32:86.
Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–6.
Xu M, Gao J, Du YQ, Gao DJ, Zhang YQ, Li ZS, et al. Reduction of pancreatic cancer cell viability and induction of apoptosis mediated by siRNA targeting DNMT1 through suppression of total DNA methyltransferase activity. Mol Med Rep. 2010;3:699–704.
Gauffin F, Diffner E, Gustafsson B, Nordgren A, Wingren AG, Sander B, et al. Expression of PTEN and SHP1, investigated from tissue microarrays in pediatric acute lymphoblastic, leukemia. Pediatr Hematol Oncol. 2009;26:48–56.
Chim CS, Fung TK, Cheung WC, Liang R, Kwong YL. SOCS1 and SHP1 hypermethylation in multiple myeloma: implications for epigenetic activation of the Jak/STAT pathway. Blood. 2004;103:4630–5.
Esposito N, Colavita I, Quintarelli C, Sica AR, Peluso AL, Luciano L, et al. SHP-1 expression accounts for resistance to imatinib treatment in Philadelphia chromosome-positive cells derived from patients with chronic myeloid leukemia. Blood. 2011;118:3634–44.
Benekli M, Xia Z, Donohue KA, Ford LA, Pixley LA, Baer MR, et al. Constitutive activity of signal transducer and activator of transcription 3 protein in acute myeloid leukemia blasts is associated with short disease-free survival. Blood. 2002;99:252–7.
Liu Q, Dong F. Gfi-1 inhibits the expression of eosinophil major basic protein (MBP) during G-CSF-induced neutrophilic differentiation. Int J Hematol. 2012;95:640–7.
Hagihara A, Miyamoto K, Furuta J, Seki S, Fukushima S, Ushijima T. Methylation-associated silencing of four genes in human pancreatic cancers. Proc Am Assoc Cancer Res. 2004;2004:1151.
Rhee YH, Jeong SJ, Lee HJ, Lee HJ, Koh W, Jung JH, et al. Inhibition of STAT3 signaling and induction of SHP1 mediate antiangiogenic and antitumor activities of ergosterol peroxide in U266 multiple myeloma cells. BMC Cancer. 2012;12:28.
Lee TL, Yeh J, Van Waes C, Chen Z. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol Cancer Ther. 2006;5:8–19.
Witzig TE, Hu G, Offer SM, Wellik LE, Han JJ, Stenson MJ, et al. Epigenetic mechanisms of protein tyrosine phosphatase 6 suppression in diffuse large B-cell lymphoma: implications for epigenetic therapy. Leukemia. 2014;28:147–54.
Zhang Q, Raghunath PN, Vonderheid E, Odum N, Wasik MA. Lack of phosphotyrosine phosphatase SHP-1 expression in malignant T-cell lymphoma cells results from methylation of the SHP-1 promoter. Am J Pathol. 2000;157:1137–46.
Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB, Tweardy DJ. Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood. 2011;117:5701–9.
Liang P, Cheng SH, Cheng CK, Lau KM, Lin SY, Chow EY, et al. Platelet factor 4 induces cell apoptosis by inhibition of STAT3 via up-regulation of SOCS3 expression in multiple myeloma. Haematologica. 2013;98:288–95.
Xiong H, Chen ZF, Liang QC, Du W, Chen HM, Su WY, et al. Inhibition of DNA methyltransferase induces G2 cell cycle arrest and apoptosis in human colorectal cancer cells via inhibition of JAK2/STAT3/STAT5 signalling. J Cell Mol Med. 2009;13:3668–79.
Shin DY, Park YS, Yang K, Kim GY, Kim WJ, Han MH, et al. Decitabine, a DNA methyltransferase inhibitor, induces apoptosis in human leukemia cells through intracellular reactive oxygen species generation. Int J Oncol. 2012;41:910–8.
Acknowledgments
This work was supported by a generous Research University’s Grant from Universiti Sains Malaysia (1001/PPSP/813050) and the Fundamental Research Grant Scheme of the Ministry of Education, Malaysia (203/PPSP/6171164) to MFJ. We thank all staffs at the Central Research and Molecular Haematology Laboratories, School of Medical Sciences; and the Craniofacial Laboratory, School of Dental Sciences, Universiti Sains Malaysia who provided technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have no conflicts of interest.
Authors’ contributions
HANA carried out the molecular, cellular, and protein studies, performed the statistical analysis and drafted the manuscript. SAMJ participated in the maintenance of the cells. MFJ conceived of the study, and participated in its design and coordination. MFJ and RH helped to draft the manuscript. All authors have read and approved the content of the manuscript.
Authors’ information
Hamid Ali Nagi Al-Jamal and Siti Asmaa Mat Jusoh are PhD students at the Department of Haematology, School of Medical Sciences, Universiti Sains Malaysia. Dr. Muhammad Farid Johan and Prof. Rosline Hassan are lecturers and scientists at the Department of Haematology, School of Medical Sciences, Universiti Sains Malaysia.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Al-Jamal, H.A.N., Mat Jusoh, S.A., Hassan, R. et al. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer 15, 869 (2015). https://doi.org/10.1186/s12885-015-1695-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-015-1695-x