
Abstract
Background
Differentiation of Parkinson’s disease (PD) from the various types of atypical parkinsonism (AP) such as multiple system atrophy (MSA), progressive supranuclear palsy (PSP), dementia with Lewy bodies (DLB), corticobasal syndrome (CBS) and vascular parkinsonism (VP), can be challenging, especially early in the disease course when symptoms overlap. A major unmet need in the diagnostic workup of these disorders is a diagnostic tool that differentiates the various disorders, preferably in the earliest disease stages when the clinical presentation is similar. Many diagnostic tests have been evaluated, but their added value was studied mostly in retrospective case-control studies that included patients with a straightforward clinical diagnosis. Here, we describe the design of a prospective cohort study in patients with parkinsonism in an early disease stage who have an uncertain clinical diagnosis. Our aim is to evaluate the diagnostic accuracy of (1) detailed clinical examination by a movement disorder specialist, (2) magnetic resonance imaging (MRI) techniques and (3) cerebrospinal fluid (CSF) biomarkers.
Methods/design
Patients with parkinsonism with an uncertain clinical diagnosis and a disease course less than three years will be recruited. Patients will undergo extensive neurological examination, brain MRI including conventional and advanced sequences, and a lumbar puncture. The diagnosis (including level of certainty) will be defined by a movement disorders expert, neuroradiologist and neurochemist based on clinical data, MRI results and CSF results, respectively. The clinical diagnosis after three years’ follow-up will serve as the “gold standard” reference diagnosis, based on consensus criteria and as established by two movement disorder specialists (blinded to the test results). Diagnostic accuracy of individual instruments and added value of brain MRI and CSF analysis after evaluation by a movement disorder expert will be calculated, expressed as the change in percentage of individuals that are correctly diagnosed with PD or AP.
Discussion
This study will yield new insights into the diagnostic value of clinical evaluation by a movement disorder specialist, brain MRI and CSF analysis in discriminating PD from AP in early disease stages. The outcome has the potential to help clinicians in choosing the optimal diagnostic strategy for patients with an uncertain clinical diagnosis.
Trial registration
NCT01249768, registered November 26 2010.
Similar content being viewed by others

Background
Differentiation between Parkinson’s Disease (PD) and the various forms of atypical parkinsonism (AP) can be difficult, especially early in the disease course. This is due to overlap in clinical symptoms, certainly when the clinical picture has not yet developed fully. Forms of AP such as multiple system atrophy (MSA), progressive supranuclear palsy (PSP) or corticobasal syndrome (CBS) follow a more malignant disease course than PD and, unlike PD, generally do not respond well to dopaminergic therapy.
While difficult, a correct diagnosis is important for adequate disease management and patient counseling, as well as for research purposes. If, in the future, any disease-modifying therapy becomes available, then reliable diagnostic tools will be essential to distinguish PD from AP at the earliest possible stage.
Because there is not yet a reliable objective test for PD and AP, expert opinion remains the gold standard for the clinical diagnosis. In the hands of a movement disorders specialist, the diagnostic accuracy is higher (90%) compared to judgment by a general neurologist (76%) [1, 2]. Although the clinical diagnosis after several years of follow-up correlates highly with the neuropathological diagnosis upon post-mortem examination, the diagnostic accuracy of the initial diagnosis varies greatly, and can be as low as 30% [1, 3,4,5]. When a clinician has little doubt about the diagnosis, ancillary investigations have hardly any diagnostic value. But when the clinical picture is puzzling, which is often the case early in the disease course, reliable biomarkers are needed for accurate and early differentiation between PD and AP.
Brain MRI is nowadays part of the diagnostic workup of patients with suspected parkinsonism, mainly to exclude secondary causes of parkinsonism (e.g. tumor, hydrocephalus, multiple sclerosis, vascular parkinsonism). Conventional MRI can demonstrate abnormalities characteristic for various AP syndromes, such as putaminal or olivopontocerebellar atrophy in MSA, midbrain atrophy in PSP and asymmetric regional cortical atrophy in CBS.
Due to limited sensitivity of these MRI abnormalities (e.g. 68% for cerebellar atrophy and 59% for putaminal atrophy), their contribution to the clinically based diagnosis is limited [6]. These abnormalities often occur late in the disease course, which make them of less diagnostic value in early disease stages. However, advances in neuroimaging techniques are promising to provide new biomarkers for improved differentiation between the different parkinsonian syndromes.
Magnetic resonance volumetry (MRV), diffusion tensor imaging (DTI), susceptibility weighted imaging (SWI) and resting state functional magnetic resonance imaging (rs-fMRI) are examples of new MR techniques which have become available [7,8,9,10]. However, these new techniques are often difficult to interpret for the individual patient, since MRI patterns are investigated on a group level. Moreover, validated diagnostic criteria are generally lacking. For example, DTI studies found alterations in fractional anisotropy and mean diffusivity in the substantia nigra in PD patients and in the cerebellum, pons, cerebellar peduncles and putamen in MSA patients. Furthermore, increased iron accumulation in the substantia nigra on SWI sequences is suggestive for PD as is increased putaminal hypointensity for MSA [7, 11, 12]. Rs-fMRI has been applied to assess functional abnormalities in neural networks of PD patients. Several studies identified PD-specific network patterns compared to healthy controls, but if these patterns differ from AP cases is yet unknown [13].
Furthermore, research on cerebrospinal fluid (CSF) biomarkers has strongly developed over the past decade. Analysis of brain specific proteins in CSF can be of value in distinguishing PD from AP. In PD patients, concentrations of brain specific proteins and neurotransmitter metabolites are generally within normal ranges, whereas in AP aberrant levels of these proteins and metabolites can be found. Currently, neurofilament light chain (NFL), total and phosphorylated tau (t-tau and p-tau), α-synuclein (α-syn) and amyloid-β42 (Aβ42) seem to be the most promising biomarkers for the differential diagnosis of parkinsonian syndromes. NFL levels are increased in AP compared with PD and healthy controls, tau proteins are elevated in CBS, while Aβ42 and tau are associated with cognitive decline (dementia) in PD and DLB [14,15,16,17,18,19,20,21].
Given the complexity of parkinsonian syndromes it is likely that neither a single clinical test nor a single biomarker can differentiate between the different parkinsonian syndromes. A combination of parameters could however improve diagnostic accuracy [14, 15, 18, 19, 21]. On the other hand, MRI is time-consuming and expensive, CSF analysis requires a lumbar puncture and many centers lack a neurologist with experience in movement disorders. More knowledge about the diagnostic value of each test could help clinicians in selecting the best available test.
With this prospective cohort study, we aim to gain insight in which single or combined ancillary investigation (including the opinion of a movement disorder specialist) will have the highest diagnostic yield for discrimination of PD and AP in early disease stage when diagnostic uncertainty is high.
Methods/design
Study design
A prospective observational cohort study to assess the diagnostic value of clinical evaluation by a movement disorder specialist, brain MRI and CSF analysis in discriminating different forms of parkinsonism early in the disease course. The Medical Ethics Committee Arnhem-Nijmegen approved the study. Written informed consent will be obtained from all patients after detailed explanation of the procedures.
Objectives
The main objective of our study is to determine the diagnostic accuracy of detailed clinical examination by a movement disorder specialist, conventional and advanced brain MRI techniques and CSF analysis to differentiate PD from AP in clinically uncertain cases. Secondary objective is to identify potential variables for differentiation between PD and AP.
Third objective is to identify potential discriminative variables between the various forms of AP.
Study population
Patients will be recruited from our outpatient movement disorder clinic and outpatient neurology clinics of referring general hospitals. Inclusion criteria are clinical signs and symptoms of parkinsonism (hypokinetic-rigid syndrome) with uncertain clinical diagnosis and disease duration less than three years. Exclusion criteria are age below 18 years and unstable co-morbidity. Inclusion and exclusion criteria are provided in Table 1.
Clinical examination
Evaluation by a movement disorder specialist
All patients will be interviewed and examined by a movement disorder specialist at the outpatient neurology department of the Radboud university medical center in Nijmegen (The Netherlands).
Structured interview
Standardized questionnaires on demographics, medical history, cardiovascular risk factors, activities in daily living, medication use, disease onset, clinical signs, most affected body site, balance and fear of falling will be administered at baseline and every year during follow-up for 3 years. In addition, the Non-Motor Symptom Scale (NMSS) [22], Hospital Anxiety and Depression Scale (HADS), Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease (QUIP) and Epworth Sleepiness Scale (ESS) will be scored [23,24,25]. A translated version of the structured questionnaires are provided as Additional file 1 with this article.
Cognitive assessment
A measurement of global cognitive function will be assessed by the Mini Mental State Examination (MMSE) [26], Frontal Assessment Battery (FAB) [27] and Cambridge Neuropsychological Automated Test Battery [28] at baseline and after three years follow-up.
Neurological examination
Standard neurological examination will be performed including testing of cranial nerves, motor and sensory function, tendon reflexes and presence or absence of primary reflexes. Horizontal and vertical blood pressure will be measured to screen for orthostatic hypotension.
In addition, the following clinimetric scales will be assessed: Unified Parkinson’s Disease rating scale (UPDRS) III and IV, Hoehn and Yahr disease severity score [29] and Scale for the assessment and rating of ataxia (SARA) [30]. The neurological examination will be performed and videotaped at baseline and after three years follow-up.
Ancillary investigations
MRI
All patients will have a brain MRI at first presentation performed on 3 Tesla MRI scanner (Siemens Magnetom Trio, Erlangen, Germany) by a standardized scanning protocol. Conventional MRI sequences will include 3D T1 MPrage, T2 turbo-spin echo, T2 fluid attenuated inversed recovery (FLAIR), and proton density sequences. Advanced MRI sequences will include diffusion weighted imaging (DWI) and diffusion tensor imaging (DTI), SWI and rs-fMRI. All MRI scans will be evaluated in a systematic fashion by two neuroradiologists blinded for clinical symptoms and outcome. The presence of the following abnormalities on conventional MRI sequences will be scored using a diagnostic workstation (Agfa-Impax version 6.5.3, Gevaert, Mortsel, Belgium): atrophy and T2 hypo-intensity of the putamen, putaminal rim sign, frontal lobe and parietal lobe atrophy, lateral, third and fourth ventricle dilatation, midbrain and pontine atrophy, hummingbird sign, atrophy of the cerebellum and cerebellar vermis, atrophy of the medulla oblongata, T2 hyper-intense signal changes of the middle cerebral peduncle and hot cross bun sign, dilated perivascular spaces, lacunar infarctions and white matter changes. Advanced imaging sequences will be analyzed by dedicated software packages, including ‘Functional MR Imaging of the Brain Software Library‘(FSL, University of Oxford, United Kingdom) and ‘Statistical Parametric Mapping’ (SPM, Trust Centre of Neuroimaging, London, United Kingdom).
CSF
The following CSF variables will be analyzed according to previously described methods [15, 31,32,33]: Aβ40, Aβ42, t-tau, p-tau (phosphorylated at Thr181), α-syn, NFL, 3-methoxy-4-hydroxyphenylethyleneglycol (MHPG), 5-hydroxyindolacetic acid (5-HIAA), homovanillic acid (HVA), blood pigments and the total cell count. The cell count and blood pigments will be analyzed within 2 h after CSF collection; α-syn will be analyzed according to previously described methods [32]; all other parameters will be analyzed within 4 weeks after CSF collection. CSF samples are collected in polypropylene tubes, centrifuged, aliquoted, and stored at − 80 °C until analysis. Because the concentrations of HVA and 5-HIAA vary in the different fractions of CSF, we aim for separate collection of the 9th–11th ml fraction for analysis of neurotransmitter metabolites, according to previously described methods [34]. Laboratory technicians blinded for clinical symptoms and outcome will perform all CSF analyses. When patients are taking dopaminergic medication at the time of lumbar puncture, the results of neurotransmitter metabolites will be excluded from the measurements.
Follow-up
During the three years after baseline examinations patients will be followed by their own neurologist. Every year patients are requested to complete a structured questionnaire. After three years, all patients will be re-examined by the study investigator (AR).
Assessment of diagnoses
Before inclusion, the referring neurologist will be asked to establish the probable diagnosis based on routine clinical examination. Specifically, a level of diagnostic (un) certainty will be indicated on a scale of 0–100. After inclusion, a movement disorder expert will also be asked for the diagnosis and level of certainty based on the clinical evaluation; a neuroradiologist makes the diagnosis based on imaging findings and a neurochemist based on CSF analysis.
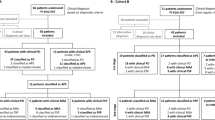
The final clinical diagnosis will be established after three years of follow-up in consensus by two independent neurologists specialized in movement disorders, and this will serve as the “silver standard” reference diagnosis. This reference diagnosis will be based on the clinical data at baseline, the disease course after 3 years follow-up and with adherence to the following international clinical criteria: Movement Disorder Society clinical diagnostic criteria for PD [35], Gilman criteria for MSA [36], the movement disorder society criteria for PSP [37], Armstrong criteria for CBS [38] and Zijlmans criteria for vascular parkinsonism [39]. These neurologists will be blinded for the results of the ancillary investigations, except for the conventional MRI because this is part of routine procedures in clinical practice. See Fig. 1 for a flowchart of the study design.

flowchart of the study design. *A translated version of the structured questionnaires are provided as Additional file 1 with this article
Outcome measures
Our primary outcome measure of interest will be the diagnosis (e.g. PD or AP) after three years of follow-up. The secondary outcome measure is the specific diagnosis (PD, MSA, PSP, etc.) after three years of follow-up.
Statistical analysis
For our first objective, the estimated initial diagnosis by the movement disorder specialist, neuroradiologist and neurochemist respectively will be compared to the reference diagnosis (after three years follow-up). Sensitivity, specificity and Kappa (k) are calculated. Next, the Net Reclassification Index (NRI) is calculated, which gives the proportion of participants correctly (re-)classified as having PD or AP. NRI values will be calculated for MRI and CSF following the classification by a movement disorder specialist [40].
For our second objective, descriptive data will be generated for all variables. Continuous variables will be presented as means ± standard deviations or medians. PD patients will be compared with AP patients using a Student’s t or Mann-Whitney U test, whichever appropriate.
Dichotomous variables will be compared using Chi-square test or Fisher exact test, whichever appropriate. A p-value of < 0.05 will be considered significant (two-sided).
For each diagnostic strategy (e.g. clinical evaluation, MRI and CSF respectively) blockwise logistic regression with stepwise forward model selection will be used to identify parameters with optimal diagnostic power. For each model ROC-curves and C-statistics are calculated.
Data management and analysis will be performed using IBM SPSS Statistics 22 (IBM Corp, Armonk, NY, USA).
Sample size calculation
This study is exploratory in nature. We estimate that a minimum of 30 AP and 30 PD patients will be needed to evaluate the diagnostic value of the three diagnostic strategies (clinical evaluation by a movement disorder specialist, MRI and CSF biomarkers). In our previous cohort study of patients with parkinsonism 60% of patients had a diagnosis of AP [34]. 30% of patients did not complete the follow-up period of 3 years, mainly because the participant had deceased or was too severely affected, the majority of which was diagnosed with AP (33 out of 46 drop-outs). Therefore, we estimate that approximately 100 participants will be needed to assess the diagnostic accuracy of the different diagnostic tests.
Loss to follow-up
In case patients opt to withdraw from the study or are no longer able to participate, the investigator will try to obtain a final interview and neurological examination if possible. When loss to follow-up is greater than 20%, analysis will be done to check for associated biased effects.
Discussion
A major unmet need in the diagnostic workup of parkinsonian disorders is a diagnostic tool to differentiate the various forms of parkinsonism when the clinically based uncertainty is high. This study is especially designed to provide directions for the neurologist in daily practice when the clinical picture of parkinsonism is puzzling.
Recently, we completed a similar prospective study of 156 consecutive patients with parkinsonism, but with an initially uncertain diagnosis [34]. All patients underwent extensive clinical testing and the following ancillary investigations: routine brain MRI; 123I-iodobenzamide single photon-emission computed tomography (IBZM-SPECT); CSF analysis; and anal sphincter electromyography (EMG).
We have shown that clinical tests such as tandem gait analysis and the axial UPDRS sub-score had a high predictive value to discriminate between PD and AP [34]. NFL and tau in CSF differentiated MSA from PD and tau and p-tau protein were elevated in CBS. Nonetheless, none of the ancillary investigations, (routine brain MRI, IBZM-SPECT, CSF analysis and anal sphincter EMG) was superior to the clinical judgment in differentiating parkinsonian syndromes [34].
However, diagnostic uncertainty in this previous cohort study was defined as being present when the neurologist who referred the patient was uncertain about the diagnosis (even when the patient fulfilled the probable criteria for one of the parkinsonian syndromes). Furthermore, patients from all disease stages were included. Therefore, the proportion of patients in an early disease stage and with an actual uncertain diagnosis was relatively small.
For that reason, the a priori chance that ancillary investigations would be of additional value was limited. Moreover, no advanced MRI techniques were used. Thus, we designed this present study to assess the diagnostic value of ancillary investigations in a population of patients with parkinsonism and an unclear clinical picture at the beginning of their disease course. We included the judgment of a movement disorder expert in the baseline examinations to compare its added diagnostic value to the various ancillary investigations. As ancillary tests, we chose brain MRI (both conventional and advanced MRI techniques) and CSF collection because of their potential to reveal new biomarkers for differentiating PD from AP based on developments over the past decade.
The gold standard for diagnosis of PD and the AP remains neuropathological confirmation of the clinical diagnosis upon post-mortem examination. However, prior research has demonstrated that neuropathological data show a high concordance between the clinical diagnosis after three years follow-up established by a movement disorder specialist and neuropathological examination post-mortem [1]. Therefore, we will use the clinical diagnosis after three years follow-up as reference diagnosis, or ‘silver standard’, with which we compare the results of ancillary investigations in individual patients. In order to minimize the potential misdiagnosis that might occur using the clinical diagnosis instead of the neuropathological diagnosis, we require the clinical diagnosis to be established in consensus by two neurologists highly experienced in movement disorders and according to the international criteria.
This study will help the general neurologist select the best available diagnostic “tool” in differentiating parkinsonian syndromes (e.g. clinical evaluation by a movement disorder specialist, brain MRI or CSF biomarkers).
Abbreviations
- AP:
-
atypical parkinsonism
- Aβ40:
-
amyloid-β40
- Aβ42:
-
amyloid-β42
- CBS:
-
corticobasal syndrome
- CSF:
-
cerebrospinal fluid
- DTI:
-
diffusion tensor imaging
- MRI:
-
magnetic resonance imaging
- MRV:
-
magnetic resonance volumetry
- MSA:
-
multiple system atrophy
- NFL:
-
neurofilament light chain
- PD:
-
Parkinson’s disease
- PSP:
-
progressive supranuclear palsy
- p-tau:
-
phosphorylated tau protein
- rs-fMRI:
-
resting state functional magnetic resonance imaging
- SWI:
-
susceptibility weighted imaging
- t-tau:
-
total tau protein
- UPDRS:
-
Unified Parkinson’s Disease rating scale
- α-syn:
-
α-synuclein
References
Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125:861–70.
Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–4.
Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9.
Litvan I, Goetz CG, Jankovic J, Wenning GK, Booth V, Bartko JJ, et al. What is the accuracy of the clinical diagnosis of multiple system atrophy? A clinicopathologic study. Arch Neurol. 1997;54:937–44.
Respondek G, Roeber S, Kretzschmar H, Troakes C, Al-Sarraj S, Gelpi E, et al. Accuracy of the National Institute for neurological disorders and stroke/Society for Progressive Supranuclear Palsy and neuroprotection and natural history in Parkinson plus syndromes criteria for the diagnosis of progressive supranuclear palsy. Mov Disord. 2013;28:504–9.
Meijer FJ, Aerts MB, Abdo WF, Prokop M, Borm GF, Esselink RA, et al. Contribution of routine brain MRI to the differential diagnosis of parkinsonism: a 3-year prospective follow-up study. J Neurol. 2012;259:929–35.
Cochrane CJ, Ebmeier KP. Diffusion tensor imaging in parkinsonian syndromes: a systematic review and meta-analysis. Neurology. 2013;80:857–64.
Broski SM, Hunt CH, Johnson GB, Morreale RF, Lowe VJ, Peller PJ. Structural and functional imaging in parkinsonian syndromes. Radiographics. 2014;34:1273–92.
Holtbernd F, Eidelberg D. The utility of neuroimaging in the differential diagnosis of parkinsonian syndromes. Semin Neurol. 2014;34:202–9.
Meijer FJ, Bloem BR, Mahlknecht P, Seppi K, Goraj B. Update on diffusion MRI in Parkinson's disease and atypical parkinsonism. J Neurol Sci. 2013;332:21–9.
Worker A, Blain C, Jarosz J, Chaudhuri KR, Barker GJ, Williams SC, et al. Diffusion tensor imaging of Parkinson's disease, multiple system atrophy and progressive supranuclear palsy: a tract-based spatial statistics study. PLoS One. 2014;9:e112638.
Berg D, Hochstrasser H. Iron metabolism in parkinsonian syndromes. Mov Disord. 2006;21:1299–310.
Tahmasian M, Bettray LM, van Eimeren T, Drzezga A, Timmermann L, Eickhoff CR, et al. A systematic review on the applications of resting-state fMRI in Parkinson's disease: does dopamine replacement therapy play a role? Cortex. 2015;73:80–105.
Abdo WF, Bloem BR, Van Geel WJ, Esselink RA, Verbeek MM. CSF neurofilament light chain and tau differentiate multiple system atrophy from Parkinson's disease. Neurobiol Aging. 2007;28:742–7.
Abdo WF, De Jong D, Hendriks JC, Horstink MW, Kremer BP, Bloem BR, et al. Cerebrospinal fluid analysis differentiates multiple system atrophy from Parkinson's disease. Mov Disord. 2004;19:571–9.
Aerts MB, Esselink RA, Abdo WF, Bloem BR, Verbeek MM. CSF alpha-synuclein does not differentiate between parkinsonian disorders. Neurobiol Aging. 2012;33:430 e1–3.
Verbeek MM, Abdo WF, De Jong D, Horstink MW, Kremer BP, Bloem BR. Cerebrospinal fluid Abeta42 levels in multiple system atrophy. Mov Disord. 2004;19:238–40. author reply 40-1
Constantinescu R, Mondello S. Cerebrospinal fluid biomarker candidates for parkinsonian disorders. Front Neurol. 2013;3:187.
Constantinescu R, Zetterberg H, Holmberg B, Rosengren L. Levels of brain related proteins in cerebrospinal fluid: an aid in the differential diagnosis of parkinsonian disorders. Parkinsonism Relat Disord. 2009;15:205–12.
Holmberg B, Rosengren L, Karlsson JE, Johnels B. Increased cerebrospinal fluid levels of neurofilament protein in progressive supranuclear palsy and multiple-system atrophy compared with Parkinson's disease. Mov Disord. 1998;13:70–7.
Aerts MB, Esselink RA, Bloem BR, Verbeek MM. Cerebrospinal fluid tau and phosphorylated tau protein are elevated in corticobasal syndrome. Mov Disord. 2011;26:169–73.
Chaudhuri KR, Martinez-Martin P, Brown RG, Sethi K, Stocchi F, Odin P, et al. The metric properties of a novel non-motor symptoms scale for Parkinson's disease: results from an international pilot study. Mov Disord. 2007;22:1901–11.
Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67:361–70.
Weintraub D, Mamikonyan E, Papay K, Shea JA, Xie SX, Siderowf A. Questionnaire for impulsive-compulsive disorders in Parkinson's disease-rating scale. Mov Disord. 2012;27:242–7.
Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14:540–5.
Folstein MF, Folstein SE, McHugh PR. "mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98.
Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a frontal assessment battery at bedside. Neurology. 2000;55:1621–6.
Robbins TW, James M, Owen AM, Sahakian BJ, McInnes L, Rabbitt P. Cambridge neuropsychological test automated battery (CANTAB): a factor analytic study of a large sample of normal elderly volunteers. Dementia. 1994;5:266–81.
Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology. 1967;17:427–42.
Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66:1717–20.
Brautigam C, Wevers RA, Jansen RJ, Smeitink JA, de Rijk-van Andel JF, Gabreels FJ, et al. Biochemical hallmarks of tyrosine hydroxylase deficiency. Clin Chem. 1998;44:1897–904.
van Geel WJ, Abdo WF, Melis R, Williams S, Bloem BR, Verbeek MM. A more efficient enzyme-linked immunosorbent assay for measurement of alpha-synuclein in cerebrospinal fluid. J Neurosci Methods. 2008;168:182–5.
Van Geel WJ, Rosengren LE, Verbeek MM. An enzyme immunoassay to quantify neurofilament light chain in cerebrospinal fluid. J Immunol Methods. 2005;296:179–85.
Aerts MB, Esselink RA, Abdo WF, Meijer FJ, Drost G, Norgren N, et al. Ancillary investigations to diagnose parkinsonism: a prospective clinical study. J Neurol. 2015;262:346–56.
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. 2015;30:1591–601.
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6.
Hoglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. 2017;32:853–64.
Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80:496–503.
Zijlmans JC, Daniel SE, Hughes AJ, Revesz T, Lees AJ. Clinicopathological investigation of vascular parkinsonism, including clinical criteria for diagnosis. Mov Disord. 2004;19:630–40.
Pencina MJ, D'Agostino RB Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med. 2011;30:11–21.
Funding
AR, MA and this study are supported by a research grant of the Stichting Parkinson Fonds and the Van Alkemade-Keuls Fonds. These non-corporate funding organizations have no role in study design or conduct of the study, data collection, data analysis and interpretation or manuscript preparation. BB is supported by a ZonMW VIDI grants (project number 91776352).
Author information
Authors and Affiliations
Contributions
AR participated in the design and coordination of the study and drafted the manuscript. MA participated in the design and coordination of the study and has been involved in revising the manuscript for important intellectual content. FM participated in the design of the study and has been involved in revising the manuscript for important intellectual content. RE conceived of the study and participated in its design and coordination and has been involved in revising the manuscript for important intellectual content. MV conceived of the study and participated in its design and coordination and has been involved in revising the manuscript for important intellectual content. BB conceived of the study and participated in its design and coordination and has been involved in revising the manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Medical Ethics Committee Arnhem-Nijmegen approved the study. Written informed consent will be obtained from all patients after detailed explanation of the procedures.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Structured questionnaire English translation. English translation of the annual questionnaire. Description of data: English translation of the annual questionnaire of the Parkinson’s Disease Diagnostic Observations Study (PADDO). (PDF 758 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
van Rumund, A., Aerts, M.B., Esselink, R.A.J. et al. Parkinson’s Disease Diagnostic Observations (PADDO): study rationale and design of a prospective cohort study for early differentiation of parkinsonism. BMC Neurol 18, 69 (2018). https://doi.org/10.1186/s12883-018-1072-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-018-1072-x