
Abstract
Background
Heterozygous mutations in COL2A1 create a spectrum of clinical entities called type II collagenopathies that range from in utero lethal to relatively mild conditions which become apparent only during adulthood. We aimed to characterize the clinical, radiological, and molecular features of a family with an atypical type II collagenopathy.
Case presentation
A family with three affected males in three generations was described. Prominent clinical findings included short stature with platyspondyly, flat midface and Pierre Robin sequence, severe dysplasia of the proximal femora, and severe retinopathy that could lead to blindness. By whole exome sequencing, a novel heterozygous deletion, c.4161_4165del, in COL2A1 was identified. The phenotype is atypical for those described for mutations in the C-propeptide region of COL2A1.
Conclusions
We have described an atypical type II collagenopathy caused by a novel out-of-frame deletion in the C-propeptide region of COL2A1. Of all the reported truncating mutations in the C-propeptide region that result in short-stature type II collagenopathies, this mutation is the farthest from the C-terminal of COL2A1.
Similar content being viewed by others

Background
Patients with COL2A1 mutations are collectively called type II collagenopathies. Missense mutations or in-frame derangement in the triple-helical region cause a phenotype on the spondyloepiphyseal dysplasia (SED) spectrum from lethal SED including achondrogenesis type II (ACG2; OMIM# 200610) and hypochodrogenesis through spondyloepiphyseal dysplasia congenita (SEDC; OMIM#183900), while mutations in the triple helical or N-propeptide regions cause Stickler syndrome type I (STL1; OMIM# 108300) or Kniest dysplasia (OMIM# 156550) [1]. Unlike mutations in the triple-helical or N-propeptide regions, those in the C-propeptide region generally produce atypical phenotypes such as platyspondylic lethal skeletal dysplasia, Torrance type (PLSDT# OMIM 151210) [2], spondyloperipheral dysplasia (SPPD) (OMIM# 271700) [3], vitreoretinopathy with phalangeal epiphyseal dysplasia (VPED) [4], avascular necrosis of the femoral head (ANFH; OMIM# 608805) [5], or early-onset osteoarthritis (OA) [6]. Here, we describe a family with atypical features of type II collagenopathies caused by a novel mutation in COL2A1. As far as we know, this truncating mutation in the C-propeptide region is the farthest one from the 3’ end of the gene that causes a disease with short stature, suggesting the existence of the mutant protein.
Case presentation
Subjects
We studied a Thai family with skeletal dysplasia who attended the Genetics Clinic at the King Chulalongkorn Memorial Hospital, Bangkok, Thailand. The medical data, pedigree, physical examinations, and laboratory results were recorded. The written informed consent and parental consent (for the proband) was obtained after explanation of the possible consequences of this study.
Genomic DNA preparation and whole-exome sequencing
To perform genetic analysis, genomic DNA was isolated from peripheral blood leukocytes using a Puregene Blood kit (Qiagen, Hilden, Germany). The genomic DNA was sent to Macrogen, Inc. (Seoul, South Korea) for whole-exome sequencing (WES). DNA was captured using a SureSelect Human All Exon version 4 kit (Agilent Technologies, Santa Clara, CA) and sequenced on a Hiseq2000 instrument. Base calling was performed and quality scores were analyzed using Real Time Analysis software version 1.7. Sequence reads were aligned against the University of California Santa Cruz human genome assembly hg19 using Burrows-Wheeler Alignment software (bio-bwa.sourceforge.net/). Single-nucleotide variants (SNVs) and insertions/deletions (Indels) were detected by SAMTOOLS (samtools.sourceforge.net/) and annotated against dbSNP & the 1000 Genomes Project. After quality filtering, we looked for variants located in the coding regions of known skeletal dysplasia genes for all potential pathogenic SNVs and Indels. Variant calling exclusion criteria were (a) coverage <10×; (b) quality score <20; (c) minor allele frequency ≥1% in the 1000 Genomes Project; and (d) non-coding variants and synonymous exonic variants. The remaining variants were subsequently filtered out if they were present in our in-house database of 165 unrelated Thai exomes. The variants were confirmed by PCR and Sanger sequencing.
Existing SNVs or known pathogenic mutations were filtered out using the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) and the Exome Aggregation Consortium database (exac.broadinstitute.org).
Results
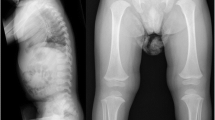
A 20 month-old male (IV:3; Fig. 1) is the first child of a non-consanguineous couple. His mother had miscarriages in the first trimester of the two previous pregnancies. The causes of both miscarriages were unknown. He was born at term by normal delivery with a birth weight of 2,850 g (10th centile) and a length of 45 cm (<3rdcentile, −4 SD). Physical examination revealed short stature, flattened face, cleft palate, micrognathia, short neck, and umbilical hernia (Fig. 2a). A radiograph obtained at age 20 months showed oval-shaped vertebral bodies (Fig. 2b). Ossification of the femoral head was absent. Long bones showed short broad tubular shape with metaphyseal flaring (Fig. 2c). Other ossification centers appeared age-appropriate (Fig. 2c and d). Hands and feet radiographs were normal (Fig. 2e and f). His eyes examination and audiometry showed no abnormality. He was given a clinical diagnosis of SEDC.

Pedigree of the index family. The arrow indicates the proband

a Photograph of the proband (IV: 3) showing flattened facial profile, micrognathia, short neck, and short trunk. b Whole spine lateral radiograph showing oval-shaped vertebral bodies. c Pelvis, femur, and tibial AP radiograph showing retarded ossification of the femoral heads while other ossification centers are age-appropriate. d Chest radiograph showing age-appropriate ossification centers at humeral heads and greater tuberosities of humeri. e, f Hand and foot AP radiographs are normal
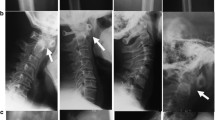
The proband’s father (III:1; Fig. 1) is a 26 year-old man. He had short trunk dwarfism with a height of 125 cm (−9 SD). He had a barrel-shaped chest, hyperlordosis of the lumbar spine, and flexion contracture of both hips (Fig. 3a). Hands and feet, including radiographs, were apparently normal (Fig. 3b and c). A radiograph of the thoracolumbar spine showed flattened vertebral bodies with kyphotic deformity (Fig. 3d). Severe dysplasia of the bilateral proximal femoral epiphyses and hip dislocation were observed (Fig. 3e). Generalized osteopenia was noted. He was blind in the left eye since he was 8 years old, while his right eye had severe myopia and retinal detachment. He also had umbilical hernia when he was young but it spontaneously resolved.

a Photograph of proband’s father (III: 1) showing disproportionate short stature and barrel-shaped chest. b, c Hand and feet radiographs are normal without brachydactyly. d Lateral radiograph of the thoracolumbar spine shows platyspondyly with kyphotic deformity. e Pelvis AP radiograph showing severe hip dysplasia and dislocation. f Thoracolumbar spine and pelvis AP radiographs of the proband’s grandfather also show platyspondyly and severe hip dysplasia and dislocation
The proband’s grandfather (II:3; Fig. 1) had short stature (−8 SD). He also had a barrel-shaped chest, hyperlordosis of the lumbar spine and flexion contracture of both hips. A radiograph of the thoracolumbar spine and pelvis showed flattened vertebral bodies, severe dysplasia of the bilateral proximal femoral epiphysis and hip dislocation (Fig. 3f). His hands and feet were normal. His eyes were reported normal but had never been formally evaluated. He died at the age of 54 because of septicemia after a nephrostomy as treatment for obstructive uropathy from ureteric stones. A blood sample was not available for mutation analysis.
Whole-exome sequencing (WES) of the proband revealed a five-nucleotide out-of-frame deletion (NM_001844.4: c.4161_4165del:p.Gln1387Hisfs*30) in COL2A1. PCR and Sanger sequencing using leukocyte-derived DNA from the proband (IV:3) and his father (III:1) confirmed that both were heterozygous for the mutation (Fig. 4a). This mutation has not been reported previously in the Human Genome Mutation Database or the Exome Aggregation Consortium database. In addition, it was not present in our in-house exome database of 165 Thai individuals.

Mutation analysis. a Direct sequencing shows that the proband is heterozygous for a five nucleotide out-of-frame deletion (NM_001844.4: c.4161_4165del:p.Gln1387His*fs30). b Map of the mutations in the C-propeptide region of COL2A1. Truncating mutations shown above the gene lead to collagenopathies with short stature, while those shown below the gene lead to disorders with normal stature. The arrowheads points to the last amino acid residue of the predicted truncated COL2A1. Green: STLI; purple: early-onset OA; red: our mutation; yellow: SPPD; blue: PLSDT. Numbers correspond to Table 2
Conclusions
Major clinical and radiographic features in our patients include short stature with platyspondyly, flat midface, Pierre Robin sequence, hip dysplasia, and retinal detachment. The major extracellular structural protein of these affected organs is collagen type II. This led us to hypothesize that the etiologic mutation was in COL2A1. WES identified a five-base pair deletion in this gene. PCR and Sanger sequencing confirmed that the proband (IV:3) and his father (III:1) harbored a heterozygous mutation, c.4161_4165del, in COL2A1. This out-of-frame deletion is in the C-propeptide region and is expected to lead to a protein truncation, p.Gln1387Hisfs*30.
Although our patients have a mutation in C-propeptide region of COL2A1, the phenotype is atypical for those described for the mutations in C-propeptide region. Previous reported mutations in the C-propeptide region of collagen type II lead to one of the six entities: PLSDT, SPPD, VPED, ANFH, STL1, or early-onset OA. However, the clinical features of this family are different from all these six diseases (Table 1).
PLSDT was excluded by absence of wafer-thin platyspondyly, small round scapulae, and brachydactyly in our patients. Moreover, PLSDT patients generally die at birth [7] but our patients live into adulthood. An important diagnostic feature of SPPD, brachydactyly [3], excluded the diagnosis of SPPD. Short stature with platyspondyly without phalangeal epiphyseal dysplasia excluded VPED from the diagnosis [4]. ANFH patients have flattened femoral heads with signs of premature osteoarthritis. However, the femoral heads of proband’s father (III:1) and grandfather (II: 3) were totally absent causing bilateral hip dysplasia and dislocation. Another important difference is the fact that ANFH patients do not have short stature, platyspondyly or retinal abnormalities [5], while our patients do. Patients with STLI usually have normal stature and premature OA of many joints [8, 9], while all our patients (III:1 and II:3) had short stature (−8 to −9 SD at adulthood). Early-onset OA patients should present with premature OA of joints without other skeletal abnormalities, while our patients had short stature and severe dysplasia of the hips.
STL1 is caused by haploinsufficiency of COL2A1. All point mutations in COL2A1 leading to STL1 are therefore expected to be subjected to nonsense-mediated mRNA decay (NMD). Heterozygous mutations leading to type II collagenopathies besides STL1 are predicted to have a dominant negative effect. Previous reports showed that truncating mutations in the C-propeptide region led either to STL1 or SPPD, when the mutant RNA did, or did not, undergo NMD, respectively (Table 2).
Interestingly, our frameshift mutation, expected to create a stop codon 69 nucleotides upstream from the exon 53–54 junction, does not lead to STL1. We therefore hypothesize that the mutant RNA transcribed from the mutant COL2A1 allele found in our patients may not undergo NMD; instead that it may produce a mutant COL2A1 protein that has a dominant negative effect, interfering with the wild-type COL2A1 and leading to the unique phenotype. In general, mRNAs harboring a premature terminal codon (PCT) 50–55 nucleotides upstream of the last exon-exon junction are efficiency degraded [10]. The NMD is signaled by the presence of the exon junction complex (EJC). However, EJCs are not equally assembled at every exon junction. Therefore, it is possible that the variation of the EJC may affect NMD efficiency and lead to efficient degradation of COL2A1 mRNAs harboring a PTC at least 70 nucleotides upstream of the final exon-exon junction. On the contrary, mutations causing premature stop codons from 69 nucleotides upstream of the exon 53–54 junction down to the 3’ end COL2A1 would probably not undergo NMD, have a dominant negative effect, and therefore cause more severe phenotypes of the spectrum such as SPPD and PLSDT [2, 3]. Unfortunately, tissues from our patients that would express COL2A1 are not available to test this hypothesis. Of all the reported mutations in the C-propeptide region that result in short-stature type II collagenopathies, our mutation is the farthest truncating mutation from the C-terminal of COL2A1 (Fig. 4b, Table 2).
Our probands and his father had some different phenotypes such as facial profile, eye involvement, and cleft palate. Intra-and extrafamilial variability of type II collagenopathies has previously been observed in patients with the same mutation. For instance, in a family with VPED, a 22-year-old patient had severe premature osteoarthritis requiring hip replacement while two older siblings had only mild osteoarthritis [4]. In another family, while the mosaic mother had SPPD, her fetus suffered from the lethal PLSDT [11]. These data suggest that genetic, epigenetic, and environmental modifiers affect the clinical presentation in type II collagenopathies. Moreover, these modifiers may be other possible explanations for the atypical phenotype of the patients.
In conclusion, we report an atypical phenotype resulting from a novel truncating mutation in the C-propeptide region of COL2A1 with prominent features including short stature, platyspondyly, hip dysplasia, and retinal detachment.
Abbreviations
- ACG2:
-
Achondrogenesis type II
- ANFH:
-
Avascular necrosis of the femoral head
- NMD:
-
Nonsense-mediated mRNA decay.
- OA:
-
Osteoarthritis
- PLSDT:
-
Platyspondylic lethal skeletal dysplasia
- SED:
-
Spondyloepiphyseal dysplasia
- SEDC:
-
Spondyloepiphyseal epiphyseal dysplasia congenital
- SPPD:
-
Spondyloperipheral dysplasia
- STL1:
-
Stickler syndrome type I
- VPED:
-
Vitreoretinopathy with phalangeal epiphyseal dysplasia
- WES:
-
Whole-exome sequencing
References
Nishimura G, Haga N, Kitoh H, Tanaka Y, Sonoda T, Kitamura M, Shirahama S, Itoh T, Nakashima E, Ohashi H, et al. The phenotypic spectrum of COL2A1 mutations. Hum Mutat. 2005;26(1):36–43.
Zankl A, Neumann L, Ignatius J, Nikkels P, Schrander-Stumpel C, Mortier G, Omran H, Wright M, Hilbert K, Bonafe L, et al. Dominant negative mutations in the C-propeptide of COL2A1 cause platyspondylic lethal skeletal dysplasia, torrance type, and define a novel subfamily within the type 2 collagenopathies. Am J Med Genet A. 2005;133A(1):61–7.
Zankl A, Zabel B, Hilbert K, Wildhardt G, Cuenot S, Xavier B, Ha-Vinh R, Bonafe L, Spranger J, Superti-Furga A. Spondyloperipheral dysplasia is caused by truncating mutations in the C-propeptide of COL2A1. Am J Med Genet A. 2004;129A(2):144–8.
Richards AJ, Morgan J, Bearcroft PW, Pickering E, Owen MJ, Holmans P, Williams N, Tysoe C, Pope FM, Snead MP, et al. Vitreoretinopathy with phalangeal epiphyseal dysplasia, a type II collagenopathy resulting from a novel mutation in the C-propeptide region of the molecule. J Med Genet. 2002;39(9):661–5.
Kannu P, O’Rielly DD, Hyland JC, Kokko LA. Avascular necrosis of the femoral head due to a novel C propeptide mutation in COL2A1. Am J Med Genet A. 2011;155A(7):1759–62.
Barat-Houari M, Sarrabay G, Gatinois V, Fabre A, Dumont B, Genevieve D, Touitou I. Mutation update for COL2A1 gene variants associated with type II collagenopathies. Hum Mutat. 2016;37(1):7–15.
Horton WA, Rimoin DL, Hollister DW, Lachman RS. Further heterogeneity within lethal neonatal short-limbed dwarfism: the platyspondylic types. J Pediatr. 1979;94(5):736–42.
Letts M, Kabir A, Davidson D. The spinal manifestations of Stickler’s syndrome. Spine. 1999;24(12):1260–4.
Rose PS, Levy HP, Liberfarb RM, Davis J, Szymko-Bennett Y, Rubin BI, Tsilou E, Griffith AJ, Francomano CA. Stickler syndrome: clinical characteristics and diagnostic criteria. Am J Med Genet A. 2005;138A(3):199–207.
Hug N, Longman D, Caceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016;44(4):1483–95.
Desir J, Cassart M, Donner C, Coucke P, Abramowicz M, Mortier G. Spondyloperipheral dysplasia as the mosaic form of platyspondylic lethal skeletal dyplasia torrance type in mother and fetus with the same COL2A1 mutation. Am J Med Genet A. 2012;158A(8):1948–52.
Hoornaert KP, Marik I, Kozlowski K, Cole T, Le Merrer M, Leroy JG, Coucke PJ, Sillence D, Mortier GR. Czech dysplasia metatarsal type: another type II collagen disorder. Eur J Hum Genet 2007, 15(12):1269–1275.
Ahmad NN, Dimascio J, Knowlton RG, Tasman WS. Stickler syndrome. A mutation in the nonhelical 3' end of type II procollagen gene. Arch Ophthalmol 1995, 113(11):1454–1457.
Annunen S, Korkko J, Czarny M, Warman ML, Brunner HG, Kaariainen H, Mulliken JB, Tranebjaerg L, Brooks DG, Cox GF et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet 1999, 65(4):974–983.
Barat-Houari M, Dumont B, Fabre A, Them FT, Alembik Y, Alessandri JL, Amiel J, Audebert S, Baumann-Morel C, Blanchet P et al. The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur J Hum Genet 2016, 24(7):992–1000.
Zabel B, Hilbert K, Stoss H, Superti-Furga A, Spranger J, Winterpacht A. A specific collagen type II gene (COL2A1) mutation presenting as spondyloperipheral dysplasia. Am J Med Genet 1996, 63(1):123–128.
Bedeschi MF, Bianchi V, Gentilin B, Colombo L, Natacci F, Giglio S, Andreucci E, Trespidi L, Acaia B, Furga AS et al. Prenatal manifestation and management of a mother and child affected by spondyloperipheral dysplasia with a C-propeptide mutation in COL2A1: case report. Orphanet J Rare Dis 2011, 6:7.
Zhang Z, Zhao SC, He JW, Fu WZ, Zhang CQ, Zhang ZL. Identification of one novel mutation in the C-propeptide of COL2A1 in a Chinese family with spondyloperipheral dysplasia. Gene 2013, 522(1):107–110.
Meredith SP, Richards AJ, Bearcroft P, Pouson AV, Snead MP. Significant ocular findings are a feature of heritable bone dysplasias resulting from defects in type II collagen. Brit J Ophth 2007, 91(9):1148–1151.
Nishimura G, Nakashima E, Mabuchi A, Shimamoto K, Shimamoto T, Shimao Y, Nagai T, Yamaguchi T, Kosaki R, Ohashi H et al. Identification of COL2A1 mutations in platyspondylic skeletal dysplasia, Torrance type. J Med Genet 2004, 41(1):75–79.
Acknowledgements
We thank the family members for participating in this study.
Funding
This study was supported by the Chulalongkorn Academic Advancement into Its 2nd Century Project and Thailand Research Fund.
Availability of data and materials
All data supporting our finding are included in the manuscript.
Authors’ contributions
AS participated in the molecular genetic studies and wrote the manuscript, CS participated in molecular genetic studies, MP participated in molecular genetic studies, KS assured the general supervision of the research group, VS assured the general supervision of the research group and raised funding. All authors revised and approved the final version of the manuscript.
Competing interest
The authors declare that they have no competing interest.
Consent for publication
Consent for publication of the cases was obtained for each participant.
Ethics approval and consent to participate
Ethical approval was obtained from the institutional review board, Faculty of Medicine, Chulalongkorn University. After explanation of the possible consequences of this study, the written informed consent and parental consent (for the proband) was obtained.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sangsin, A., Srichomthong, C., Pongpanich, M. et al. Short stature, platyspondyly, hip dysplasia, and retinal detachment: an atypical type II collagenopathy caused by a novel mutation in the C-propeptide region of COL2A1: a case report. BMC Med Genet 17, 96 (2016). https://doi.org/10.1186/s12881-016-0357-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-016-0357-4