
Abstract
The present study utilized network pharmacology to identify therapeutic targets and mechanisms of Rehmannia glutinosa in sepsis treatment. RNA-sequencing was conducted on peripheral blood samples collected from 23 sepsis patients and 10 healthy individuals. Subsequently, the RNA sequence data were analyzed for differential expression. Identification of active components and their putative targets was achieved through the HERB and SwissTarget Prediction databases, respectively. Functional enrichment analysis was performed using GO and KEGG pathways. Additionally, protein-protein interaction networks were constructed and survival analysis of key targets was conducted. Single-cell RNA sequencing provided cellular localization data, while molecular docking explored interactions with central targets. Results indicated significant involvement of identified targets in inflammation and Th17 cell differentiation. Survival analysis linked several targets with mortality rates, while molecular docking highlighted potential interactions between active components and specific targets, such as rehmaionoside a with ADAM17 and rehmapicrogenin with CD81. Molecular dynamics simulations confirmed the stability of these interactions, suggesting Rehmannia glutinosa’s role in modulating immune functions in sepsis.
Similar content being viewed by others


Introduction
Sepsis is a complex organismal response to infectious agents. It is characterized by a systemic inflammatory response syndrome (SIRS) triggered by infection and is the leading cause of death from infections globally [1]. Current Western medicine approaches to sepsis treatment face challenges in providing specific and effective therapies. Therefore, timely recognition and intervention are crucial for patient survival. Extensive research has revealed the multifaceted effects of traditional Chinese medicine (TCM, shown in Table 1) in treating sepsis, including anti-inflammatory, antimicrobial, and immunomodulatory properties [2]. This has led to significant interest in TCM as a potential anti-infective therapy. A classic TCM text, the “Item Differentiation of Warm Febrile Diseases”, describes the efficacy of Qing-Ying-Tang in clearing heat, promoting detoxification, and nourishing yin. Primarily used for cases involving heat affecting the body’s yin aspect, Rehmannia glutinosa is a key component of Qing-Ying-Tang. Within the TCM framework, Rehmannia glutinosa is believed to play a vital role in reducing mortality and inflammation and improving coagulation factors in sepsis patients [3]. Several key constituents of Rehmannia glutinosa have been identified, including rehmaionoside a, rehmapicrogenin, Rehmaglutin C, and diincarvilone A. These components exhibit a wide range of pharmacological activities, including antitumor, immunomodulatory, neuroprotective, hypoglycemic, cardioprotective, and antioxidant effects [4].
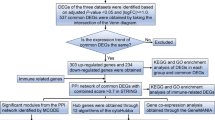
While the potential benefits of Rehmannia glutinosa for sepsis prognosis have been documented, the precise mechanisms underlying its efficacy remain unclear. Network pharmacology offers a powerful approach to shed light on the complex interplay between the various components of herbal medicines and their biological targets. This method integrates concepts from systems biology and computational techniques to map intricate networks of interactions within biological systems. By incorporating diverse omics data, network pharmacology provides a comprehensive understanding of the molecular mechanisms governing drug action and disease processes [5]. Single-cell sequencing technologies have revolutionized our understanding of cellular heterogeneity and the dynamic nature of the immune landscape in various diseases, including sepsis. These technologies enable detailed profiling of individual cells, revealing cellular diversity and specific cell states critical for disease progression [6]. Molecular docking and molecular dynamics (MD) simulations are established tools in the biomedical field, providing insights that are often difficult to obtain through traditional biochemical or pathology experiments [7, 8]. This study employs these advanced techniques to elucidate the potential targets and mechanisms of Rehmannia glutinosa in sepsis treatment. The findings will provide valuable insights to guide future research efforts, as illustrated in Fig. 1.

Experiment flowchart of this study. Network pharmacology and RNA-seq technology were applied to reveal potential effective targets and mechanisms of Rehmannia glutinosa in sepsis therapy
Methods
Data sources
Raw data from sepsis patients were obtained from the Chinese National GeneBank (CNGN) at https://db.cngb.org/search/project/CNP0002611/ (Dataset number: CNP0002611). The dataset comprises information on 23 sepsis patients admitted to the Emergency Intensive Care Unit of the Affiliated Hospital of Southwest Medical University between February 2019 and December 2020, with venous blood samples collected. Additionally, venous blood samples were taken from 10 healthy volunteers to serve as a control group. For analysis based on the sepsis 3.0 criteria (infection + ΔSOFA score ≥ 2), as defined by the Society of Critical Care Medicine in collaboration with the European Society of Intensive Care Medicine in 2016, the GSE65682 dataset and prognostic data were downloaded from the publicly available GEO database for survival analysis.
RNA-seq
Total RNA was extracted from blood samples with the Trizol reagent. RNA quality and quantity were assessed using the Agilent 2100 bioanalyzer (Thermo Fisher Scientific, MA, USA). Ribosomal RNA (rRNA) was removed using an enzyme H reagent targeting rRNA-specific sequences. The purified RNA was then fragmented into small pieces using SPRI beads and bivalent cations under high temperatures. First-strand cDNA was synthesized from the RNA fragments using reverse transcriptase and random primers. Second-strand cDNA synthesis employed DNA polymerase I and RNase H. The Agilent 2100 bioanalyzer was again used to measure the size distribution of the cDNA fragments. Library pool quantification was performed using qPCR analysis. Following the manufacturer’s protocol, eligible library pools were sequenced on the BGISEQ-500/MGISEQ-2000 system (BGI-Shenzhen, China). Low-quality reads, adapter sequences, and reads with unknown base N content exceeding 5% were removed from the obtained data. Low-quality reads were defined as those with a quality value less than 10 and where more than 20% of the bases fell below this threshold. Clean reads were filtered using SOAPnuke software for reliability and saved in FASTQ format. These clean reads were aligned to the reference genome using HISAT and Bowtie2 software.
Screening of differentially expressed RNA
Data quality was rigorously assessed using the online tool iDEP96 (http://bioinformatics.sdstate.edu/idep96/) [9]. Box plots were employed to assess dataset consistency and reliability. To ensure data integrity, principal component analysis (PCA) was employed to identify and remove outlier samples. Prior to analysis, data were standardized using the prcomp function in R4.2.1 with centering and scaling applied (center = TRUE and scale.=TRUE). Outliers were identified based on the scatter plot of the first two principal components and subsequently removed. Differential expression analysis was conducted using the DEseq2 method, with a minimum fold change threshold of 2 and a false discovery rate (FDR) of less than 0.05.
Exploration of active components and targets of Rehmannia Glutinosa
The HERB Database [10] (http://herb.ac.cn/), a centralized repository for TCM information, offers a comprehensive collection of herbal medicines and their constituents. This invaluable resource facilitates pharmacological studies within modern TCM drug discovery efforts. We utilized the HERB database to acquire information on the active components of Rehmannia glutinosa (ID: HERB001783). For target prediction, the species was designated as human, and only targets with a probability greater than 0.01 (P > 0.01) were included. We subsequently utilized the SwissTargetPrediction database [11] (http://www.swisstargetprediction.ch/) for target prediction related to the identified active components of Rehmannia glutinosa. Following the removal of duplicate targets associated with both Rehmannia glutinosa and sepsis, the overlapping genes were identified. A Venn diagram was generated to visualize these results using http://www.liuxiaoyuyuan.cn/.
Construction of the “active component-target-disease” network
To establish a network of active component targets for sepsis therapy, network construction was performed using Cytoscape 3.7.2. The identified overlapping genes were imported into Cytoscape to build the network. The built-in network visualization tools were then employed to map the interactions between the active components and their predicted targets. Next, the constructed network was analyzed using the Network Analyzer plugin. This plugin calculates topological parameters such as node degree, betweenness centrality, and clustering coefficient, which characterize the interconnectedness within the network. The size of each node was positively correlated with its degree of connectivity, indicating its importance within the network. Nodes with a higher degree value exhibited increased connectivity, suggesting their potential importance within the network.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis
For gene function annotation and enrichment analysis, we employed Metascape [12] (http://metascape.org/), a robust tool integrating databases like GO, KEGG, and UniProt. GO annotations encompass three main categories: biological processes, cellular components, and molecular functions, providing a comprehensive understanding of gene functions. KEGG pathway information is also accessible through Metascape, allowing analysis across different species [13,14,15]. The intersecting genes were uploaded to Metascape and enrichment analysis was performed using default settings, including a significance threshold of P ≤ 0.01 and a minimum count of 3 genes per pathway. Results with a P-value ≤ 0.01 were considered significant. Chord diagrams and other visualization tools, including SRplot (http://www.bioinformatics.com.cn/srplot), were employed to depict the top 20 most significant pathways based on P-value and enrichment score.
Protein-protein interaction (PPI) analysis
To identify potential key targets, we performed protein-protein interaction network analysis using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (www.string-db.org) [16]. Interacting proteins within the network indicate potential functional relationships. Proteins with a higher number of interactions become more indicative of core genes. Species selection was set to “Homo sapiens” and a confidence score threshold of 0.7 was applied. Nodes with no connections were excluded from the network. The 135 intersecting genes were uploaded to the STRING database to construct the PPI network. A heatmap was then generated to visualize the core genes within the network using the Xiantao platform (https://www.xiantaozi.com/). The visualization was performed in R version 4.2.1 using the ComplexHeatmap package [17].
Survival curve analysis
The relationship between key targets and clinical prognosis was assessed using survival analysis with gene expression data from the GSE65682 dataset [18] and 28-day clinical prognosis data. This dataset, obtained from a public database, encompasses peripheral blood samples from 478 patients with confirmed sepsis, along with gene expression profiles and clinical prognosis information. We used the gene expression data for each key target to categorize patients into high and low-expression groups. The categorization was based on the median expression value of each gene; patients with expression values below the median were placed in the low-expression group, while those with values above the median were placed in the high-expression group. The survival analysis was conducted using the Kaplan-Meier method, and the log-rank test was employed to compare survival curves between the high and low-expression groups. Statistical significance was established using a P-value < 0.05 in the log-rank test.
Single-cell sequencing analysis
Five peripheral blood mononuclear cell (PBMC) samples were collected for 10× scRNA-seq analysis. These samples comprised two from healthy volunteers, one from a patient with systemic inflammatory response syndrome (SIRS), and two from septic patients.
Cell Ranger (utilizing STAR14 software) was employed for data quality assessment and read alignment to the reference genome during single-cell RNA sequencing. This technique incorporates transcript sequences with Unique Molecular Identifiers (UMIs) and Cell Barcodes, allowing absolute quantification of each transcript. Dimensionality reduction, a process that simplifies high-dimensional data for better visualization and analysis, was performed using Mutual Nearest Neighbors (MNN) and t-distributed Stochastic Nearest Neighbor Embedding (t-SNE) algorithms. The results of MNN-based dimensionality reduction were visualized using t-SNE to identify distinct cell populations. Marker genes, in this context, are a set of genes with high expression within a specific cell type and minimal expression in others. They also exhibit significant upregulation in that specific cell population compared to other cells. Based on marker gene expression, cell populations were manually identified to construct sepsis-related single-cell libraries. Transcriptome sequencing of the five cell samples revealed that among these populations, groups 3 and 5 represented macrophages, group 4 represented natural killer cells, and groups 1, 2, 6, and 8 were T cells. Additionally, group 7 represented B cells, and group 9 represented platelets [19](Fig. 8A).

The evaluation of data quality and the analysis of differential RNA expression. (A) Box plots show that after normalization, the data in each sample exhibited a consistent distribution, making them comparable. (B) PCA illustrates the significant differences between the two cohorts and the lack of atypical data points. (C) Volcano plots showing downregulated (blue) and upregulated (red) genes. Fold Change values are plotted on the horizontal axis, and P values are plotted on the vertical axis. (D) Histograms showing upregulated (red) and down-regulated (blue) genes screened under FDR < 0.05 and FC ? 2. The horizontal axis indicates the number of differentially expressed RNAs; the vertical axis indicates groups
Molecular docking
Based on the results of the PPI analysis, the “active component-target-disease” network, and survival curves, core proteins were selected for further investigation. Autodock Vina 1.2.2, a widely used molecular docking software, assessed the binding energy and interaction mode between the core proteins and Rehmannia glutinosa active components. Protein-ligand docking is a computational method that simulates the interaction between a small molecule (ligand) and a protein target. The 3D structure of each active component was retrieved from the PubChem database [20] (https://pubchem.ncbi.nlm.nih.gov/), while the protein structures were obtained from the Protein Data Bank database [21] (https://www.rcsb.org/). In preparation for docking simulations, protein and ligand files were converted to the PDBQT format. This involved standard protein preparation steps such as eliminating water molecules and adding polar hydrogen atoms. The structural domains of each protein were then enclosed by cubic lattice boxes, allowing ligand molecules to explore the binding site freely. The docking pocket was defined as a 30 Å × 30 Å × 30 Å box with a grid spacing of 0.05 nm. Docking simulations were performed using Autodock Vina 1.2.2 (http://autodock.scripps.edu/) to determine the binding energies of the ligand-protein complexes. The results were visualized using PyMOL 2.4.1 to analyze the binding interactions and conformations. These visualizations included 3D structural representations, highlighting key interactions such as hydrogen bonds, hydrophobic contacts, and pi-pi stacking interactions between the active components and the target proteins.
Molecular dynamics simulation
To further validate the reliability of the docking protocol, this study employed GROMACS 2022.4 software to perform all-atom molecular dynamics simulations on the protein-ligand complexes obtained from molecular docking [22]. The protein component was parameterized using the Amber14SB force field. Topology files for the small molecule ligands were generated using ACPYPE and Antechamber programs. A cubic solvent box with a minimum distance of 1 nm from the system’s edges to the complex was employed. The TIP3P water model was used, and sodium and chloride ions were added to neutralize the overall charge. The energy minimization process utilized the steepest descent method. Temperature (300 K) and pressure (101.325 kPa) were controlled using NVT and NPT ensembles, respectively, to ensure system stability throughout the simulations. For each equilibrated system, a 100 ns MD simulation was conducted at 300 K, resulting in 10,000 frames of simulated trajectory data. Key parameters such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and total number of hydrogen bonds between protein and ligand were analyzed using the trajectory files obtained from the simulations.
Results
Screening of differentially expressed genes
This investigation into differentially expressed RNAs employed both mRNA localization and principal component analysis. Notably, the analysis of sepsis and control samples revealed consistent patterns with no outliers identified, as illustrated in Fig. 2A-D. Consequently, this analysis identified 4501 differentially expressed genes, including 2447 upregulated and 2054 downregulated genes.

Screening of active component targets of Rehmannia glutinosa. (A) In this Venn diagram, purple shading indicates 567 active targets, while blue indicates 4329 differentially expressed RNAs associated with sepsis. The central region consists of 135 overlapping targets representing potentially effective targets of Rehmannia glutinosa for treating sepsis. (B) In this network, the circular nodes on the left side represent the 135 cross-targets, the right diamond nodes represent the 32 active compounds of Rehmannia glutinosa, the inverted triangle nodes represent sepsis, and the lines indicate the interactions of the components with the targets
Exploration of the active components and targets of Rehmannia glutinosa
Our investigation into the active components and potential therapeutic targets of Rehmannia glutinosa for sepsis began by identifying its bioactive components. We retrieved information on Rehmannia glutinosa from the HERB database, identifying a total of 33 bioactive components. Next, we employed the SwissTargetPrediction database to predict potential target proteins associated with these bioactive components. This comprehensive analysis yielded a total of 1095 candidate target proteins. Following the removal of duplicate entries, a final set of 567 potential targets specific to Rehmannia glutinosa was established. In parallel, we obtained a list of 4329 targets associated with sepsis through independent means. To identify potential therapeutic targets at the intersection of Rehmannia glutinosa bioactivity and sepsis, a Venn diagram was generated (Fig. 3A). This analysis revealed 135 genes representing the overlap between the 567 targets linked to Rehmannia glutinosa’s active components and the 1069 targets associated with sepsis. These co-occurring genes were then analyzed and presented alongside their corresponding active components and properties in Table 2.

GO enrichment analysis of cross-targets. (A) Intersected targets are intersected targets primarily engaged in biological processes such as phosphorylation, modulation of inflammatory response, and inflammatory response. (B) The color shade of each node reflects its P-value magnitude. (C) Different colors of each node correspond to different biological processes (cluster ID)
Construction of a network of sepsis target active compounds
A PPI network was constructed using Cytoscape (version 3.7.2) to visualize the interactions between the 135 genes common to Rehmannia glutinosa and sepsis, along with their corresponding active compounds. Circular nodes represented these common genes, diamond nodes denoted active compounds of Rehmannia glutinosa, and inverted triangle nodes indicated genes associated with sepsis. Network analysis was performed using the Network Analyzer plugin. The size of each node was proportional to its degree of connectivity, with a higher number of connections indicating greater connectivity. Notably, ADORA2A, CA1, and AR emerged as the intersecting genes with the highest degree values, suggesting their potential importance in sepsis treatment. Among the active compounds, diincarvilone A exhibited the highest degree value, suggesting its potential significance as a key active component of Rehmannia glutinosa (Fig. 3B).
GO and KEGG enrichment analysis
A comprehensive functional profile was compiled based on the GO annotation of the 5799 identified genes. Four thousand four hundred seventy-one genes were associated with biological processes (BP), four hundred sixty-one with cellular components (CC), and eight hundred sixty-seven with molecular functions (MF). Key biological processes included phosphorylation, modulation of inflammatory response, and inflammatory response itself (Fig. 4A). To further elucidate the relationships between these GO terms, a network diagram was generated. Terms with a similarity score exceeding 0.3 were connected by lines. The most statistically significant p-values from 20 clusters were selected, with each cluster limited to no more than 15 terms, ensuring no more than 250. The network was visualized using Cytoscape, with nodes representing GO terms. Initially, these nodes were colored by their cluster ID (Fig. 4C) and subsequently by their p-value (Fig. 4B). KEGG pathway analysis revealed that these genes were primarily involved in crucial pathways such as those associated with cancer, neuroactive ligand-receptor interactions, lipids and atherosclerosis, cellular senescence, and various signaling pathways (Fig. 5A). Five of these important pathways were visualized for a clearer understanding (Fig. 5B).

KEGG pathway analysis of cross-targets. The pathway data in the figure were obtained from the KEGG database (Kanehisa Laboratories: www.kegg.jp/kegg/kegg1.html). (A) the y-axis indicates the P value, and the x-axis indicates gene counts. The cross-targets are mainly involved in various signaling pathways such as cancer pathways, neuroactive ligand-receptor interactions, lipids and atherosclerosis, and cellular senescence, etc. (B) Five different colors on the right side represent the different P values corresponding to 5 signaling pathways, and different colors on the left side correspond to the FC values of the genes included in the pathways

PPI network of proteins. (A) CD81, LCK, PNP, ADAM17, IMPDH1, and PRKCD represent the central nodes. (B) Heatmap of six potential hub genes in the center of the PPI network (CD81, LCK, PNP, ADAM17, IMPDH1, and PRKCD). Higher and lower expression were indicated by red and green colors, respectively
PPI analysis
Following the removal of irrelevant data points, the PPI network was reduced to 103 nodes (Fig. 6A). This complex network identified CD81, LCK, PNP, ADAM17, IMPDH1, and PRKCD as central nodes. A heatmap (Fig. 7B) visualizes the expression levels of these six hub genes across different samples. Notably, these genes are associated with the regulation of inflammatory response and cellular senescence, suggesting their potential as therapeutic targets for Rehmannia glutinosa in treating sepsis.

Survival analysis for the 6 key targets. The plot shows survival time in days on the x axis and survival rate on the y axis. The green line corresponds to low mRNA samples, while the red line represents high mRNA samples. (A-F) In the group with high expression of LCK (A), IMPDH1 (B), ADAM17 (C), CD81 (D), PRKCD (E), and low expression of PNP (F), the survival rate was significantly higher, indicating a superior prognosis (P < 0.05)
Survival curve analysis
To assess the correlation between patient prognosis and the 135 cross-targets, this study employed the GSE65682 dataset [18] for prognostic analysis. Log-rank tests revealed that the expression levels of six genes (LCK, IMPDH1, ADAM17, CD81, PRKCD, and PNP) were associated with prognosis (Fig. 7A-F). The survival curves depict a more significant change in patient prognosis when the disparity in gene expression is greater. Notably, the group with high expression of LCK, IMPDH1, ADAM17, CD81, and PRKCD, and low expression of PNP, exhibited a significantly higher survival rate, indicating a superior prognosis. Therefore, LCK, IMPDH1, ADAM17, CD81, PRKCD, and PNP were identified as potential key targets for Rehmannia glutinosa therapy in sepsis.

Cell lineage localization of key genes. (A) Groups 3 and 5 are macrophages; group 4 represents natural killer cells; groups 1, 2, 6, and 8 are T cells. Groups 7 and 9 represent B cells and platelets, respectively. (B-G) ADAM17 and CD81 are widely distributed in immune cells. PRKCD, PNP, and IMPDH1 were mainly enriched in groups 3 and 5 (macrophage lineage). LCK was mainly enriched in group 6 (T-cell lineage). (H) The expression abundance values and proportions of each hub gene in different cell populations are shown in bubble plots
Single-cell RNA sequencing
Six key genes (LCK, CD81, PRKCD, IMPDH1, ADAM17, and PNP) were analyzed in single-cell libraries to determine their cellular localization patterns. The results revealed distinct cell type distributions (Fig. 8B-H). ADAM17 and CD81 exhibited widespread expression across various immune cell types. In contrast, PRKCD, PNP, and IMPDH1 were primarily enriched in macrophage lineages, specifically Groups 3 and 5. LCK, on the other hand, was predominantly localized in T-cell lineages, particularly Group 6.
Molecular docking
Following the preceding analyses, the major active components of Rehmannia glutinosa, including rehmaionoside a, rehmapicrogenin, diincarvilone A, trans-p-hydroxy cinnamic acid methyl ester, and Rehmaglutin C, were selected for molecular docking simulations. Docking simulations were performed against ADAM17 (Fig. 9A), PNP (Fig. 9E), CD81 (Fig. 9B), IMPDH1 (Fig. 9C), LCK (Fig. 9D), and PRKCD (Fig. 9F), respectively. In these figures, affinity values below − 4.25 kcal/mol indicate potential binding activity, values below − 5.0 kcal/mol indicate strong binding activity, and values below − 7.0 kcal/mol suggest significant docking interactions between the ligand and the target protein [23]. The detailed molecular docking results for these active components and target proteins are presented in Table 3.

Molecular docking results. (A) rehmaionoside a binds to ADAM17 with a binding affinity of − 5.775 kcal-mol− 1. (B) rehmapicrogenin binds to CD81 with a binding affinity of − 5.074 kcal-mol− 1. (C) diincarvilone A binds to IMPDH1 with a binding affinity of − 6.916 kcal-mol− 1. (D) trans-p-hydroxy cinnamic acid methyl ester has a binding affinity of − 7.641 kcal-mol− 1 for LCK. (E) rehmaionoside a has a binding affinity for PNP of -6.437 kcal-mol− 1. (F) Rehmaglutin C has a binding affinity for PRKCD of -4.327 kcal-mol− 1
Molecular dynamics simulation
Following a 20 ns molecular dynamics simulation, the RMSD-time curve plateaus, indicating stabilization (Fig. 10A). The RMSD values for rehmaionoside a-ADAM17, rehmapicrogenin-CD81, trans-p-hydroxy cinnamic acid methyl ester-LCK, diincarvilone A-IMPDH1, and rehmaionoside a-PNP all stabilized between 0 and 0.5 nm. The RMSD value of Rehmaglutin C-PRKCD fluctuated within a range of 0.5 to 1.2 nm. The Rg values of all complexes exhibited approximately horizontal lines in Fig. 10B, signifying relative stability throughout the simulation. During the simulation process, most complexes displayed fluctuations in their RMSF values within a range of 0 to 1 nm (Fig. 11A-F). We observed dynamic formation and breaking of hydrogen bonds throughout the simulations. However, most complexes maintained a consistent presence of hydrogen bonds, with rehmaionoside a-ADAM17 and trans-p-hydroxy cinnamic acid methyl ester-LCK exhibiting the most stable hydrogen bonding patterns (Fig. 12A-F). Overall, these results strongly support the effectiveness of the docking protocols and suggest favorable conformational stability for these six complexes.

RMSD and Rg during Molecular Dynamics Simulations. (A) RMSD can indicate structural conformational changes and systemic stability of complexes. (B) The Rg value can indicate the tightness of the complex system, which can reflect the degree of protein folding

RMSF-Residue number curve (A-F) RMSF can be used to indicate the fluctuation of the complex at the residue level, reflecting the flexibility of the protein

Hydrogen bond number-time curve. (A-F) Hydrogen bond (H-bond) is a strong non-covalent interaction that can, to some extent, reflect the binding strength between the ligand and the receptor
Discussion
Sepsis is a life-threatening condition characterized by organ dysfunction arising from a dysregulated host response. This response can progress to severe sepsis and septic shock [24]. Western medicine recognizes that sepsis-induced immunosuppression results from disrupted immune homeostasis. This disruption manifests as the release of anti-inflammatory cytokines, excessive expansion of immunosuppressive cells, abnormal death of immune effector cells, and the expression of immune checkpoints [25]. For over 2,000 years, Traditional Chinese Medicine (TCM) has been vital in managing infectious diseases in Eastern cultures. Scientific evidence increasingly demonstrates that various TCM agents can attenuate sepsis-induced organ dysfunction through mechanisms including inhibiting inflammatory responses, enhancing immunity, reducing oxidative stress, and maintaining cellular homeostasis [26]. This study employed an integrative approach that combined network pharmacology, single-cell sequencing, molecular docking, and molecular dynamics simulation. We aimed to screen the active components of Rehmannia glutinosa and identify potential therapeutic targets for sepsis. By unraveling the immunomodulatory effects of Rehmannia glutinosa, we hope to gain insights and directions for novel drug design and development. This, in turn, could pave the way for personalized treatment strategies for sepsis and facilitate a deeper understanding of how the active components of Rehmannia glutinosa interact with their targets. Ultimately, this study sought to elucidate the therapeutic mechanisms of Rehmannia glutinosa in treating sepsis. By providing a crucial scientific basis and theoretical foundation, we hope to contribute to personalized treatment strategies, drug development, and successful clinical translation of this traditional medicine.
Current scientific research suggests that Rehmannia glutinosa exerts beneficial effects for various significant diseases. It enhances the intestinal epithelial barrier, induces dendritic cell maturation, inhibits ferroptosis, and activates the PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathways. These mechanisms contribute to preventing colitis, exhibiting anticancer properties, and improving cognitive dysfunction after cerebral ischemia [27,28,29]. Rehmannia glutinosa also suppresses the expression of pro-inflammatory genes (TNF-α, MCP-1, IP-10, COX-2, and iNOS) and promotes the activation of natural killer cells in mice [30, 31], suggesting its efficacy in anti-inflammation and immunomodulation, potentially applicable in the treatment and prognosis improvement of sepsis. Previous ex vivo and in vivo studies support the role of Rehmannia glutinosa in inflammatory response and immune modulation. For instance, Feng et al. demonstrated that oral administration of its polysaccharides significantly improved immune response and antioxidant capacity in common carp, enhancing their resistance to infection [32]. Similarly, another study demonstrated the protective effects of rhizoma geophyllum oligosaccharides (RGO) against intestinal inflammation and barrier damage in mice [33].These findings, highlighting modulation of gut flora, reduction of inflammatory factors, and enhanced antioxidant capacity, align with our results and further support the potential of Rehmannia glutinosa in sepsis treatment.
However, scientific research on treating sepsis with Rehmannia glutinosa is relatively scarce, and the mechanism by which it improves prognosis remains unclear. This study identified the effective components of Rehmannia glutinosa for sepsis treatment through public databases, potentially paving the way for new directions in the clinical management of sepsis.
ADAM17 considered the first line of defense against injury, promotes inflammation and subsequent repair of skin and intestinal barriers. It maintains epidermal barrier function by releasing tumor necrosis factor-alpha to induce inflammatory responses and epidermal growth factor receptor ligands [34]. ADAM17 cleaves substrates like interleukin-6 receptor, heparin-binding EGF-like growth factor, and hepatocyte growth factor receptor c-Met [35]. Interestingly, survival curve analysis indicated a correlation between increased ADAM17 expression and a better prognosis for sepsis patients, suggesting that elevated ADAM17 expression may be favorable in this population. RNA-seq analysis revealed widespread distribution of ADAM17 across different immune cells. Furthermore, network pharmacological evaluation identified rehmaionoside a, an active component of Rehmannia glutinosa, as a potential binder to ADAM17 and a compound with antimicrobial properties.
CD81, a regulator of immune synapses, receptor aggregation, and signaling, also mediates both adaptive and innate immunosuppression [36]. In vitro studies have demonstrated that CD81 overexpression in human CD4+ T cells promotes the development of Th9 cells [37]. Survival curve analysis indicated that increased CD81 expression in the group with a better prognosis correlates with longer survival, suggesting that elevated CD81 expression may be beneficial in sepsis. RNA-seq analysis further revealed that CD81 is widely expressed in different immune cells. Network pharmacological evaluation identified rehmapicrogenin, an active component of Rehmannia glutinosa, as a compound that targets CD81 and exhibits antimicrobial properties.
Inosine monophosphate dehydrogenase 1 (IMPDH1), a rate-limiting enzyme crucial for guanine nucleotide synthesis via conversion of 5’-inosine to xanthine, plays an essential role in lymphocyte antigen antagonism. IMPDH1 is typically associated with immune cells, immune checkpoints, and immune-related genes and pathways within the tumor immune microenvironment [38]. Interestingly, survival curve analysis revealed upregulated IMPDH1 expression in the group with a better prognosis, suggesting potential benefits in sepsis. RNA-seq analysis localized IMPDH1 enrichment primarily within the macrophage lineage. Network pharmacological analysis identified diincarvilone A, an active ingredient of Rehmannia glutinosa, as a potential binder to IMPDH1 and a compound with antimicrobial properties.
Purine nucleoside phosphorylase (PNP) is an intracellular enzyme that converts dGTP to guanine. Mutations in the PNP gene are known to cause immunodeficiency [39]. It also plays a crucial role in the purine salvage pathway, and its deficiency can result in severe combined immunodeficiency [40]. Interestingly, survival curve analysis in our study indicated that reduced PNP expression was associated with a better prognosis in sepsis, suggesting that decreased PNP expression may be beneficial. RNA-seq analysis revealed that PNP is mostly localized in the macrophage lineage. Network pharmacological evaluation identified rehmaionoside a, an active ingredient of Rehmannia glutinosa, as a potential target for PNP and a compound with antibacterial properties.
Protein kinase C delta (PRKCD), a member of a novel class of calcium-dependent PKCs, can promote the development of chronic obstructive pulmonary disease by inducing mitochondrial dysfunction, inflammatory responses, apoptosis, and MUC5AC hypersecretion. Additionally, PRKCD plays a crucial role in diseases like X-linked agammaglobulinemia and mediates platelet-induced tumor progression [41, 42]. In the present study, survival analysis demonstrated that increased PRKCD expression was associated with a better prognosis, suggesting a potentially beneficial role in sepsis. RNA-seq analysis revealed that PRKCD is predominantly localized in the macrophage cell lineage. Rehmaglutin C, an active ingredient of Rehmannia glutinosa, was found to target PRKCD and exhibit antimicrobial properties according to network pharmacological analysis.
Lymphocyte-specific protein tyrosine kinase (LCK) serves as a positive regulator of inflammatory signaling [43]. Research has identified LCK as a potential new biomarker for sepsis based on ROC analysis [44]. Survival analysis indicated increased LCK expression was associated with a better prognosis, suggesting a favorable role. Furthermore, RNA-seq analysis revealed that LCK was predominantly localized in natural killer and T cells. Network pharmacological analysis identified trans-p-hydroxy cinnamic acid methyl ester, an active component of Rehmannia glutinosa, as capable of targeting LCK and exhibiting antimicrobial properties.
LCK, CD81, PRKCD, IMPDH1, ADAM17, and PNP, identified through our analysis, are involved in various biochemical reactions critical for immunity, inflammation, and metabolism, playing a role in the development and progression of several disease categories like immune disorders, inflammatory conditions, and metabolic imbalances. The mechanisms by which these genes influence sepsis remain unclear, and limited research has explored their specific roles. Our study employed single-cell sequencing technology to determine the cellular localization of these genes, revealing their high expression in immune cells. Furthermore, we analyzed public databases and validated the correlation between their expression and clinical prognosis in sepsis patients. These findings highlight LCK, CD81, PRKCD, IMPDH1, ADAM17, and PNP as potential therapeutic targets for sepsis and pave the way for further investigation into their mechanisms of action in this disease.
The molecular docking results showed binding affinities below − 4.25 kcal/mol for several complexes: trans-p-hydroxy cinnamic acid methyl ester-LCK, rehmapicrogenin-CD81, diincarvilone A-IMPDH1, rehmaionoside a-ADAM17, and rehmaionoside a-PNP, as well as Rehmaglutin C-PRKCD. Among these, trans-p-hydroxy cinnamic acid methyl ester-LCK displayed the lowest binding energy (-7.641 kcal·mol⁻¹). Furthermore, molecular dynamics simulations revealed excellent stability for trans-p-hydroxy cinnamic acid methyl ester-LCK, rehmaionoside a-ADAM17, and rehmaionoside a-PNP. These complexes exhibited consistently low RMSD, RMSF, and Rg values while maintaining stable and continuous hydrogen bonds. No dissociation of the complexes was observed during the simulations. These results strongly suggest robust and stable binding between these active compounds and their targets, providing a solid foundation for further investigating these complexes.
This study employed single-cell sequencing technology to gain a deeper understanding of individual differences and dysregulated immune responses in sepsis patients. This information can be used to develop personalized treatment strategies, potentially improving treatment efficacy and prognosis. Additionally, network pharmacology analysis identified potential targets and signaling pathways of Rehmannia glutinosa, supporting its potential for clinical application. Furthermore, molecular docking and dynamics simulations provided novel insights into the binding between Rehmannia glutinosa and its targets, shedding light on the pathogenesis of sepsis and offering new avenues for disease treatment and prevention. By integrating these advanced technologies, this study presents promising prospects for future research and clinical applications.
This study acknowledges limitations inherent to the network pharmacology method, which relies on bioinformatic analysis from publicly available databases. Clinical trial validation and evidence-based medicine studies are necessary to fully understand the pharmacological effects of Rehmannia glutinosa. Additionally, further mechanistic cellular experiments are warranted to verify the inhibitory or promotional effects of small molecules binding to these key targets.
Conclusions
-
1.
Elevated levels of LCK, CD81, PRKCD, IMPDH1, and ADAM17, along with decreased levels of PNP, are associated with improved survival in septic patients.
-
2.
The expression of LCK, IMPDH1, CD81, PRKCD, ADAM17, and PNP is influenced by trans-p-hydroxy cinnamic acid methyl ester, diincarvilone A, rehmapicrogenin, Rehmaglutin C, and rehmaionoside a.
-
3.
Our findings suggest that compounds like diincarvilone A, rehmapicrogenin, Rehmaglutin C, trans-p-hydroxy cinnamic acid methyl ester, and rehmaionoside a may improve survival and exhibit antimicrobial properties in septic patients.
-
4.
Strong binding was observed between trans-p-hydroxy cinnamic acid methyl ester and LCK, rehmapicrogenin and CD81, diincarvilone A and IMPDH1, rehmaionoside a and ADAM17, and rehmaionoside a and PNP, indicating potential interactions. Rehmaglutin C also exhibited binding activity with PRKCD, further supporting its potential therapeutic role.
Data availability
The CNGBdb repository contains the datasets that were analyzed for this study, (https://db.cngb.org/search/project/CNP0002611/).
References
Huang M, Cai S, Su J. The pathogenesis of Sepsis and potential therapeutic targets. Int J Mol Sci 2019, 20(21).
Fan T, Cheng B, Fang X, Chen Y, Su F. Application of Chinese Medicine in the management of critical conditions: a review on Sepsis. Am J Chin Med. 2020;48(06):1315–30.
Z ZG, DI H R Hgyjlbhfct, Q FS. Research progress of traditional Chinese medicine inprevention and treatment of sepsis. China J Chin Materia Med. 2017;42(8):1423–9.
Bian Z, Zhang R, Zhang X, Zhang J, Xu L, Zhu L, Ma Y, Liu Y. Extraction, structure and bioactivities of polysaccharides from Rehmannia glutinosa: a review. J ETHNOPHARMACOL. 2023;305:116132.
Yang AL, Wu Q, Hu ZD, Wang SP, Tao YF, Wang AM, Sun YX, Li XL, Dai L, Zhang J. A network pharmacology approach to investigate the anticancer mechanism of cinobufagin against hepatocellular carcinoma via downregulation of EGFR-CDK2 signaling. Toxicol Appl Pharm. 2021;431:115739.
Cheng C, Chen W, Jin H, Chen X. A review of single-cell RNA-Seq annotation, integration, and cell-cell communication. Cells-Basel 2023, 12(15).
Wu X, Xu LY, Li EM, Dong G. Application of molecular dynamics simulation in biomedicine. CHEM BIOL DRUG DES. 2022;99(5):789–800.
Stanzione F, Giangreco I, Cole JC. Use of molecular docking computational tools in drug discovery. Prog Med Chem. 2021;60:273–343.
Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics. 2018;19(1):524–34.
Fang S, Dong L, Liu L, Guo J, Zhao L, Zhang J, Bu D, Liu X, Huo P, Cao W, et al. HERB: a high-throughput experiment- and reference-guided database of traditional Chinese medicine. Nucleic Acids Res. 2021;49(D1):D1197–206.
Gfeller D, Grosdidier A, Wirth M, Daina A, Michielin O, Zoete V. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids RES 2014, 42(Web Server issue):W32–8.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587–92.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51.
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–12.
Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–9.
Scicluna BP, Klein Klouwenberg PMC, van Vught LA, Wiewel MA, Ong DSY, Zwinderman AH, Franitza M, Toliat MR, Nürnberg P, Hoogendijk AJ, et al. A Molecular Biomarker to Diagnose Community-acquired Pneumonia on Intensive Care Unit Admission. Am J Resp Crit Care. 2015;192(7):826–35.
Wang C, Li S, Shen Y, Li Y, Chen M, Wang Y, Lan Y, Hu Y. Mechanisms of Panax Ginseng on treating Sepsis by RNA-Seq technology. Infect Drug Resist. 2022;15:7667–78.
Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Zhou Z, Han L, Karapetyan K, Dracheva S, Shoemaker BA, et al. PubChem’s BioAssay Database. Nucleic Acids Res. 2012;40(D1):D400–12.
Xu W, Velankar S, Patwardhan A, Hoch JC, Burley SK, Kurisu G. Announcing the launch of Protein Data Bank China as an associate member of the Worldwide Protein Data Bank Partnership. Acta Crystallogr D. 2023;79(Pt 9):792–5.
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–18.
Hsin K, Ghosh S, Kitano H. Combining machine Learning systems and multiple docking Simulation packages to improve docking prediction reliability for Network Pharmacology. Plos One. 2014;8(12):e83922.
Srzić I. Sepsis definition: what’s new in the Treatment guidelines. Acta Clin Croat; 2022.
Liu D, Huang SY, Sun JH, Zhang HC, Cai QL, Gao C, Li L, Cao J, Xu F, Zhou Y, et al. Sepsis-induced immunosuppression: mechanisms, diagnosis and current treatment options. Military Med Res. 2022;9(1):56.
Song Y, Lin W, Zhu W. Traditional Chinese medicine for treatment of sepsis and related multi-organ injury. Front Pharmacol. 2023;14:1003658.
Wang Y, Kwak M, Lee PC, Jin JO. Rehmannia Glutinosa polysaccharide promoted activation of human dendritic cells. Int J Biol Macromol. 2018;116:232–8.
Lv H, Jia H, Cai W, Cao R, Xue C, Dong N. Rehmannia Glutinosa polysaccharides attenuates colitis via reshaping gut microbiota and short-chain fatty acid production. J Sci Food Agr. 2023;103(8):3926–38.
Fu C, Wu Y, Liu S, Luo C, Lu Y, Liu M, Wang L, Zhang Y, Liu X. Rehmannioside A improves cognitive impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway after ischemia. J Ethnopharmacol. 2022;289:115021.
Xu L, Zhang W, Zeng L, Jin JO. Rehmannia Glutinosa polysaccharide induced an anti-cancer effect by activating natural killer cells. Int J Biol Macromol. 2017;105(Pt 1):680–5.
Baek GH, Jang YS, Jeong SI, Cha J, Joo M, Shin SW, Ha KT, Jeong HS. Rehmannia Glutinosa suppresses inflammatory responses elicited by advanced glycation end products. Inflammation. 2012;35(4):1232–41.
Feng JC, Cai ZL, Zhang XP, Chen YY, Chang XL, Wang XF, Qin CB, Yan X, Ma X, Zhang JX, et al. The effects of oral Rehmannia Glutinosa Polysaccharide Administration on Immune responses, antioxidant activity and Resistance against Aeromonas hydrophila in the common carp, Cyprinus carpio L. Front Immunol. 2020;11:904.
Li X, Gui R, Wang X, Ning E, Zhang L, Fan Y, Chen L, Yu L, Zhu J, Li Z, et al. Oligosaccharides isolated from Rehmannia Glutinosa protect LPS-induced intestinal inflammation and barrier injury in mice. Front Nutr. 2023;10:1139006.
Matthews AL, Noy PJ, Reyat JS, Tomlinson MG. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets 2017, 28(4):333–341.
Al-Salihi M, Bornikoel A, Zhuang Y, Stachura P, Scheller J, Lang KS, Lang PA. The role of ADAM17 during liver damage. Biol Chem. 2021;402(9):1115–28.
Vences-Catalán F, Duault C, Kuo C, Rajapaksa R, Levy R, Levy S. CD81 as a tumor target. Biochem Soc T. 2017;45(2):531–5.
Zhao W, Tan C, Yu X, Yu R, Mei Q, Cheng Y. A 7-Amino Acid Peptide Mimic from Hepatitis C Virus Hypervariable Region 1 Inhibits Mouse Lung Th9 Cell Differentiation by Blocking CD81 Signaling during Allergic Lung Inflammation. J Immunol Res 2020, 2020:4184380.
Liu C, Zhang W, Zhou X, Liu L. IMPDH1, a prognostic biomarker and immunotherapy target that correlates with tumor immune microenvironment in pan-cancer and hepatocellular carcinoma. Front Immunol. 2022;13:983490.
Davenne T, Rehwinkel J. PNP inhibitors selectively kill cancer cells lacking Samhd1. Mol Cell Oncol. 2020;7(6):1804308.
Torun B, Bilgin A, Orhan D, Gocmen R, Kılıc SS, Kuskonmaz B, Cetinkaya D, Tezcan I, Cagdas D. Combined immunodeficiency due to purine nucleoside phosphorylase deficiency: outcome of three patients. Eur J Med Genet. 2022;65(3):104428.
Li X, Zhao K, Lu Y, Wang J, Yao W. Genetic analysis of platelet-related genes in Hepatocellular Carcinoma reveals a Novel Prognostic signature and determines PRKCD as the potential Molecular Bridge. Biol Proced Online. 2022;24(1):22.
Li S, Huang Q, Zhou D, He B. PRKCD as a potential therapeutic target for chronic obstructive pulmonary disease. Int Immunopharmacol. 2022;113:109374.
Kim DH, Park JW, Jeong HO, Lee B, Chung KW, Lee Y, Jung HJ, Hyun MK, Lee AK, Kim BM, et al. Novel role of Lck in Leptin-Induced inflammation and implications for renal aging. Aging Dis. 2019;10(6):1174–86.
Gong F, Ji R, Wang Y, Yang Z, Chen Y, Mao E, Chen E. Identification of potential biomarkers and Immune features of Sepsis using Bioinformatics Analysis. Mediat Inflamm. 2020;2020:1–12.
Acknowledgements
Not applicable.
Funding
Not applicable.
Ethics declarations
Human Ethics approval and consent to participate
Each patient and their family members voluntarily participated in this study and signed an informed consent form. The study was approved by the Ethics Committee of the Affiliated Hospital of Southwest Medical University (No. 1. ky2018029), Clinical Trial No.:ChiCTR1900021261,Registration Date: February 4, 2019.The research was conducted in accordance with the Declaration of Helsinki.
Consent for publication
All authors agreed to publish the study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Topic: Luzhou-Southwest Medical University joint project (2021LZXNYD-J13); Funding Support for the Construction of Key Clinical Specialties in Sichuan Province.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, H., Laram, Y., Hu, L. et al. Exploring the potential mechanisms of Rehmannia glutinosa in treating sepsis based on network pharmacology. BMC Infect Dis 24, 893 (2024). https://doi.org/10.1186/s12879-024-09796-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-024-09796-x